

Abstract
There is strong evidence for the use of angiotensin converting enzyme inhibitors and beta‐blockers to reduce morbidity and mortality in patients with myocardial infarction (MI), whereas the effect of angiotensin receptor blockers is less clear. We evaluated the effects of an angiotensin receptor blocker losartan and a beta‐blocker metoprolol on left ventricular (LV) remodeling, c‐kit+ cells, proliferation, fibrosis, apoptosis, and angiogenesis using a model of coronary ligation in rats. Metoprolol treatment for 2 weeks improved LV systolic function. In contrast, losartan triggered deleterious structural remodeling and functional deterioration of LV systolic function, ejection fraction being 41% and fractional shortening 47% lower in losartan group than in controls 2 weeks after MI. The number of c‐kit+ cells as well as expression of Ki‐67 was increased by metoprolol. Losartan‐induced thinning of the anterior wall and ventricular dilation were associated with increased apoptosis and fibrosis, while losartan had no effect on the expression of c‐kit or Ki‐67. Metoprolol or losartan had no effect on microvessel density. These results demonstrate that beta‐blocker treatment attenuated adverse remodeling via c‐kit+ cells and proliferation, whereas angiotensin receptor blocker‐induced worsening of LV systolic function was associated with increased apoptosis and fibrosis in the peri‐infarct region.
Keywords: myocardial infarction, heart failure, remodeling, cells
Introduction
The activation of the renin–angiotensin–aldosterone system and sympathetic hyperactivation play central roles in pathogenesis of postinfarction left ventricular (LV) remodeling and heart failure. This understanding has provided strong therapeutic rationale for using angiotensin converting enzyme (ACE) inhibitors and beta‐blockers in patients with myocardial infarction (MI). 1 , 2 However, data supporting the use of angiotensin receptor blockers (ARBs) for the primary prevention of myocardial infarction are generally weak and, in some cases, negative. 3 , 4 , 5 In the VALUE trial in hypertensive patients, the ARB valsartan‐based regimen had a significantly higher (19%, p < 0.02) incidence of myocardial infarction than the calcium channel blocker amlodipine‐based regimen. 6 In the recently published ONTARGET trial in patients with vascular disease or high‐risk diabetes, myocardial infarction occurred in 440 patients (5.2%) in the ARB telmisartan group and in 413 patients (4.8%) in the ACE inhibitor ramipril group (relative risk, 1.07, 0.94–1.22), telmisartan being equivalent to ramipril for the prespecified primary outcome of death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure. 7 As compared with a beta‐blocker in the LIFE study, myocardial infarction occurred in 198 ARB losartan‐treated and 188 beta‐blocker atenolol‐treated hypertensive patients (relative risk, 1.07, 0.88–1.31), while the stroke outcome was highly in favor of losartan, showing a 24.9% relative risk reduction compared with atenolol. 8
In view that the randomized, controlled trials have convincingly demonstrated that beta‐blockers reduce rates of myocardial infarction and improve LV structure and function after MI, 1 , 2 , 5 we evaluated the effects of a beta‐blocker, metoprolol, and an ARB, losartan, on the progression of LV remodeling and function in rats subjected to MI by ligating the left anterior descending artery (LAD). Since these experiments revealed that early treatment with metoprolol improved and losartan worsened LV systolic function, we tested the hypothesis that metoprolol and losartan may divergently affect apoptosis, proliferation, fibrosis, angiogenesis, or the number of cardiac stem cells (CSCs) in the LV after MI. Our results demonstrate that the impairment of cardiac function and structure with the ARB was associated with increased apoptosis and fibrosis whereas beta‐blocker treatment attenuated adverse LV remodeling via c‐kit+ cells and cellular proliferation.
Methods
Myocardial infarction, drug treatments with osmotic minipumps and echocardiography
MI was produced by ligation of the LAD. 9 Beta‐blocker metoprolol (1.5 mg/kg/h) or angiotensin II type 1 (AT1) receptor antagonist losartan (400 μg/kg/h) was administered via osmotic minipumps. Transthoracic echocardiography was performed 1 day, 2 weeks or 4 weeks after operation using Acuson Ultrasound System (Sequoia™ 512) and a 15‐MHz linear transducer (15L8) (Acuson, Mountain View, CA). A group of rats was sacrificed at each time point, hearts were removed and the chambers separated. LV tissue samples were fixed in 10% neutral buffered formalin, embedded in paraffin, cut into 5 μm sections and mounted on slides. Methods are described in detail in the online‐only Supporting Information.
Results
Metoprolol but not losartan improves LV function and remodeling after MI
To compare the effect of metoprolol and losartan on postinfarction remodeling, the LAD was ligated in rats. MI progressively decreased systolic function and caused dilatation of the left ventricle, as assessed by echocardiography during the 4‐week follow‐up period (Supporting Table 1). The LV dilatation, thinning of the anterior wall, and hypertrophy of the posterior wall were also seen in hematoxylin‐eosin stained myocardial sections (Supporting Figure 1A). The size of the infarcted area, measured from histological sections stained with Masson's trichrome as area of fibrosis of LV circumference (Supporting Figure 1B), was 32 ± 9% (n= 3), 40 ± 2% (n= 5) and 40 ± 4% (n= 5) 1 day, 2 weeks and 4 weeks after MI, respectively. These functional and morphological changes were accompanied by an increase in A‐type and B‐type natriuretic peptide gene expression in the left ventricle (data not shown).
Metoprolol treatment for 2 weeks improved LV systolic function ( Figure 1A‐C ). LV ejection fraction and fractional shortening increased 32% (p < 0.05) and 47% (p < 0.05), respectively, in metoprolol‐treated rats compared with the vehicle‐treated animals. In contrast, treatment with losartan resulted in a significant functional deterioration of LV systolic function, ejection fraction being 41% (p < 0.01) and fractional shortening 47% (p < 0.05) lower in losartan group than in controls 2 weeks after MI ( Figure 1A‐C ). Consistently with the opposite effects on LV systolic function, losartan treatment intensified the dilatation of the left ventricle (p < 0.05), whereas metoprolol treatment attenuated (p < 0.05) it, as assessed by hematoxylin‐eosin stained myocardial sections ( Figure 2A ) or echocardiography ( Figure 2B ). Furthermore, the posterior wall was significantly thinner in losartan‐treated rats compared with metoprolol‐treated rats 2 weeks after MI (p < 0.05, Figure 2C ). Metoprolol and losartan treatments had no significant effect on infarct size, the area of fibrosis of LV circumference being 43 ± 3% (n= 6), 44 ± 3% (n= 5) and 40 ± 2% (n= 5) in metoprolol, losartan and vehicle groups, respectively ( Figure 2D ). Interestingly, the percentage of fibrotic area measured from infarct border zone was 2.3 fold higher (p < 0.05) in the losartan‐treated animals than in metoprolol‐treated animals ( Figure 2E ).
Figure 1.
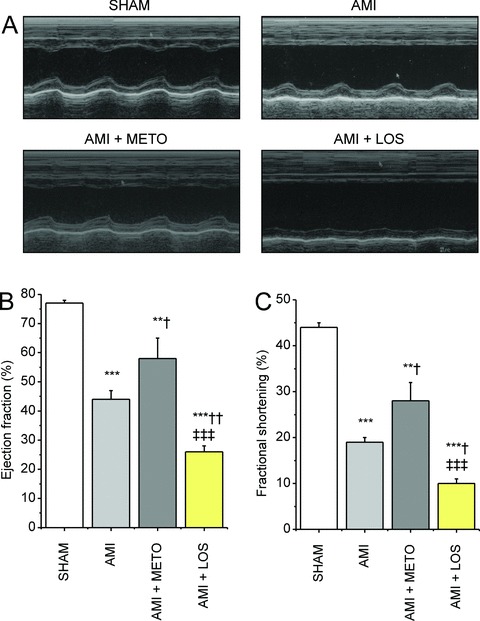
Metoprolol but not losartan improves LV systolic function after MI. Effects of drug treatments on cardiac function were examined by echocardiography 2 weeks after MI. (A) Representative M‐mode images from each experimental group are shown. (B) LV ejection fraction and (C) fractional shortening were significantly improved by metoprolol treatment. Results are mean ± SEM (n= 6/group). Data were analyzed by one‐way ANOVA followed with LSD post hoc test. AMI = acute myocardial infarction; METO = metoprolol; LOS = losartan.**p < 0.01,***p < 0.001 versus sham;† p < 0.05,†† p < 0.01 versus AMI;‡‡‡ p < 0.001 versus AMI + METO.
Figure 2.
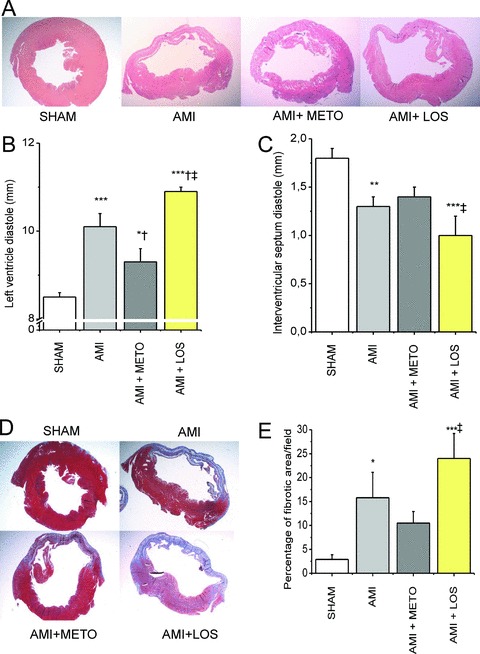
Metoprolol attenuates and losartan augments LV remodeling after MI. (A) LV sections stained with hematoxylin‐eosin. (B) LV diastolic diameter was significantly greater and (C) interventricular septum diastolic diameter smaller in losartan‐treated rats compared to vehicle‐treated rats. (D) Masson's trichrome technique was used to measure the size of MI and (E) define the area of fibrosis by analyzing the extent of positive staining. Results are mean ± SEM (n= 6/group). Data were analyzed by one‐way ANOVA followed with LSD post hoc test. AMI = acute myocardial infarction; METO = metoprolol; LOS = losartan.*p < 0.05,**p < 0.01,***p < 0.001 versus sham;† p < 0.05 versus AMI.‡ p < 0.05 versus AMI + METO.
Increased apoptosis in infarcted hearts with losartan treatment
Because losartan treatment augmented the deterioration of cardiac function, we analysed apoptosis in response to metoprolol and losartan treatments. By TUNEL analysis, the number of apoptotic cells and bodies in the LV increased 1.5‐fold at 1 day, 4.5‐fold (p < 0.05) at 2 weeks and 5.7‐fold (p < 0.01) at 4 weeks after MI ( Figure 3A‐B ). The results of TUNEL were supported by immunostaining for activated caspase‐3, which showed a similar increase in the number of apoptotic cells ( Figure 3C‐D ). A significant additional increase (p < 0.05) in apoptosis was observed with 2 weeks losartan treatment in infarcted hearts, as assessed by TUNEL ( Figure 4A‐C ). Apoptotic cells were not positive for cardiomyocyte marker alpha‐actinin ( Figure 4C ). There was also a tendency for the increase of activated caspase‐3 in the LV by losartan treatment, however, this change was not statistically significant ( Figure 4D‐E ). On the contrary, metoprolol treatment for 2 weeks had no effect on apoptosis ( Figure 4 ).
Figure 3.
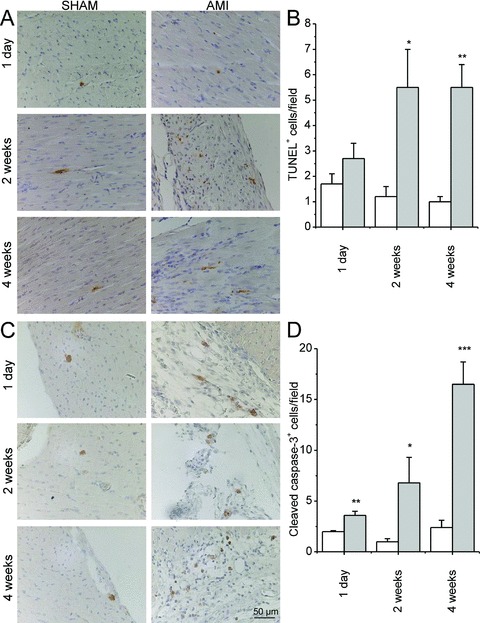
Apoptosis was assessed by TUNEL (A) and immunostaining for cleaved caspase‐3 (C) 1 day, 2 weeks and 4 weeks after MI. (B) Number of TUNEL+ cells/high power field. (D) Number of cleaved caspase‐3+ cells/high power field. White bars indicate sham and gray bars AMI group. Results are mean ± SEM (n= 5–6/group). AMI = acute myocardial infarction.*p < 0.05,**p < 0.01,***p < 0.001 versus AMI.
Figure 4.
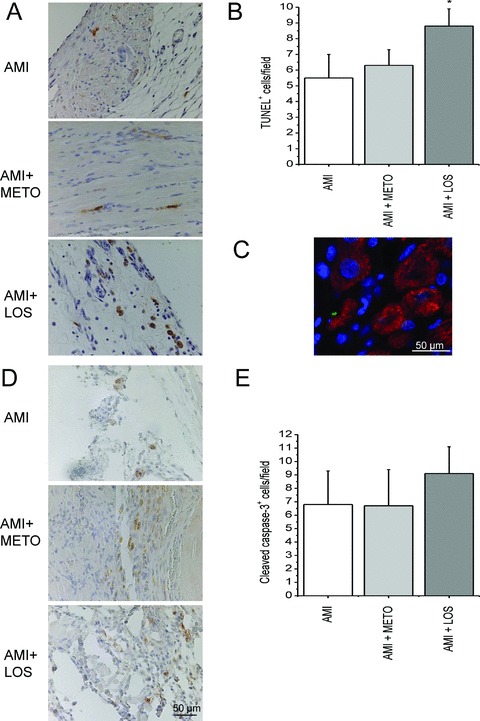
Losartan treatment increases apoptosis after MI. Apoptosis was assessed by TUNEL (A, B, C) and immunostaining for cleaved caspase‐3 (D, E) 2 weeks after MI. The apoptotic cells and bodies were counted in 5 high power fields (40× objective) from the peri‐infarct regions choosing hot spot areas in each sample. Peri‐infarct zone is an ischemic area adjacent to infarct and hot spot areas are areas where the number of apoptotic cells appeared maximal. (C) Immunofl uorescence staining showing that apoptotic cells are not cardiomyocytes (green; TUNEL, blue; DAPI, red; alpha‐actinin). AMI = acute myocardial infarction; METO = metoprolol; LOS = losartan. Results are mean ± SEM (n= 6/group). *p < 0.05 versus AMI.
Metoprolol increases the number of c‐kit positive cells after MI
We next tested the hypothesis that metoprolol and losartan treatments may have divergent effects on c‐kit+ cells during postinfarction LV remodeling. The number of c‐kit+ cells increased in the LV 3.5‐fold (p < 0.01) at 1 day, 4.2‐fold (p < 0.01) at 2 weeks and 13.3‐fold (p < 0.05) at 4 weeks after MI ( Figure 5A‐C ). Interestingly, 2 weeks metoprolol treatment further increased the number of c‐kit+ cells in the LV (1.9‐fold, p < 0.01), whereas it did not differ in infarcted hearts between losartan‐ and vehicle‐treated rats ( Figure 5D‐G ). Immunohistochemistry using c‐kit‐specific antibody showed that the number of c‐kit+ cells increased particularly in the border zone of the infarction by metoprolol treatment ( Figure 5H ).
Figure 5.
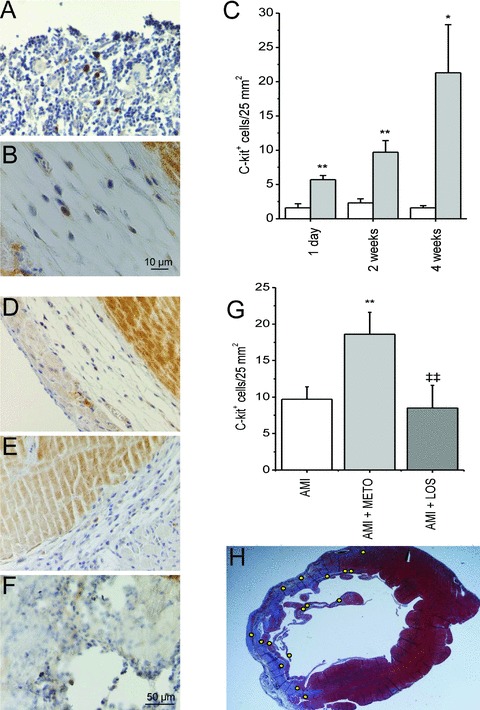
Metoprolol treatment for 2 weeks increases the number of c‐kit + cells in the border zone of the infarction. (A) c‐kit+ cells in rat sternum (positive control). (B) Single c‐kit+ cell in peri‐infarct area of the left ventricle. (C) Number of c‐kit+ cells in the anterior wall of LV of sham‐operated and AMI groups during the 4‐week follow‐up period. The area of counted section was determined by computerized methods and a number of positively staining cells was related to the area (cells/25 mm2). White bars indicate sham and gray bars AMI.*p < 0.05,**p < 0.01 vs. sham. Section of the left ventricle 2 weeks after AMI (D), AMI with metoprolol (E) and AMI with losartan (F). (G) Metoprolol treatment further increased the number of c‐kit+ cells in the LV.**p < 0.01 versus AMI,‡‡ p < 0.01 versus AMI + METO. (H) Section of LV 2 weeks after AMI with metoprolol treatment, yellow dots representing localization of c‐kit+ cells in the infarct border zone. Results are mean ± SEM (n= 5–6/group). AMI = acute myocardial infarction; METO = metoprolol; LOS = losartan.
To characterize further the potential mechanism by which metoprolol may influence LV remodeling and function, we performed immunohistochemical staining of the infarcted region against leukocyte common antigen CD45 and Ki‐67 antigen. The number of CD45+ cells was significantly increased at 2 and 4 weeks after MI (Supporting Table 2). Similarly to c‐kit+ cells, metoprolol but not losartan treatment for 2 weeks further increased the number of CD45+ cells in the LV compared with control rats (2.5 fold, p < 0.01, Figure 6A‐B ). Moreover, Ki‐67 immunostaining showed a higher number of Ki‐67+ nuclei in the LV wall of the metoprolol‐treated hearts compared with vehicle‐treated hearts (2.1‐fold, p < 0.01), while losartan treatment had no effect on the number of Ki‐67+ cells ( Figure 6C‐D ). Staining of Ki‐67+ proliferative cells was not different between control and infarcted hearts 1 day, 2 weeks or 4 weeks after MI (Supporting Table 2).
Figure 6.
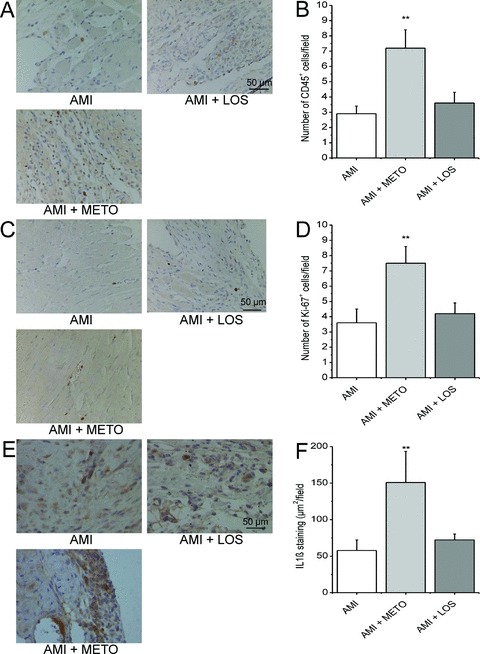
Treatment with metoprolol for 2 weeks after MI increases the number of hematopoietic and proliferative cells, and the expression of IL1β in the LV. Sections of LV were stained for CD45+ cells (A), Ki‐67+ cells (C) and IL1β (E). The number of CD45+ cells/high power field (B) and Ki‐67+ cells/high power field (D), and IL1β staining (μm2/field) (F) in each experimental group. Results are mean ± SEM (n= 5–6/group). AMI = acute myocardial infarction; METO = metoprolol; LOS = losartan.**p < 0.01 versus AMI.
Increase in the expression of IL1β by metoprolol treatment after MI
Metoprolol treatment for 2 weeks significantly increased the expression of interleukin‐1β (IL1β) in infarcted hearts (2.6‐fold, p < 0.01), whereas losartan treatment had no effect ( Figure 6E‐F ). The levels of stromal cell‐derived factor‐1α (SDF1α) and SDF1β as well as tumor necrosis factor‐α (TNFα) were not significantly altered by drug treatments after MI (Supporting Figure 2).
Angiogenesis in the peri‐infarct region of metoprolol‐ and losartan‐treated hearts
Finally, we counted the capillary density of the peri‐infarct area in order to evaluate the degree of angiogenesis in the LV in response to drug treatments. Capillary density in the peri‐infarct region decreased within 2 weeks after MI (111 ± 2/field, n= 6 vs. 73 ± 7/field, n= 6, p < 0.001), as shown earlier. 9 Treatment with metoprolol or losartan for 2 weeks had no effect on microvessel density, as assessed by immunostaining of endothelial cells with primary antibodies for Pecam‐1 and von Willebrand factor (Supporting Figure 3A‐B). Metoprolol and losartan treatments for 2 weeks had no effect on hypoxia‐inducible factor‐1α (HIF1α) protein expression. Perfusion of rat hearts for 4 hours, starting with 30 minutes ischemia, was used as a positive control for induction of HIF1α expression (Supporting Figure 3C).
Discussion
Left ventricular remodeling remains an important treatment target in patients after myocardial infarction and chronic heart failure. 10 Accumulating evidence has supported the concept that beneficial effects of current pharmacological treatments, such as beta‐blockers, are, at least in part, due to their effects on LV remodeling and dysfunction. 1 , 2 A key finding of our present study using rat model of coronary ligation is that beta‐blocker metoprolol elicited beneficial structural remodeling and subsequent improvement of LV systolic function, whereas ARB losartan had adverse effects on LV remodeling during development of post‐infarction heart failure. Both treatments had no effect on the scar area, a hallmark of late postinfarction remodeling, as shown earlier by others. 11 , 12 We evaluated several potential mechanisms triggering the divergent changes in LV function and structure by metoprolol and losartan after MI. These studies showed that metoprolol treatment attenuated adverse remodeling via c‐kit+ cells and proliferation, whereas losartan‐induced worsening of LV systolic function was associated with increased apoptosis and fibrosis in the peri‐infarct region.
Cardiac stem and progenitor cells, expressing characteristic surface antigens such as c‐kit, have been identified in adult myocardium. 13 , 14 Mobilization of bone marrow cells and transfusion of progenitor cells reduce infarct size and improve LV function after MI 15 , 16 and intramyocardial injection of c‐kit+ CSCs can result in reconstitution of the infarcted heart. 17 Several clinical studies suggest that cell therapy reduces the infarct size and improves cardiac contractile function. 18 In this study, we observed an increase in the number of c‐kit+ cells in LV after MI, in agreement with previous studies. 19 , 20 The number of c‐kit+ cells remained elevated for the entire duration of the study (4 weeks). Previously, the level of c‐kit+ cells have been shown to decline to essentially undetectable by 8 weeks after MI. 21
A major finding of our study is that metoprolol but not losartan treatment increased the number of c‐kit+ cells, in addition to Ki‐67+ and CD45+ cells, in infarcted LV. The cell surface glycoprotein CD45 is expressed on almost all hemopoietic lineage cells, also on hematopoietic stem cells, 22 while Ki‐67 is considered as a marker of cellular proliferation. 23 The precise mechanism(s) for the increase in number of c‐kit+ cells as well as proliferative Ki‐67+ cells and hematopoietic CD45+ cells by metoprolol treatment may be due to the tissue's diminished need for oxygen. Beta‐blockers reduce heart rate, lower blood pressure and cardiac contractility 24 leading to diminished oxygen consumption of the surrounding tissue, likely resulting in more intense proliferation. The proliferative changes in the peri‐infarct area may in turn elicit the metoprolol‐induced improvement in function and structure. This is in agreement with the notion of worsening in cardiac dilatation and function in c‐kit‐deficient mice. 25 To our knowledge the effect of metoprolol or losartan on CSCs after MI has not been studied earlier. The number of c‐kit+ cells, however, was limited considering the extent of myocardial damage by infarction, and thus the changes in c‐kit+ cells may be an indication of an overall phenotypic change caused by beta‐blocker treatment in addition to or instead of a distinct regenerative process.
The observed effect of beta‐blocker treatment on LV remodeling and proliferation, on the other hand, could also be explained by factors involved in mobilization of cells and engraftment into the heart. Recent studies suggest a requirement for cooperation between a combination of factors that regulate homing (chemokines, selectins, and integrins) and engraftment (matrix metalloproteases and cathepsins) to determine the specific cell types recruited to ischemic tissue. 26
It is well established that stress signals such as tissue injury or inf ammation cause upregulation of SDF1α, which promotes the recruitment of stem cells into heart. 27 The fact that the expression of SDF1α and SDF1β remained unaltered with metoprolol treatment suggests that the increase in c‐kit+ and Ki‐67+ cells was a local phenomenon, rather than an indication of mobilization of cells from bone marrow. However, the increase in the number of CD45+ cells suggests that cells could be derived from the circulation and thus the true nature of the c‐kit+ cells requires further studies. Despite the origin of c‐kit+ cells, they clearly are associated with better cardiac performance in post‐MI rats. IL1β has previously been shown to reinitiate myocyte DNA synthesis 28 and we observed a simultaneous induction of IL1β and proliferation with metoprolol treatment. Interestingly, IL1β plays a protective role for the heart in the acute phase of MI, 29 suggesting that IL1β could contribute to the increased proliferation and protection of the post‐MI heart by metoprolol treatment.
Myocardial infarction results in hypertrophy of the viable myocardial tissue bordering the infarct zone. Because angiogenesis within the infarcted tissue is unable to maintain tissue growth and support the tissue with required oxygen and nutrients, the viable myocardium undergoes apoptosis. 16 Mobilization of bone‐marrow‐derived angioblasts has been shown to prevent cardiomyocyte apoptosis, reduce remodeling and improve cardiac function in a rat MI model. 16 On the other hand, a phase of apoptosis a few days after pressure overload 30 and upregulation of apoptosis‐related genes as a response to MI 31 implies that apoptosis is rather a signal indicating the phenotypic changes involved in the initiation of cardiac hypertrophy and remodeling. Since losartan treatment after LAD‐ligation caused adverse effects on LV remodeling, we tested the hypothesis that losartan may increase apoptosis in the peri‐infarct region. In previous studies, AT1 receptor blockade by losartan has been connected to decreased apoptosis in cultured ventricular myocytes 32 and in MI model 1 day post‐MI. 33 It is important to note that in the present study, losartan treatment was started simultaneously with the ligation of LAD and continued for 2 weeks. Increased apoptosis and fibrosis with the thinning of the anterior wall as well as the posterior wall of the LV was observed. TUNEL positive cells did not express alpha‐actinin, suggesting that they were not cardiomyocytes. However, this cannot be excluded, because cardiomyocytes may not necessarily express cytoskeletal proteins 2 or more weeks after the onset of the apoptotic program. The dilatation of LV was increased with losartan treatment, similarly to a study, in which rabbits were administered losartan 3 hours after MI for 35 days. 34 Thus, losartan treatment at the time or immediately after MI seems to be detrimental for subsequent LV remodeling process at least partly via apoptotic effect.
In addition to stem cell recruitment, apoptosis, and fibrosis, also angiogenesis in the ischemic border zone of the infarct may affect the remodeling process. 10 Angiogenesis is induced by bradycardia, and reduction of heart rate by beta‐blockers has been shown to promote growth of arterioles in post‐MI heart. 35 ARBs, on the other hand, have no such effect on heart rate and this could explain the worsening of LV systolic function after losartan treatment. By using a rat model of coronary ligation, we observed, however, that metoprolol and losartan treatments did not affect angiogenesis in the peri‐infarct region. In agreement with this, metoprolol treatment after MI did not inf uence the density of vessels in the peri‐infarct area in nude rats. 36 On the other hand, several reports indicate that angiotensin II can induce neovascularization in experimental systems due to the expression of different growth factors, nitric oxide synthase, metalloproteinases, and inf ammation. 37 , 38 In agreement with this, a significant decrease in vascular density in rat glioma after losartan treatment was observed, 39 but also a stimulatory effect with losartan on angiogenesis in rat hearts 40 and in cerebral cortex 41 have been reported. Taken together, ARBs may have angiogenic or antiangiogenic effects, or no effect on the number of capillaries depending on experimental conditions.
One reason for the detrimental effects of losartan treatment on the remodeling process might be the activation of AT2‐receptors, because blockade of AT1 ‐receptors results in increased plasma angiotensin II levels. 42 Stimulation of AT2 ‐receptors has beneficial, but also—especially in chronic stimulation—deleterious effects including antiangiogenesis and stimulation of apoptosis. 42 , 43 Although the precise role of AT2 ‐receptors in myocardial remodeling is not clear, they could, in part, explain the differences in the effects of ACE inhibitors and ARBs. 44 Furthermore, some progenitor cells, e.g., endothelial progenitor cells, express both AT1‐ and AT2‐receptors 45 and CSCs are known to express AT1‐receptor subtype. 46 In the present study, AT1 ‐receptor blockade by losartan had no effect on the number of c‐kit+ cells. ARBs and beta‐blockers have different effect on peripheral resistance, 5 , 24 but this difference unlikely contributes to their divergent effects on LV remodeling, because one would expect losartan by decreasing peripheral resistance and ventricular afterload to attenuate adverse remodeling better than metoprolol.
The size of the infarcted area and the extent of postinfarction LV remodeling are major factors that determine the prognosis of patients after MI. 47 Activation of the renin–angiotensin–aldosterone system and sympathetic hyperactivation, which occur early after myocardial injury, play central roles in the pathogenesis of cardiac structural and functional abnormalities. 1 , 2 , 10 Consistent with this, a substudy of the CAPRICORN trial suggests a beneficial effect of beta‐blocker carvedilol on LV remodeling. 48 Yet, while beta‐blocker and ACE inhibitor trials have found that these treatments significantly reduce the risk of myocardial infarction and cardiovascular death, several meta‐analyses have found no reduction in the risk of myocardial infarction with ARBs. 3 , 4 , 5 Our results on the effects of losartan on postinfarction LV remodeling suggest that the use of ARBs immediately after or at the time of myocardial infarction in primary prevention might be disadvantageous. Furthermore, the proliferative changes including the increase in number of c‐kit+ cells could contribute to the positive outcome of beta‐blockers in primary prevention.
In the present study, we specifically aimed to mimic the design of VALUE, 6 LIFE, 8 and ONTARGET 7 trials in which patients were on ARB treatment at the time when the first myocardial infarction occurs, i.e., during the very early phase of remodeling process.
VALUE, LIFE and ONTARGET trials enrolled patients with hypertension and/or high‐risk individuals and diabetics, but without previous heart failure and myocardial infarction within the previous 6 months or post uncomplicated MI. 6 , 7 , 8 In contrast, VALIANT 49 and OPTIMAAL 50 studies enrolled patients with evidence of heart failure or LV dysfunction, including patients with MI who developed signs of heart failure or evidence of LV dysfunction within 10 days of hospitalization. It is also possible that the potential adverse effects of the ARBs observed in the present study may be blunted by concomitant beta‐blockade, since the majority of patients with heart failure are treated with both agents. However, this hypothesis is not supported by OPTIMAAL, 50 VALIANT, 49 and ONTARGET trials 7 reporting that the results for major outcomes were consistent in analyses that were adjusted for the patients’ use of various concomitant drugs including beta‐blockers. Moreover, in LIFE study, 8 patients with no previous or concomitant beta‐blocker treatment were enrolled whereas patients receiving antihypertensive treatment discontinued taking previous drugs in VALUE trial before valsartan or amlodipine treatments were started. 6
Conclusion
In summary, the major finding of the present study is that angiotensin receptor blocker losartan had adverse effects on LV remodeling during development of postinfarction heart failure. Our rat model of coronary ligation demonstrates that losartan triggered deleterious structural remodeling characterized by ventricular dilation, infarct wall thinning and corresponding impairment of contractile function. It is likely that the pronounced apoptotic and profibrotic effects of losartan disturbed healing of the infarcted area, whereas the beneficial effects of beta‐blocker metoprolol on the LV structure and function were associated with the increase in number of c‐kit+ cells and cellular proliferation ( Figure 7 ). These results suggest that combining drugs that stimulate CSCs to promote myocardial repair with stem‐cell therapies may further enhance the efficacy of cell therapy. Furthermore, the present studies also warrant further analysis on the effects of early treatment of angiotensin receptor blockers on left ventricular remodeling.
Figure 7.
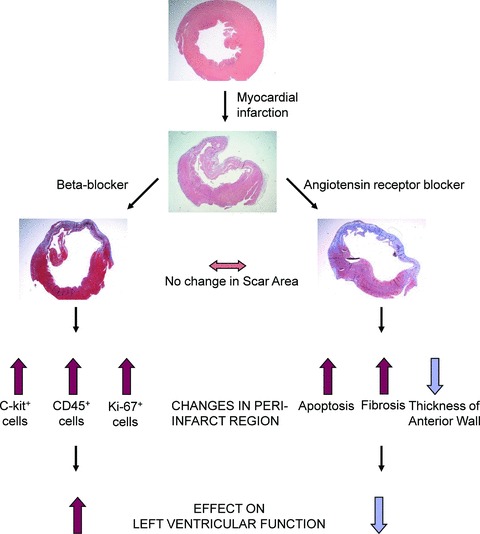
A schematic diagram summarizing the effects of metoprolol and losartan treatments on LV function and remodeling after MI. Metoprolol treatment attenuated adverse remodeling via c‐kit+ and CD45+ cells and proliferation, whereas losartan‐induced worsening of LV systolic function was associated with the increased apoptosis in the peri‐infarct region.
Sources of funding
This work was supported by grants from the Academy of Finland, (Center of Excellence, 1105818, 1211096), Finnish Foundation for Cardiovascular Research, and the Sigrid Juselius Foundation.
Supporting information
Figure S1. Effect of coronary ligation on LV remodeling. Rats were killed 1 day (n = 3) and 2 (n = 5) and 4 (n = 5) weeks after myocardial infarction, and paraffin‐embedded histological sections were cut. LV sections were consequently stained with hematoxylin‐eosin (A) and Masson's trichrome (B) to reveal fibrosis. AMI = acute myocardial infarction.
Figure S2. Effect of metoprolol and losartan treatments for 2 weeks after MI on the the expression of stromal cell‐derived factor (SDF)‐1 and tumor necrosis factor‐α (TNFα) in the left ventricle. Sections of the left ventricle were stained for SDF‐1α, SDF‐1β and TNFα. (A) SDF1α staining (μm2/field), (B) SDF1β staining (μm2/field) and (C) TNFα staining (μm2/field) in each experimental group. Results are mean ± SEM (n = 6/group). AMI = acute myocardial infarction; METO = metoprolol; LOS = losartan.
Figure S3. Effects of metoprolol and losartan treatments on markers of myocardial angiogenesis. (A) Pecam‐1 stained sections of the ischemic border zone of infarcted LV area 2 weeks after MI in each experimental group. (B) Number of capillaries per high power (40×) field. Results are mean ± SEM (n = 6/group).***,†††,‡‡‡ p < 0.001 versus SHAM. (C) Sections stained for HIF1α. Perfusion of rat hearts by the Langendorf 's technique for 4 hours with 30 minutes ischemia in the beginning was used as a positive control for HIF1α induction. AMI = acute myocardial infarction; METO = metoprolol; LOS = losartan.
Table S1. Effects of ligating the left anterior descending artery (LAD) on left ventricular structure and systolic function, as analyzed by echocardiography.
Table S2. Number of CD45+ and Ki‐67+ cells in left ventricle after AMI measured by immunohistochemistry.
Please note: Wiley‐Blackwell Publishing is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
We thank Tuulikki Kärnä, Sirpa Rutanen and Mirja Ahvensalmi for their expert technical assistance and Ulla Hirvonen and Tuula Mäkinen for expert animal care.
References
- 1. Jessup M, Brozena S. Heart failure. N Engl J Med. 2003; 348: 2007–2018. [DOI] [PubMed] [Google Scholar]
- 2. McMurray J, Pfeffer M. Heart failure. Lancet. 2005; 365: 1877–1889. [DOI] [PubMed] [Google Scholar]
- 3. Strauss MH, Hall AS. Angiotensin receptor blockers may increase risk of myocardial infarction: unraveling the ARB‐MI paradox. Circulation. 2006; 114: 838–854. [DOI] [PubMed] [Google Scholar]
- 4. Tsuyuki RT, McDonald MA. Angiotensin receptor blockers do not increase risk of myocardial infarction. Circulation. 2006; 114: 855–860. [DOI] [PubMed] [Google Scholar]
- 5. Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BMW. Renin–angiotensin system and cardiovascular risk. Lancet. 2007; 369: 1208–1219. [DOI] [PubMed] [Google Scholar]
- 6. Julius S, Kjeldsen SE, Weber M, Brunner HR, Ekman S, Hansson L, Hua T, Laragh J, McInnes GT, Mitchell L, Plat F, Schork A, Smith B, Zanchetti A; VALUE trial group . Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. Lancet 2004; 363: 2022–2031. [DOI] [PubMed] [Google Scholar]
- 7. ONTARGET Investigators , Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, Schumacher H, Dagenais G, Sleight P, Anderson C. Telmisartan, ramipril, or both in patients at high risk for vascular events. New Engl J Med. 2008; 358: 1547–1559. [DOI] [PubMed] [Google Scholar]
- 8. Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, De Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe‐Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H. LIFE Study Group. Cardiovascular morbidity and mortality in the losartan intervention for endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002; 359: 995–1003. [DOI] [PubMed] [Google Scholar]
- 9. Tenhunen O, Soini Y, Ilves M, Rysä J, Tuukkanen J, Serpi R, Pennanen H, Ruskoaho H, Leskinen H. p38 Kinase rescues failing myocardium after myocardial infarction: evidence for angiogenic and anti‐apoptotic mechanisms. FASEB J. 2006; 20: E1276–E1286. [DOI] [PubMed] [Google Scholar]
- 10. Sutton MG, Sharpe N. Left ventricular remodelling after myocardial infarction: pathophysiology and therapy. Circulation. 2000; 101: 2981–2988. [DOI] [PubMed] [Google Scholar]
- 11. Wei S, Chow L T C, Sanderson JE. Effect of carvedilol in comparison with metoprolol on myocardial collagen postinfarction. J Am Coll Cardiol. 2000; 36: 276–281. [DOI] [PubMed] [Google Scholar]
- 12. Jain M, Liao R, Ngoy S, Whittaker P, Apstein CS, Eberli FR. Angiotensin II receptor blockade attenuates the deleterious effects of exercise training on post‐MI ventricular remodelling in rats. Cardiovasc Res. 2000; 46: 66–72. [DOI] [PubMed] [Google Scholar]
- 13. Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA. 2003; 100: 12313–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang X, Gurel O, Mendiaz EA, Stearns GW, Clogston CL, Lu HS, Osslund TD, Syed RS, Langley KE, Hendrickson WA. Structure of the active core of human stem cell factor and analysis of binding to its receptor kit. EMBO J. 2000; 19: 3192–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Assmus B, Schachinger V, Teupe C, Britten M, Lehmann R, Dobert N, Frunwald F, Aicher A, Urbich C, Martin H, Hoelzer D, Dimmeler S, Zeiher AM. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction (TOPCARE‐AMI). Circulation. 2002; 106: 3009–3017. [DOI] [PubMed] [Google Scholar]
- 16. Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM, Itescu S. Neovascularization of ischemic myocardium by human bone‐marrow‐derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med. 2001; 7: 430–436. [DOI] [PubMed] [Google Scholar]
- 17. Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal‐Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003; 114: 763–776. [DOI] [PubMed] [Google Scholar]
- 18. Dimmeler S, Burchfield J, Zeiher AM. Cell‐based therapy of myocardial infarction. Arterioscler Thromb Vasc Biol. 2008; 28: 208–216. [DOI] [PubMed] [Google Scholar]
- 19. Urbanek K, Torella D, Sheikh F, De Angelis A, Nurzynska D, Silvestri C, Beltrami A, Bussani R, Beltrami AP, Quaini F, Bolli R, Leri A, Kajstura J, Anversa P. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci USA. 2005; 102: 8692–8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, Weisel RD, Keating A, Li R‐K. Cardioprotective c‐kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest. 2006; 116: 1865–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lehrke S, Mazhari R, Durand DJ, Zheng M, Bedja D, Zimmet JM, Schuleri KH, Chi AS, Gabriel‐son KL, Hare JM. Aging impairs the beneficial effect of granulocyte colony‐stimulating factor and stem cell factor on post‐myocardial infarction remodeling. Circ Res. 2006; 99: 553–560. [DOI] [PubMed] [Google Scholar]
- 22. Dahlke MH, Larsen SR, Rasko JEJ, Schlitt HJ. The biology of CD45 and its use as a therapeutic target. Leuk Lymphoma. 2004; 45: 229–236. [DOI] [PubMed] [Google Scholar]
- 23. Brown DC, Gatter KC. Ki67 protein: the immaculate deception? Histopathology. 2002; 40: 2–11. [DOI] [PubMed] [Google Scholar]
- 24. Cruickshank JM. The beta 1 hyperselectivity in beta‐blocker treatment. J Cardiovasc Pharmacol. 1995; 25(Suppl 1): S35–S46. [DOI] [PubMed] [Google Scholar]
- 25. Ayach BB, Yoshimitsu M, Dawood F, Sun M, Arab S, Chen M, Higuchi K, Siatskas C, Lee P, Lim H, Zhang J, Cukerman E, Stanford WL, Medin JA, Liu PP. Stem cell factor receptor induces progenitor and natural killer cell‐mediated cardiac survival and repair after myocardial infarction. Proc Natl Acad Sci USA. 2006; 103: 2304–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smart N, Riley PR. Stem cell movement. Circ Res. 2008; 102: 1155–1168. [DOI] [PubMed] [Google Scholar]
- 27. Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M, Rovner A, Ellis SG, Thomas JD, DiCorleto PE, Topol EJ, Penn MS. Effect of stromal‐cell‐derived factor 1 on stem‐cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet. 2003; 362: 697–703. [DOI] [PubMed] [Google Scholar]
- 28. Palmer JN, Hartogensis WE, Patten M, Fortuin FD, Long CS. Interleukin‐1β induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest. 1995; 95: 2555–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hwang M‐W, Matsumori A, Furukawa Y, Ono K, Okada M, Iwasaki A, Hara M, Miyamoto T, Touma M, Sasayama S. Neutralization of interleukin‐1β in the acute phase of myocardial infarction promotes the progression of left ventricular remodeling. J Am Coll Cardiol. 2001; 38: 1546–1553. [DOI] [PubMed] [Google Scholar]
- 30. Teiger E, Dam T‐V, Richard L, Wisnewsky C, Tea B‐S, Gaboury L, Tremblay J, Schwartz K, Hamet P. Apoptosis in pressure overload‐induced heart hypertrophy in the rat. J Clin Invest. 1996; 97: 2891–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. LaFramboise WA, Bombach KL, Dhir RJ, Muha N, Cullen RF, Pogozelski AR, Turk D, George JD, Guthrie RD, Magovern JA. Molecular dynamics of the compensatory response to myocardial infarct. J Mol Cell Cardiol. 2005; 38: 103–117. [DOI] [PubMed] [Google Scholar]
- 32. Kajstura J, Cigola E, Malhotra A, Li P, Cheng W, Meggs LG, Anversa P. Angiotensin II induces apoptosis of adult ventricular myocytes in vitro . J Mol Cell Cardiol. 1997; 29: 859–870. [DOI] [PubMed] [Google Scholar]
- 33. El‐Adawi H, Ding L, Tramontano A, Smith S, Mascareno E, Ganguly K, Castillo R, El‐Sherif N. The functional role of the JAK‐STAT pathway in post‐infarction remodelling. Cardiovasc Res. 2003; 57: 129–138. [DOI] [PubMed] [Google Scholar]
- 34. Gonzalez GE, Palleiro J, Monroy S, Perez S, Rodriquez M, Masucci A, Gelpi RJ, Morales C. Effects of the early administration of losartan on the functional and morphological aspects of postmyocardial infarction ventricular remodeling in rabbits. Cardiovascular Pathol. 2005; 14: 88–95. [DOI] [PubMed] [Google Scholar]
- 35. Dedkov EI, Christensen LP, Weiss RM, Tomanek RJ. Reduction of heart rate by chronic beta1‐adrenoceptor blockade promotes growth of arterioles and preserves coronary perfusion reserve in postinfarcted heart. Am J Physiol Heart Circ Physiol. 2005; 288(6): H2684–H2693. [DOI] [PubMed] [Google Scholar]
- 36. Boyle AJ, Schuster M, Witkowski P, Xiang G, Seki T, Way K, Itescu S. Additive effects of endo‐thelial progenitor cells combined with ACE inhibition and β‐blockade on left ventricular function following acute myocardial infarction. J Renin Angiotensin Aldosterone Syst 2005; 6: 33–37. [DOI] [PubMed] [Google Scholar]
- 37. Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II‐mediated cardiovascular and renal diseases. Pharmacol Rev. 2000; 52: 11–34. [PubMed] [Google Scholar]
- 38. Escobar E, Rodriquez‐Reyna TS, Arrieta O, Sotelo J. Angiotensin II, cell proliferation and an‐giogenesis regulator: biologic and therapeutic implications in cancer. Curr Vasc Pharmacol. 2004; 2: 385–399. [DOI] [PubMed] [Google Scholar]
- 39. Rivera E, Arrieta O, Guevara P, Rojo‐Duarte A, Sotelo J. AT1 receptor is present in glioma cells; its blockage reduces the growth of rat glioma. Br J Cancer. 2001; 85(9): 1396–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. De Boer RA, Pinto YM, Suurmeijer AJH, Pokharel S, Scholtens E, Humler M, Saavedra JM, Boomsma F, Van Gilst WH, Van Veldhuisen DJ. Increased expression of cardiac angiotensin II type 1 (AT1) receptors decreases myocardial microvessel density after experimental myocardial infarction. Cardiovasc Res. 2003; 57: 434–442. [DOI] [PubMed] [Google Scholar]
- 41. Munzenmaier DH, Greene AS. Chronic angiotensin II AT1 receptor blockade increases cerebral cortical microvessel density. Am J Physiol Heart Circ Physiol. 2006; 290: H512–H516. [DOI] [PubMed] [Google Scholar]
- 42. Levy BI. Can angiotensin II type 2 receptors have deleterious effects in cardiovascular disease? Circulation. 2004; 109: 8–13. [DOI] [PubMed] [Google Scholar]
- 43. Schneider MD, Lorell BH. A T 2, judgment day. Which angiotensin receptor is the culprit in cardiac hypertrophy? Circulation. 2001; 104: 247–248. [DOI] [PubMed] [Google Scholar]
- 44. Verma S, Strauss M. Angiotensin receptor blockers and myocardial infarction. BMJ. 2004; 329: 1248–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu Y, Fukuda N, Yao E‐H, Matsumoto T, Kobayashi N, Suzuki R, Tahira Y, Ueno T, Matsumoto K. Effects of an ARB on endothelial progenitor cell function and cardiovascular oxidation in hypertension. Am J Hypertens. 2008; 21: 72–77. [DOI] [PubMed] [Google Scholar]
- 46. Leri A, Kajstura J, Anversa P, Frishman WH. Myocardial regeneration and stem cell repair. Curr Probl Cardiol. 2008; 33: 91–153. [DOI] [PubMed] [Google Scholar]
- 47. Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990; 81: 1161–1172. [DOI] [PubMed] [Google Scholar]
- 48. Doughty RN, Whalley GA, Walsh HA, Gamble GD, Lopez‐Sendon J, Sharpe N, for the CAPRICORN Echo Substudy Investigators . Effects of carvedilol on left ventricular remodeling after acute myocardial infarction: the CAPRICORN Echo Substudy. Circulation. 2004; 109: 201–206. [DOI] [PubMed] [Google Scholar]
- 49. Pfeffer MA, McMurray JJV, Velazquez EJ, Rouleau J‐L, Køber L, Maggioni AP, Solomon SD, Swedberg K, Van de Werf F, White H, Leimberger JD, Henis M, Edwards S, Zelenkofske S, Sellers MA, Califf RM; Valsartan in Acute Myocardial Infarction Trial Investigators . Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Eng J Med. 2003; 349(20): 1893–1906. [DOI] [PubMed] [Google Scholar]
- 50. Dickstein K, Kjekshus J, and the OPTIMAAL Steering Committee for the OPTIMAAL Study Group . Effects of losartan and captopril on mortality and morbidity in high‐risk patients after acute myocardial infarction: the OPTIMAAL randomised trial. Lancet. 2005; 360: 752–760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of coronary ligation on LV remodeling. Rats were killed 1 day (n = 3) and 2 (n = 5) and 4 (n = 5) weeks after myocardial infarction, and paraffin‐embedded histological sections were cut. LV sections were consequently stained with hematoxylin‐eosin (A) and Masson's trichrome (B) to reveal fibrosis. AMI = acute myocardial infarction.
Figure S2. Effect of metoprolol and losartan treatments for 2 weeks after MI on the the expression of stromal cell‐derived factor (SDF)‐1 and tumor necrosis factor‐α (TNFα) in the left ventricle. Sections of the left ventricle were stained for SDF‐1α, SDF‐1β and TNFα. (A) SDF1α staining (μm2/field), (B) SDF1β staining (μm2/field) and (C) TNFα staining (μm2/field) in each experimental group. Results are mean ± SEM (n = 6/group). AMI = acute myocardial infarction; METO = metoprolol; LOS = losartan.
Figure S3. Effects of metoprolol and losartan treatments on markers of myocardial angiogenesis. (A) Pecam‐1 stained sections of the ischemic border zone of infarcted LV area 2 weeks after MI in each experimental group. (B) Number of capillaries per high power (40×) field. Results are mean ± SEM (n = 6/group).***,†††,‡‡‡ p < 0.001 versus SHAM. (C) Sections stained for HIF1α. Perfusion of rat hearts by the Langendorf 's technique for 4 hours with 30 minutes ischemia in the beginning was used as a positive control for HIF1α induction. AMI = acute myocardial infarction; METO = metoprolol; LOS = losartan.
Table S1. Effects of ligating the left anterior descending artery (LAD) on left ventricular structure and systolic function, as analyzed by echocardiography.
Table S2. Number of CD45+ and Ki‐67+ cells in left ventricle after AMI measured by immunohistochemistry.
Please note: Wiley‐Blackwell Publishing is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item