

Abstract
Potential biomarkers were identified for in vitro sensitivity to the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib in head and neck cancer. Gefitinib sensitivity was determined in cell lines, followed by transcript profiling coupled with a novel pathway analysis approach. Eleven cell lines were highly sensitive to gefitinib (inhibitor concentration required to give 50% growth inhibition [GI50] < 1 μM), three had intermediate sensitivity (GI50 1–7 μM), and six were resistant (GI50 > 7 μM); an exploratory principal component analysis revealed a separation between the genomic profiles of sensitive and resistant cell lines. Subsequently, a hypothesis‐driven analysis of Affymetrix data (Affymetrix, Inc., Santa Clara, CA, USA) revealed higher mRNA levels for E‐cadherin (CDH1); transforming growth factor, alpha (TGF‐α); amphiregulin (AREG); FLJ22662; EGFR; p21‐activated kinase 6 (PAK6); glutathione S‐transferase Pi (GSTP1); and ATP‐binding cassette, subfamily C, member 5 (ABCC5) in sensitive versus resistant cell lines. A hypothesis‐free analysis identified 46 gene transcripts that were strongly differentiated, seven of which had a known association with EGFR and head and neck cancer (human EGF receptor 3 [HER3], TGF‐α, CDH1, EGFR, keratin 16 [KRT16], fibroblast growth factor 2 [FGF2], and cortactin [CTTN]). Polymerase chain reaction (PCR) and enzyme‐linked immunoabsorbant assay analysis confirmed Affymetrix data, and EGFR gene mutation, amplification, and genomic gain correlated strongly with gefitinib sensitivity. We identified biomarkers that predict for in vitro responsiveness to gefitinib, seven of which have known association with EGFR and head and neck cancer. These in vitro predictive biomarkers may have potential utility in the clinic and warrant further investigation.
Keywords: epidermal growth factor receptor tyrosine kinase inhibitor (EGFR‐TKI), gefitinib, personalized medicine, biomarker, FISH
Introduction
The erbB receptor family is composed of four closely related receptor tyrosine kinases: epidermal growth factor receptor (EGFR; also known as HER1 [human EGF receptor 1] and erbB1), HER2 (erbB2, or HER2/neu), HER3 (erbB3), and HER4 (erbB4). Aberrant functioning and overexpression of the erbB family of receptor tyrosine kinases have been postulated to be involved in the development and progression of various tumors. EGFR has been shown to be overexpressed in lung, head and neck, breast, colon, prostate, kidney, ovary, brain, pancreas, and bladder solid tumors, 1 and the critical role of EGFR in a variety of cancers has prompted the development of a number of small‐molecule‐targeted therapies, 2 including the EGFR tyrosine kinase inhibitor (EGFR‐TKI) gefitinib (IRESSA, AstraZeneca, Macclesfield, UK). In vitro, gefitinib disrupts EGFR‐mediated kinase activity by binding within the catalytic domain for adenosine triphosphate (ATP), thereby blocking EGFR autophosphorylation in tumor cells. 3
In patients with non‐small‐cell lung cancer (NSCLC), clinical studies confirmed that EGFR mutations resulting in the activation of the antiapoptotic protein Akt and/or high EGFR gene copy number are associated with a clinical response to gefitinib. 4 , 5 Data from a phase III study suggest that EGFR gene copy number predicts benefit in terms of overall survival for patients with pretreated advanced NSCLC receiving gefitinib compared with placebo; 6 response to the TKI erlotinib is also predicted by EGFR expression. 7 Additionally, increased HER2 gene copy number or HER3 overexpression has been correlated with response to gefitinib therapy in EGFR‐positive patients with NSCLC. 8 , 9
Reports that somatic mutations of EGFR have been identified in tumor tissues derived from patients with squamous cell carcinoma of the head and neck (SCCHN) 10 , 11 and evidence that EGFR overexpression in SCCHN tumors correlates with poor prognosis 12 have prompted clinical investigations into the role of EGFR genes in SCCHN. Protein and mRNA analyses have shown that EGFR and HER2 are expressed at much higher levels in SCCHN tumors than in normal epithelial tissue. 10 , 12 , 13 Because of its efficacy in patients with advanced NSCLC, 14 , 15 and aberrant EGFR functioning, 16 gefitinib was assessed in phase I–III studies in patients with recurrent or metastatic SCCHN and has demonstrated antitumor activity. 17 , 18 , 19 A number of other erbB receptor ligands have also been implicated in SCCHN, including transforming growth factor, alpha (TGF‐α), heregulin, and amphiregulin (AREG). 13 , 20 , 21 , 22
To further our understanding of the clinical response to the EGFR‐TKI gefitinib, a panel of predominantly SCCHN tumor cell lines were selected as preclinical models to identify potential biomarkers that predict for in vitro gefitinib sensitivity. However, the ability to interpret gene lists by highlighting functional roles and the involvement in disease processes is a continuing challenge in the scientific community with no globally accepted methodology. In this study, gene expression data generated using Affymetrix technology were analyzed by statistical methods and a novel pathway analysis approach (Ingenuity pathway analysis [IPA] software [Ingenuity, Redwood City, CA, USA] coupled with an in‐house data‐mining tool) in order to identify biomarkers with linkage to EGFR biology and head and neck cancer that predict for in vitro responsiveness to gefitinib.
Materials and Methods
Cell lines
A panel of 20 head and neck tumor cell lines was used in this study, which originated from a range of tissues, including the tongue, pharynx, larynx, esophagus, and oral cavity. KYSE‐30, OE21, PE/CA‐PJ15, PE/CA‐PJ34 (clone C12), PE/CA‐PJ41 (clone D2), PE/CA‐PJ49, and DOK cell lines were obtained from the European Collection of Cell Cultures (ECACC; Sigma, Poole, UK); Detroit562, RPMI2650, SCC‐4, SCC‐9, SCC‐25, CAL 27, SW579, FaDu, Hs 840.T, and KB cell lines were obtained from the American Type Culture Collection (ATCC; Teddington, UK). The KYSE‐450, HEp‐2, and HN5 cell lines were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ; Braunschweig, Germany), Gibco BRL Ltd. (Paisley, UK), and the Ludwig Institute for Cancer Research (London, UK), respectively. KYSE‐450 is an EGFR‐expressing esophageal cell line with a gefitinib‐sensitizing mutation. 23
All cells were seeded and maintained in a growth medium (1:1 mix of Dulbecco's modified Eagle's medium [DMEM]/ Ham's F‐12 [phenol red‐free; Sigma, Paisley, UK] containing 10% fetal calf serum [FCS; Sigma, Paisley, UK; F7524] and 1% L‐glutamine [Gibco BRL Ltd.; 25030‐024]) at 37°C in 7.5% CO2 and harvested after 4 days incubation or at 70–80% confluence. The identity of each cell line was blinded before being assayed to determine mutation status, gene copy number, or protein expression.
In vitro drug sensitivity testing
The cells (100 μL in culture media) were seeded into 96‐well plates (Costar, Fisher Scientific, Loughborough, UK; no. 3595) at a predetermined optimal density (500–5,500 cells/well) and incubated for 4 hours at 37°C/7.5% CO2 prior to dosing with gefitinib (0.001–10 μM; 100 μL/well). Following a 96‐hour incubation with gefitinib, viable cell number (GI50[inhibitor concentration required to give 50% growth inhibition]) was determined by the addition of 40 μL of MTS Colorimetric Assay reagent, as per the manufacturer's instructions (no. G1111, Promega, Madison, WI, USA), and the plates were incubated for a further 4 hours. Absorbance was measured at 490 nm on a spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Each experiment was carried out in triplicate for each gefitinib concentration, and data are presented as geometric means. Sensitivity groupings of GI50 data were: <1 μM classed as sensitive, 1–7 μM as intermediate, and >7 μM as resistant.
Affymetrix profiling
RNA was isolated from all 20 cell lines using the RNeasy kit, (Qiagen, Crawley, UK; no. 74104), as per the manufacturer's instructions, to provide a baseline expression profile of untreated cells. The quality of RNA for Affymetrix profiling was determined using an Agilent BioAnalyzer (Santa Clara, CA, USA). Total RNAs (4 μg) were converted to cDNA and transcribed using T7 polymerase. The resulting biotinylated transcripts were fragmented to an average size of approximately 50 bp and hybridized to Affymetrix Human Genome U133 Plus 2 GeneChips, as per the manufacturer's protocol, using an Affymetrix GeneChip Hybridization Oven 640. After staining with streptavidin–phycoerythrin (Molecular Probes, Paisley, UK; no. S‐866) and washing chips at the recommended stringency (Affymetrix GeneChip Fluidics Station 450), the f uorescent hybridization signal was captured using a laser scanner (Affymetrix GeneChip Scanner 3000). The data passed the in‐house quality checks.
Biotinylated DNA from each cell line was hybridized to one Affymetrix Human Genome U133 Plus 2 GeneChip each, and the full genome data set was then interrogated using a hypothesis‐driven analysis, followed by a hypothesis‐free analysis.
Analysis of affymetrix data
The exploratory analysis included principal component analysis (PCA) based on all 54,675 probe sets measured, covering 38,500 well‐characterized human genes. This exploratory analysis showed a separation in terms of genomic profile between cell lines that demonstrated high/intermediate sensitivity and cell lines resistant to gefitinib. The separation did not appear to be attributable to any other potentially confounding factors such as the time of experiment or tissue origin of the cell line. t‐tests were performed on each of the 54,675 probe sets to identify individual probe sets that had significant differences in the mean levels between the sensitive and the resistant lines. The nonparametric Wilcoxon rank‐sum test was also used to provide an alternative assessment.
Hypothesis‐driven analysis
Initially, the hypothesis‐driven analysis focused on 119 genes (123 probe sets) that were preselected based on biological information and encompassed (i) erbB receptors or ligands, (ii) downstream or associated genes (including genes in the public domain that were associated with disease types other than SCCHN), or (iii) genes identified from previous preclinical and clinical studies as well as from the literature ( Table 1 ). A subset of corresponding gene transcripts were prioritized using Affymetrix data based on their fold change (≤0.5 or ≥2, with the fold change defined as the ratio of the geometric means between sensitive and resistant cell lines) and their p value ranking on the parametric and nonparametric tests applied. These were subsequently analyzed using TaqMan® (Applied Biosystems, Warrington, UK).
Table 1.
List of genes that were preselected for analysis based on biological information and encompassing erbB receptors or ligands, downstream or associated genes (including genes in the public domain that were associated with disease types other than SCCHN), or genes identified from previous preclinical and clinical studies as well as from the literature.
| Gene | Gene name |
|---|---|
| ABCA2 | ATP‐binding cassette, subfamily A, member 2 |
| ABCB1 | ATP‐binding cassette, subfamily B, member 1 |
| ABCC2 | ATP‐binding cassette, subfamily C, member 2 |
| ABCC3 | ATP‐binding cassette, subfamily C, member 3 |
| ABCC4 | ATP‐binding cassette, subfamily C, member 4 |
| ABCC5 | ATP‐binding cassette, subfamily C, member 5 |
| ABCG2 | ATP‐binding cassette, subfamily G, member 2 |
| AC0×2 | Acyl‐coenzyme A oxidase 2 |
| ADAM10 | a disintegrin and metalloproteinase domain 10 (= Kuzbanian) |
| ADAM9 | a disintegrin and metalloproteinase domain 9 |
| AKT1 | v‐akt murine thymoma viral oncogene homolog 1 |
| AKT2 | v‐akt murine thymoma viral oncogene homolog 2 |
| AKT3 | v‐akt murine thymoma viral oncogene homolog 3 (protein kinase B, gamma) |
| AREG | Amphiregulin |
| AVEN | Apoptosis, caspase activation inhibitor |
| BAD | BCL2 antagonist of cell death |
| BAG1 | BCL2‐associated athanogene |
| BCL2 | BCL2/adenovirus E1B, 19 kD, protein‐interacting protein 3a |
| BCL2L1 | Apoptosis regulator Bcl‐x (BCL2‐like protein) |
| BCLW | Apoptosis regulator Bcl‐w (BCL2‐like protein) |
| BRAF | v‐raf murine sarcoma viral oncogene homolog B1 |
| BTC | Beta‐cellulin |
| CBL | Cas‐Br‐M (murine) ecotropic retroviral transforming sequence |
| CDH1 | E‐cadherin |
| CDK4 | Cyclin‐dependent kinase 4 |
| CDK6 | Cyclin‐dependent kinase 6 |
| CHST7 | Carbohydrate (N‐acetylglucosamine 6‐O) sulfotransferase 7 |
| C0L4A3B | Collagen, type IV, alpha 3‐binding protein |
| C0R01C | Coronin, actin‐binding protein, 1C |
| CSNK2A1 | Casein kinase 2, alpha 1 polypeptide |
| DAPK1 | Death‐associated protein kinase 1 |
| DAPK2 | Death‐associated protein kinase 2 |
| DUSP3 | Dual‐specificity phosphatase 3 |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor (erythroblastic leukemia viral [v‐erb‐b] oncogene homolog, avian) |
| ELK1 | ELK1, member of ETS oncogene family |
| EMP1 | Carbohydrate (N‐acetylglucosamine 6‐O) sulfotransferase 7 |
| ERBB2 | v‐erb‐b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma‐derived oncogene homolog (avian) |
| ERBB3 | v‐erb‐b2 erythroblastic leukemia viral oncogene homolog 3 (avian) |
| ERCC1 | Excision‐repair cross‐complementing rodent repair deficiency |
| EREG | Epiregulin |
| ESR1 | Estrogen receptor 1 |
| FLJ22662 | Hypothetical protein FLJ22662 |
| FOSB | FBJ murine osteosarcoma viral oncogene homolog B |
| FOSL2 | FOS‐like antigen 2 |
| FOXF1 | Forkhead |
| FRAP1 | FK506‐binding protein 12‐rapamycin‐associated protein 1 |
| Fos | v‐fos FBJ murine osteosarcoma viral oncogene homolog |
| GAK | Cyclin G‐associated kinase |
| GCLC | Glutamate‐cysteine ligase, catalytic subunit |
| GSH | Glutathione synthetase |
| GSK3A | Glycogen synthase kinase 3 alpha |
| GSK3B | Glycogen synthase kinase 3 beta |
| GSPT2 | Gl to S phase transition 2 |
| GSTP1 | Glutathione S‐transferase Pi |
| HB‐EGF | Heparin‐binding EGF |
| HRAS | v‐Ha‐ras Harvey rat sarcoma viral oncogene homolog |
| IGF1 | Insulin‐like growth factor 1 |
| IGF1R | Insulin‐like growth factor 1 receptor |
| IGF2 | Insulin‐like growth factor 2 |
| JUN | v‐jun sarcoma virus 17 oncogene homolog (avian) |
| JUNB | Jun B proto‐oncogene |
| JUND | Jun D proto‐oncogene |
| KRAS2 | v‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog |
| MAP2K1 | Mitogen‐activated protein kinase kinase 1 (MEK) |
| MAP2K2 | Mitogen‐activated protein kinase kinase 2 |
| MAP3K1 | Mitogen‐activated protein kinase kinase kinase 1 (MEKK) |
| MAPK1 | Mitogen‐activated protein kinase 1 |
| MAPK3 | Mitogen‐activated protein kinase 3 |
| MCL1 | Myeloid leukemia 1 |
| MGMT | O‐6‐methylguanine DNA methyltransferase |
| MOSPD1 | Motile sperm domain‐containing 1 |
| MVP | Major vault protein |
| NES | Nestin |
| NOTCH3 | Notch homolog 3 |
| NPAS2 | Neuronal PAS domain protein 2 |
| NRAS | Neuroblastoma RAS viral (v‐ras) oncogene homolog |
| NRG1 | Neuregulin 1 |
| OSMR | Oncostatin M receptor |
| PAK1 | p21/Cdc42/Racl‐activated kinase 1 |
| PAK2 | p21 (CDKNlA)‐activated kinase 2 |
| PAK4 | p21 (CDKNlA)‐activated kinase 4 |
| PAK6 | p21 (CDKNlA)‐activated kinase 6 |
| PDPK1 | 3‐phosphoinositide‐dependent protein kinase‐1 |
| PGM1 | Phosphoglucomutase 1 |
| PHLDA2 | Pleckstrin homology‐like domain, family A, member 2 |
| PIK3CA | Phosphoinositide‐3‐kinase, catalytic, alpha polypeptide |
| PIK3CB | Phosphoinositide‐3‐kinase, catalytic, beta polypeptide |
| PIK3R1 | Phosphoinositide‐3‐kinase, regulatory subunit, polypeptide 1 (p85, alpha) |
| PIK3R2 | Phosphoinositide‐3‐kinase, regulatory subunit, polypeptide 2 (p85, beta) |
| PIK3R3 | Phosphoinositide‐3‐kinase, regulatory subunit, polypeptide 3 (p55, gamma) |
| PLCG1 | Phospholipase C, gamma 1 (formerly subtype 148) |
| RAF1 | v‐raf‐1 murine leukemia viral oncogene homolog 1 |
| RASGRP1 | RAS guanyl‐releasing protein 1 (calcium‐ and DAG‐regulated) |
| RBM7 | RNA‐binding motif protein 7 |
| RPS6KA1 | Ribosomal protein S6 kinase, 90 kD, polypeptide 1 |
| RPS6KA2 | Ribosomal protein S6 kinase, 90 kD, polypeptide 2 |
| RPS6KA3 | Ribosomal protein S6 kinase, 90 kD, polypeptide 3 |
| RPS6KA4 | Ribosomal protein S6 kinase, 90 kD, polypeptide 4 |
| RPS6KB1 | Ribosomal protein S6 kinase, 70 kD, polypeptide 1 |
| RPS6KB2 | Ribosomal protein S6 kinase, 70 kD, polypeptide 2 |
| SHC1 | SHC (src homology 2 domain‐containing) transforming protein 1 |
| SIAT9 | ST3 beta‐galactoside alpha‐2,3‐sialyltransferase 5 |
| SLC20A1 | Solute carrier family 20 (phosphate transporter), member 1 |
| SOS | Son of sevenless protein homolog 1 |
| SPRY2 | Sprouty homolog 2 |
| SRC | v‐src sarcoma (Schmidt‐Ruppin A‐2) viral oncogene homolog (avian) |
| STAT1 | Signal transducer and activator of transcription 1, 91 kD |
| STAT3 | Signal transducer and activator of transcription 3 (acute‐phase response factor) |
| STAT5A | Signal transducer and activator of transcription 5A |
| STAT5B | Signal transducer and activator of transcription 5B |
| STK12 | Serine/threonine kinase 12 = Aurora B |
| STK6 | Serine/threonine kinase 6 = Aurora A |
| TACE | Tumor necrosis factor, alpha, converting enzyme |
| TGF‐α | Transforming growth factor alpha |
| TNNC1 | Troponin C, slow |
| VEGF | Vascular endothelial growth factor A |
| p27KIP | Cyclin‐dependent kinase inhibitor 1B |
Hypothesis‐free analysis
For use in the pathway analysis, a list of 823 probe sets (587 annotated gene transcripts) were selected from the total of 54,675 probe sets based on a fold change of expression in sensitive versus resistant lines of ≤0.5 or ≥2, together with a statistical cutof of p < 0.01 from the t‐test analysis and a filter to ensure that the average signal was above background noise.
In order to interpret gene lists with functional roles and the involvement in disease processes, several tools were used to develop a novel method and analyze the 823 probe sets. Data were analyzed through the use of IPA 24 and the in‐house data‐mining tool, Gene Catalogue Literature Mining. 25 The criteria used for gene selection using these tools, listed in order of importance, were: (i) SCCHN and EGFR association, (ii) SCCHN association alone, (iii) EGFR association alone, and (iv) hypothesis‐free (novel findings).
Profiling for EGFR and HER2 tyrosine kinase domain and BRAF, HRAS, NRAS, and KRAS mutation status
Genomic DNA was extracted from tumor cell lines using the Puregene method (Gentra Systems, Crawley, UK). Primers were designed around exons 18–24 for both EGFR and HER2, tagged with M13, and the region was amplified using the polymerase chain reaction (PCR, 12 mL reaction; final concentrations: 1x GeneAmp buffer [Life Technologies, Carlsbad, CA, USA], 2 mM MgCl2, 10 mM dNTPs, 0.2 mM of each primer, 1 unit of AmpliTaq Gold DNA polymerase [Life Technologies, Carlsbad, CA, USA], and 25 ng of genomic DNA). Cycling was carried out on an MJ Tetrad thermal cycler as follows: 95°C for 10 minutes; 40 cycles at 95°C for 30 seconds, 63°C for 30 seconds, and 72°C for 1 minute; and a final extension step at 72°C for 10 minutes. Following amplification, excess primer and dNTPs were removed by treatment with ExoSAP‐IT (USB, Cleveland, OH, USA). The products were sequenced using BigDye terminator cycle sequencing kit (v3.1, Applied Biosystems) on a 3730 DNA Analyzer (Applied Biosystems). Sequence trace analysis was performed using Phred, Phrap, Consed, and PolyPhred (Genome Sciences Department, University of Washington, Washington, USA) for mutation determination.
Additionally, v‐raf murine sarcoma viral oncogene homolog B1 (BRAF) was analyzed within exon 15, and v‐Ha‐ras Harvey rat sarcoma viral oncogene homolog (HRAS), v‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS), and neuroblastoma ras viral (v‐ras) oncogene homolog (NRAS) were analyzed within exons 2 and 3.
Analysis of EGFR, HER2, and HER3 gene copy number by fluorescence in situ hybridization
The cell cultures were harvested after incubation with colcemide (0.05 μg/mL) for 2 hours. Hypotonization was performed with KCl (0.075 M) at 37°C for 12 minutes and cell pellets were fixed in methanol:glacial acetic acid (3:1). Fixed‐cell suspensions were dropped onto slides, air‐dried, and stored at ‐20°C for 1–3 weeks. Prior to the fluorescence in situ hybridization (FISH) assays, the slides were thawed in a dessicator, incubated in 2× SSC buffer (3 M NaCl, 0.3 M Na3 C6H507·2H2O) at 37°C for 3 hours, washed for 20 seconds in 70% glacial acetic acid, and rinsed in PBS for 2 minutes. Subsequently, the slides were dehydrated in ethanol (in a series of concentrations from 70% to 85% and then to 100%), incubated in 0.008% pepsin/0.01 M HC1 at 37°C for 5 minutes and in 1% formaldehyde for 10 minutes at room temperature, and dehydrated with ethanol.
Dual‐color FISH assays were carried out using one of the following probe sets: LSI EGFR/chromosome enumeration probe (CEP) 7 (Vysis/Abbott Molecular, Des Plaines, IL, USA), PathVysion HER2/CEP17 (Vysis/Abbott Molecular, Ontario, Canada), or HER3/CEP12 (QBiogene, Illkirch, France). The probe sets were applied and chromosomal DNA was co‐denatured for 8 minutes at 80°C; hybridization was allowed to occur at 37°C for 20 hours. Posthybridization washes were performed in 2× SSC/0.3% NP‐40 at 72°C for 2 minutes and in 2× SSC for 1 minute, followed by ethanol dehydration (as previously described). Chromatin was counterstained with 4’,6‐diamidino‐2‐phenylindole (DAPI; 0.15 μg/mL in Vectashield Mounting Medium; Vector Laboratories, Burlingame, CA, USA). Analysis was performed on epifluorescence microscopes using single interference filter sets for green (fluorescein‐5‐isothiocyanate [FITC]), red (Texas Red), and blue (DAPI) as well as dual (red/green) and triple (red, green, blue) band‐pass filters. Representative images were acquired for individual channels using a Photometrics charge‐coupled device (CCD) camera and merged using the CytoVision software (Applied Imaging, Inc., Genetix Limited, Hampshire, UK).
Metaphase analysis was performed in at least 20 spreads selected on the basis of good morphology, spreading, and signal intensity. Interphase analysis was performed in at least 100 consecutive nuclei per specimen. The number of red (EGFR, HER2, and HER3) and green (CEP7, CEP17, and CEP12) signals was scored in individual cells, and abnormal chromosomes were documented and tentatively identified. Genomic gain was defined as either gene amplification (clusters of gene signals) or gene overrepresentation (genomic gain relative to the cell ploidy).
A Fisher's exact test was performed to evaluate the association between sensitivity to gefitinib and EGFR gene overrepresentation/amplification at the p= 0.05 significance level.
Baseline protein profile of pharmacodynamic biomarkers determined by ELISA
To determine the baseline protein levels connected with response to gefitinib, pharmacodynamic biomarkers associated with EGFR activity, survival, proliferation, and cell cycle progression were analyzed across the head and neck tumor cell line panel by enzyme‐linked immunosorbent assay (ELISA), according to the manufacturer's instructions. Cell pellets of all 20 head and neck tumor cell lines were lysed in radioimmunoprecipitation buffer and profiled using various ELISA kits. A two‐tailed t‐test was performed, assuming unequal variance, on the log‐transformed data. The following ELISA kits were used in this study: EGF (no. DY236), TGF‐α (no. DY239), AREG (no. DY262), NRG1 (no. DY377), EGFR (no. DYC1854), HER2 (no. DYC1129), HER3 (no. DYC234), and E‐cadherin (CDH1; no. DY648). All ELISA kits were supplied by R&D Systems (Abingdon, UK). Prior to utilization in this study, the ELISA kits were validated using a variety of biochemical techniques, including Western blotting.
Baseline gene expression profile of pharmacodynamic biomarkers determined by mRNA analysis
Relative quantitation was performed by two‐step TaqMan® real‐time reverse transcriptase polymerase chain reaction (RT‐PCR) using primers and probes designed in‐house. TATA box‐binding protein was selected as a housekeeping gene, and cell line KYSE‐450 was the calibrator sample for comparative results. The comparative threshold cycle (comparative CT) method, which uses the arithmetic formula 2‐DDCT, was used to calculate fold differences in target gene levels between the samples. Briefly, RNA was isolated from cellular lysates using Qiagen RNeasy kits, according to the manufacturers’ instructions, and cDNA synthesis was achieved by incubating 4 μg RNA per sample with Applied Biosystems TaqMan® reagents for 10 minutes at room temperature, followed by 30 minutes at 48°C and 5 minutes at 95°C. The RT‐PCR reaction was then set up using Applied Biosystems TaqMan® PCR reagents and primers and probes, which were designed in‐house. The samples were analyzed using an ABI Prism 7700 Sequence Detector (Life Technologies Corp, Carlsbad, CA, USA) with initial incubations of 2 minutes at 50°C and 10 minutes at 95°C and a subsequent 50 cycles of 0.15 minutes at 95°C and 1 minute at 60°C The samples were run in duplicate, and the mean CT values were calculated.
Results
In vitro drug sensitivity testing
In 11 of the 20 head and neck tumor cell lines, a high level of sensitivity to gefitinib (GI50 < 1 μM) was observed, intermediate sensitivity (GI50 between 1 and 7 μM) was found in 3 cell lines, and resistance (GI50 > 7 μM) was seen in 6 cell lines ( Figure 1 ).
Figure 1.
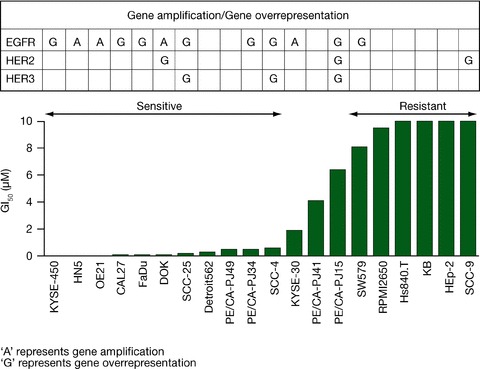
Correlation of genomic gain with sensitivity to gefitinib in head and neck cancer cell lines.
The most sensitive cell lines to gefitinib were KYSE‐450 (GI50= 1 nM) and HN5 (GI50= 3 nM).
Gene transcripts associated with sensitivity to gefitinib
There were many probe sets that statistically separated between the sensitive and the resistant cell lines; for example, 2,855 probe sets (1 666 genes) had a t‐test p value of ≤0.01.
In the hypothesis‐driven analysis, nine probe sets were associated with sensitivity to gefitinib, mapping to CDH1 (2 probe sets); TGF‐α; AREG; hypothetical protein FLJ22662 (FLJ22662); EGFR; p21‐activated kinase 6 (PAK6); glutathione S‐transferase Pi (GSTP1); and ATP‐binding cassette, subfamily C, member 5 (ABCC5) ( Table 2 ). Affymetrix data were supported by TaqMan® analysis for all eight gene transcripts ( Figure 2A ).
Table 2.
Gene transcripts associated with sensitivity to gefitinib, identified from the hypothesis‐driven gene set. Genes were preselected for analysis based on biological information and encompassed erbB receptors or ligands, downstream or associated genes (including genes that were associated with disease types other than SCCHN), or genes identified from previous preclinical and clinical studies as well as from the literature.
| Gene | Gene name | Fold change in sensitive vs. resistant cell lines | p value |
|---|---|---|---|
| CDH1 | Cadherin 1, type‐1 preproprotein | 156.12 | <0.001 |
| TGF‐α | Transforming growth factor, alpha | 13.15 | <0.001 |
| AREG | Amphiregulin | 9.40 | 0.001 |
| FLJ22662 | Hypothetical protein FLJ22662 | 8.34 | 0.006 |
| EGFR | Epidermal growth factor receptor (erythroblastic leukemia viral [v‐erb‐b] oncogene homolog, avian) | 7.48 | 0.009 |
| PAK6 | p21‐activated kinase 6 | 5.81 | <0.001 |
| GSTP1 | Glutathione s‐transferase Pi | 4.72 | 0.004 |
| ABCC5 | ATP‐binding cassette, subfamily c, member 5 isoform 1 | 3.32 | <0.001 |
Figure 2.
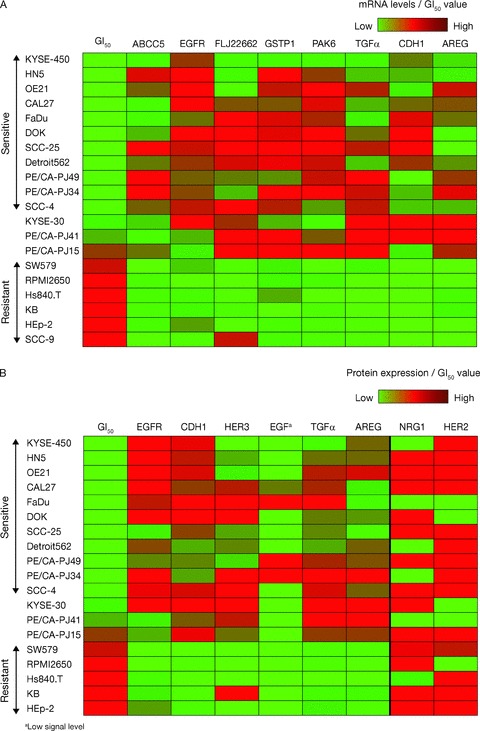
Baseline mRNA and protein expression levels and association with sensitivity to gefitinib. (A) Association between mRNA levels of ABBC5, EGFR, FLJ22662, GSTP1, PAK6, TGF‐α, CDH1, and AREG and gefitinib sensitivity. (B) Left panel: significant association between protein levels of EGFR, CDH1, HER3, EGF, TGF‐α, and AREG and gefitinib sensitivity; right panel: no statistically signifi cant association at the 5% level between protein levels of NRG1 and HER2 and gefitinib sensitivity.
In the hypothesis‐free analysis genome‐wide approach, 587 annotated gene transcripts were identified based on the predefined criteria. The pathway analysis identified seven of these gene transcripts that had a known association with both SCCHN and EGFR expression: HER3, TGF‐α, CDH1, EGFR, keratin (KRT) 16, fibroblast growth factor (FGF) 2, and cortactin (CTTN) ( Figure 3 ).
Figure 3.
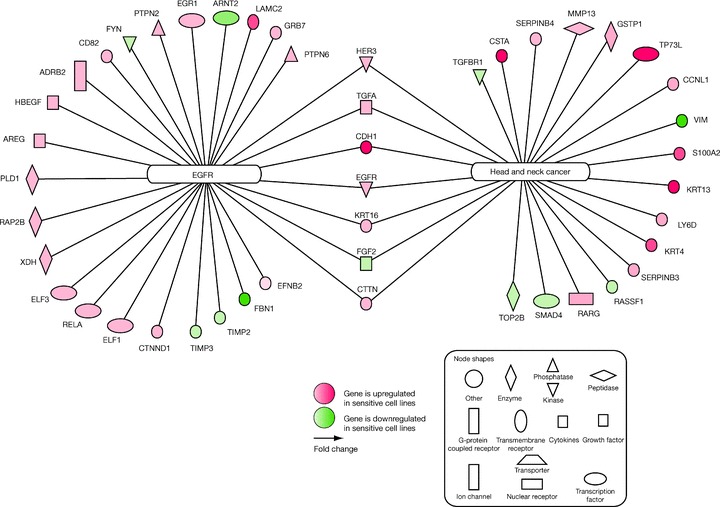
Gene transcripts associated with sensitivity to gefitinib and EGFR and/or head and neck cancer, identified from the genome‐wide statistical analysis, followed by pathway analysis. ADRB2 = adrenergic, beta‐2‐, receptor, surface; AREG = amphiregulin; ARNT2 = aryl‐hydrocarbon receptor nuclear translocator 2; CCNL1 = cyclin L1; CD82 = CD82 molecule; CDH1 = E‐cadherin; CSTA = cystatin A; CTNND1 = catenin; CTTN = cortactin; EFNB2 = ephrin‐B2; EGFR = epidermal growth factor receptor; EGR1 = early growth response 1; ELF1 = E74‐like factor 1; ELF3 = E74‐like factor 3; FBN1 = fibrillin 1; FGF2 = fibroblast growth factor 2; FYN = FYN oncogene related to SRC; GRB7 = growth factor receptor‐bound protein 7; GSTP1 = glutathione S‐transferase pi; HBEGF = heparin‐binding EGF‐like growth factor; HER3 = v‐erb‐b2 avian erythroblastic leukemia viral oncogene homolog 3; KRT4 = keratin 4; KRT13 = keratin 13; KRT16 = keratin 16; LAMC2 = laminin; LY6D = lymphocyte antigen 6 complex; MMP13 = matrix metallopeptidase 13; PLD1 = phospholipase D1; PTPN2 = protein tyrosine phosphatase, non‐receptor type 2; PTPN6 = protein tyrosine phosphatase, non‐receptor type 6; RAP2B = member of RAS oncogene family; RARG = retinoic acid receptor, gamma; RASSF1 = Ras association (RalGDS/AF‐6) domain family 1; RELA = v‐rel reticuloendotheliosis viral oncogene homolog A; S100A2 = S100 calcium‐binding protein A2; SERPINB3 = serpin peptidase inhibitor, clade B (ovalbumin), member 3; SERPINB4 = serpin peptidase inhibitor, clade B (ovalbumin), member 4; SMAD4 = SMAD, mothers against decapentaplegic homolog 4; TGFA = transforming growth factor, alpha; TGFBR1 = transforming growth factor, beta receptor I; TIMP2 = TIMP metallopeptidase inhibitor 2; TIMP3 = TIMP metallopeptidase inhibitor 3; TOP2B = topoisomerase (DNA) II beta; TP73L = tumor protein p73‐like; VIM = vimentin; XDH = xanthine dehydrogenase.
Profiling for EGFR and HER2 tyrosine kinase domain and BRAF, HRAS, NRAS, and KRAS mutational status
KYSE‐450 was confirmed as having a somatic mutation in the EGFR tyrosine kinase domain (exon 20; base change: G2303T; amino acid change: S768I). Furthermore, this cell line was the most sensitive (lowest GI50) to gefitinib and was also positive for EGFR genomic gain. No somatic mutations were detected in the HER2 tyrosine kinase domain in any of the cell lines. One cell line, CAL27, showed two putative somatic mutations in NRAS, both of which were in exon 3 (base changes: G203C, G247A; amino acid changes: R68T, D29N, respectively), but was wild‐type at the commonly mutated codons 12, 13, and 61. Somatic mutations were not detected in BRAF or KRAS genes; however, KYSE‐30 was found to express mutant HRAS (Q61L [exon 3; base change: A182T]).
Analysis of EGFR, HER2, and HER3 gene copy number
Twelve of the 20 head and neck tumor cell lines had genomic gain of EGFR ( Figure 1 ); of these, 8 resulted from gene overrepresentation and 4 from gene amplification. Genomic gain of EGFR was observed in 9 of the 11 cell lines sensitive to gefitinib (6 via gene overrepresentation; 3 via gene amplification) and in 2 out of 3 cell lines with intermediate sensitivity compared with 1 out of the 6 resistant cell lines ( Figure 1 ). The examples of no EGFR genomic gain are shown in Figure 4A , unbalanced EGFR gain based on cell ploidy in Figure 4B , and EGFR gene amplification in Figure 4C . Evidence of a significant association between sensitivity to gefitinib and EGFR gene overrepresentation/amplification was determined (Fisher's exact test, p= 0.035).
Figure 4.
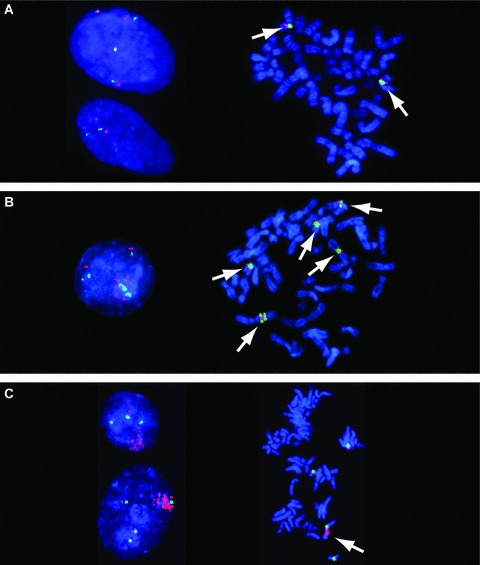
Profiling for EGFR genomic gain. Examples of (A) no EGFR genomic gain, (B) unbalanced EGFR gain based on cell ploidy, and (C) EGFR gene amplification. (A) FISH images showing no EGFR genomic gain. Cell line Hs840.T has a low EGFR gene copy number. Interphase nuclei (left image) show two and four copies of each DNA target. Metaphase spread (right image) shows two chromosomes harboring EGFR (red signals) and CEP7 (green signals), as indicated by arrows. (B) FISH images showing unbalanced EGFR gain based on cell ploidy. Cell line KYSE‐450 has chromosomal aneuploidy, leading to gain of EGFR gene copy numbers. The interphase nucleus (left image) shows numerous copies of EGFR (red signals). Metaphase analysis (right image) identified this line as hyperdiploid, with, on average, five chromosomes carrying EGFR sequences (as indicated by arrows). (C) FISH images showing EGFR gene amplification. Cell line HN5: interphase nuclei (left image) show large clusters of EGFR (red signals). The cluster was mapped to a chromosome 7 derivative (as indicated by arrow) by metaphase analysis (right image).
Genomic gains of HER2 and HER3 were both reported in three cell lines ( Figure 1 ), but there was no apparent association with sensitivity to gefitinib.
Association of sensitivity to gefitinib with baseline protein and gene expression profile of pharmacodynamic biomarkers determined by ELISA
The sensitivity to gefitinib (GI50 < 1 μM) was associated with EGFR protein expression and also with HER3 and CDH1 expression ( Figure 2B ); the p values for differences in the mean protein levels between sensitive and resistant cell lines were: EGFR: p= 0.041; HER3: p= 0.030; CDH1: p= 0.045. Protein levels of the EGFR‐activating ligands TGF‐α and AREG were also higher in sensitive cell lines compared with resistant cell lines (p= 0.010 and P= 0.003, respectively); protein signal levels for EGF were only detected in four cell lines and were low, so no formal statistical test was carried out, but all four were sensitive cell lines ( Figure 2B ). Although NRG1 protein levels tended to be higher in sensitive cell lines compared with resistant lines, statistical significance at the 5% level was not achieved (p= 0.059; Figure 2B ). There was a 2‐fold difference in geometric mean of HER2 protein levels between sensitive and resistant cell lines, but statistical significance was not reached ( Figure 2B ).
Overall, the proteomic data compare well with genomic data; however, ELISA kits were not available to confirm the protein levels of the remaining genes identified. The results of the mRNA TaqMan® ( Figure 2A ) were also in agreement with the ELISA assays.
Discussion
Here, we report the results of investigations in a panel of head and neck tumor cell lines that aimed to identify potential biomarkers for in vitro sensitivity to the EGFR‐TKI gefitinib. The functional classification of the cell lines as sensitive or resistant to gefitinib allowed the molecular characteristics of the cell lines to be compared using Affymetrix mRNA gene expression analysis, TaqMan® mRNA analysis, FISH analysis, and ELISA. EGFR gene mutation, amplification, and overrepresentation correlated significantly with sensitivity to gefitinib; the seven most sensitive head and neck tumor cell lines each displayed at least one of these characteristics, whereas only one of the six resistant cell lines did so.
Of the 119 genes selected for initial focus using predefined biologically relevant hypotheses, transcripts from eight genes (CDH1, TGF‐α, AREG, FLJ22662, EGFR, PAK6, GSTP1, and ABCC5) were identified by Affymetrix as being expressed at significantly higher levels in sensitive versus resistant head and neck tumor cell lines (p < 0.01). The TaqMan® analysis confirmed elevated mRNA levels in sensitive lines for all eight of these genes (CDH1, TGF‐α, AREG, FLJ22662, EGFR, PAK6, GSTP1, and ABCC5) and protein analysis by ELISA also supported higher expression levels of CDH1, TGF‐α, AREG, and EGFR in cell lines that were sensitive to gefitinib ( Figure 2B ). High levels of protein expression of TGF‐α and EGFR in SCCHN are supported by other studies. 13 Some genes known to predict for gefitinib response preclinically (e.g., CDH1) 26 , 27 were also identified in this study; therefore, the additional genes identified may play a role in conferring sensitivity to gefitinib.
Using a genome‐wide analysis of the data (nonhypothesis‐driven and novel pathway approach), seven of these were associated with both the EGFR pathway and SCCHN: HER3, TGF‐α, CDH1, EGFR, KRT16, FGF2, and CTTN. A biological hypothesis for three of these gene transcripts (TGF‐α, CDH1, and EGFR) had already been identified. Affymetrix profiling showed higher levels of HER3 mRNA in head and neck tumor cell lines that were sensitive to gefitinib than in resistant cell lines, and ELISA data confirmed higher levels of HER3 protein expression in sensitive cell lines ( Figure 2B ). To date, the small number of studies reporting data for HER3 expression in SCCHN have suggested that it is overexpressed in oral squamous cell carcinomas and is associated with malignant progression. 28 , 29
These gene lists provide a range of biomarkers that may have potential utility in the clinic to select patients most likely to respond to gefitinib.
The cell line KYSE‐30 was found to express mutant HRAS (Q61L). HRAS is involved in the downstream signaling pathway of EGFR; therefore, an activating mutation could potentially explain the intermediate sensitivity observed in KYSE‐30 cells to gefitinib compared with other cell lines in which genomic gain of EGFR was observed. Data from this study confirm the S768I EGFR mutation in the tyrosine kinase domain of KYSE‐450 reported by Guo et al. 23 In NSCLC, the mutations located in exon 20 of the tyrosine kinase domain are not necessarily identified as activating mutations. 30 However, this mutation does seem to be associated with sensitivity to gefitinib in the KYSE‐450 SCCHN cell line, but it is also worth noting that this cell line also demonstrated genomic gain. High EGFR gene copy number is a biomarker that has been clinically shown to identify NSCLC patients most likely to have improved survival following treatment with gefitinib 31 or erlotinib 32 compared with placebo. These preclinical studies suggest that a similar patient selection effect could be achieved for gefitinib when treating SCCHN. This in vitro study aimed to identify biomarkers to predict sensitivity to gefitinib in head and neck cancer following reports from phase I/II trials of gefitinib effcacy and tolerability in patients with SCCHN. 17 , 18 A recent phase III study compared gefitinib (250 mg/day or 500 mg/day) with methotrexate (40 mg/m2 weekly) in patients with recurrent SCCHN. Neither dose of gefitinib demonstrated an advantage over methotrexate for the primary endpoint of overall survival; however, more patients experienced an improvement in quality of life with gefitinib (250 mg/day or 500 mg/day) than with methotrexate. 19 A numerically greater proportion of patients with high EGFR gene copy number, determined by FISH, experienced an objective response to gefitinib compared with patients with low EGFR gene copy number, although a significant difference in survival was not detected between these two groups in any treatment arm.
There are a number of merits to integrating a variety of techniques to generate gene data associated with sensitivity to gefitinib when compared with using any one of these techniques in isolation. When >50,000 genes are analyzed, the probability of finding genes whose expression correlates with gefitinib sensitivity is very high. 33 However, by utilizing the Ingenuity Systems pathway sof ware to identify gene transcripts known to have an association with EGFR and/or SCCHN, coupled with tools such as the in‐house gene annotation Gene Catalogue Literature Mining sof ware, the likelihood of false‐positives is reduced. Additionally, in order to allow for changes in cell lines that occur during cell culture, this study used resistant cell lines as an internal control. Nonetheless, it is also recommended that the identification of putative genetic biomarkers for response to chemotherapeutic agents should be reinforced with in vitro data that show correlation with drug response.
In summary, this study used statistical analysis, in conjunction with two diverse bioinformatics tools, to develop a novel method of interpreting gene lists and thus to identify potential biomarkers for in vitro sensitivity to gefitinib. Data presented here are in some cases confirmatory but have additionally identified a range of highly statistically relevant gene transcripts with no previously known association with gefitinib response, despite identified associations with EGFR or head and neck cancer. These putative biomarkers as predictors of sensitivity to gefitinib in head and neck cancer warrant further investigation.
Conflict of Interest
Fred R. Hirsch has received consultancy funding from Lilly Oncology and research support from AstraZeneca, Genetech, OSI, Syndax, Ventana‐Roche, and Merck. MarileilaVarella‐Garcia has received research funding from AstraZeneca. Paul A. Bunn has received consultancy funding from Bayer Pharmaceuticals Corporation, Eli Lilly & Co., and Glaxo Smith‐Kline and is an Amgen, AstraZeneca, Bayer Pharmaceuticals Corporation, Bristol Myers‐Squibb, Eli Lilly & Co., Genentech, Glaxo Smith‐Kline, Hoffman La Roche Ltd., ImClone Systems, Novartis Pharmaceuticals Corporation, OSI Pharmaceuticals, Inc., Pfizer, Inc., Sanof ‐Aventis, and Schering‐Plough Corporation advisory committee member. Gayle B. Marshall, Garry J. Beran, Elizabeth A. Mills, Marie C. South, Geal McWalter, Andrew M. Cassidy, Tim French, Rose M. McCormack, and Georgina Speake are salaried employees of AstraZeneca. D. Mark Hickinson, Brian R. Holloway, and Alex Graham were formerly salaried employees of AstraZeneca. Garry J. Beran, Elizabeth A. Mills, Marie C. South, Rose M. McCormack, Georgina Speake, and Gael McWalter have equities in AstraZeneca. Kerry L. Acheson declared no potential conflict of interest relevant to this article.
Acknowledgments
The authors thank the University of Colorado Cancer Center Cytogenetics Core for their technical assistance. We thank Dr. Kim Croskery, from Complete Medical Communications, who provided medical writing support funded by AstraZeneca. This research was funded by AstraZeneca. IRESSA is a trademark of the AstraZeneca group of companies.
References
- 1. Kim ES, Khuri FR, Herbst RS. Epidermal growth factor receptor biology (IMC‐C225). Curr Opin Oncol. 2001; 13: 506–513. [DOI] [PubMed] [Google Scholar]
- 2. Boschelli DH. Small molecule inhibitors of receptor tyrosine kinases. Drugs Future. 1999; 24: 515–537. [Google Scholar]
- 3. Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002; 62: 5749–5754. [PubMed] [Google Scholar]
- 4. Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I, Ludovini V, Magrini E, Gregorc V, Doglioni C, Sidoni A, Tonato M, Franklin WA, Crino L, Bunn PA Jr, Varella‐Garcia M. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non‐small‐cell lung cancer. J Natl Cancer Inst. 2005; 97: 643–655. [DOI] [PubMed] [Google Scholar]
- 5. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib‐sensitizing EGFR mutations in lung cancer activate anti‐apoptotic pathways. Science. 2004; 305: 1163–1167. [DOI] [PubMed] [Google Scholar]
- 6. Hirsch FR, Varella‐Garcia M, Bunn PA Jr, Franklin WA, Dziadziuszko R, Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, Von Pawel J, Watkins C, Flannery A, Ellison G, Donald E, Knight L, Parums D, Botwood N, Holloway B. Molecular predictors of outcome with gefitinib in a phase III placebo‐controlled study in advanced non‐small‐cell lung cancer. J Clin Oncol. 2006; 24: 5034–5042. [DOI] [PubMed] [Google Scholar]
- 7. Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel‐Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, Da Cunha Santos G, Lagarde A, Richardson F, Seymour L, Whitehead M, Ding K, Pater J, Shepherd FA. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med. 2005; 353: 133–144. [DOI] [PubMed] [Google Scholar]
- 8. Cappuzzo F, Varella‐Garcia M, Shigematsu H, Domenichini I, Bartolini S, Ceresoli GL, Rossi E, Ludovini V, Gregorc V, Toschi L, Franklin WA, Crino L, Gazdar AF, Bunn PA Jr, Hirsch FR. Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor‐positive non‐small‐cell lung cancer patients. J Clin Oncol. 2005; 23: 5007–5018. [DOI] [PubMed] [Google Scholar]
- 9. Cappuzzo F, Toschi L, Domenichini I, Bartolini S, Ceresoli GL, Rossi E, Ludovini V, Cancellieri A, Magrini E, Bemis L, Franklin WA, Crino L, Bunn PA Jr, Hirsch FR, Varella‐Garcia M. HER3 genomic gain and sensitivity to gefitinib in advanced non‐small‐cell lung cancer patients. Br J Cancer. 2005; 93: 1334–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Willmore‐Payne C, Holden JA, Layfield LJ. Detection of EGFR‐ and HER2‐activating mutations in squamous cell carcinoma involving the head and neck. Mod Pathol. 2006; 19: 634–640. [DOI] [PubMed] [Google Scholar]
- 11. Lee JW, Soung YH, Kim SY, Nam HK, Park WS, Nam SW, Kim MS, Sun DI, Lee YS, Jang JJ, Lee JY, Yoo NJ, Lee SH. Somatic mutations of EGFR gene in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2005; 11: 2879–2882. [DOI] [PubMed] [Google Scholar]
- 12. Santini J, Formento JL, Francoual M, Milano G, Schneider M, Dassonville O, Demard F. Characterization, quantifi cation, and potential clinical value of the epidermal growth factor receptor in head and neck squamous cell carcinomas. Head Neck. 1991; 13: 132–139. [DOI] [PubMed] [Google Scholar]
- 13. Rubin Grandis J, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, Drenning SD, Tweardy DJ. Levels of TGF‐α and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998; 90: 824–832. [DOI] [PubMed] [Google Scholar]
- 14. Kris MG, Natale RB, Herbst RS, Lynch TJ Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ, Kay AC. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non‐small cell lung cancer. A randomized trial. JAMA. 2003; 290: 2149–2158. [DOI] [PubMed] [Google Scholar]
- 15. Thatcher N, Chang A, Parikh P, Rodrigues PJ, Ciuleanu T, Von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V, Carroll K. Gefitinib plus best supportive care in previously treated patients with refractory advanced non‐small‐cell lung cancer: results from a randomised, placebo‐controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet. 2005; 366: 1527–1537. [DOI] [PubMed] [Google Scholar]
- 16. Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, Peyton M, Juroske D, Huang Y, Stuart SJ, Kim YH, Pollack JR, Yanagisawa K, Gazdar A, Minna JD, Kurie JM, Carbone DP. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005; 65: 226–235. [PubMed] [Google Scholar]
- 17. Cohen EEW, Rosen F, Stadler WM, Recant W, Stanson K, Huo D, Vokes EE. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2003; 21: 1980–1987. [DOI] [PubMed] [Google Scholar]
- 18. Herbst RS, Maddox AM, Rothenberg ML, Small EJ, Rubin EH, Baselga J, Rojo F, Hong WK, Swaisland H, Averbuch SD, Ochs J, LoRusso PM. Selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 is generally well‐tolerated and has activity in non‐small‐cell lung cancer and other solid tumors: results of a phase I trial. J Clin Oncol. 2002; 20: 3815–3825. [DOI] [PubMed] [Google Scholar]
- 19. Stewart S, Cohen E, Licitra L, Van Herpen CML, Khorprasert C, Soulieres D, Vodvarka P, Rischin D, Garin AM, Hirsch FR, Ghiorghiu S, Hargreaves L, Speake G, Armour A, Vokes E. A phase III randomized parallel‐group study of gefitinib (IRESSA) versus methotrexate (IMEX) in patients with recurrent squamous cell carcinoma of the head and neck. Poster 3522. Proceedings of the American Association for Cancer Research; 2007; Los Angeles , CA , USA .
- 20. Charoenrat P, Rhys‐Evans P, Eccles S. Expression and regulation of c‐ERBB ligands in human head and neck squamous carcinoma cells. Int J Cancer. 2000; 88: 759–765. [DOI] [PubMed] [Google Scholar]
- 21. Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res. 2001; 7: 2958–2970. [PubMed] [Google Scholar]
- 22. Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res. 2003; 284: 31–53. [DOI] [PubMed] [Google Scholar]
- 23. Guo M, Liu S, Herman JG, Zhuang H, Lu F. Gefitinib‐sensitizing mutation in esophageal carcinoma cell line Kyse450. Cancer Biol Ther. 2006; 5: 152–155. [DOI] [PubMed] [Google Scholar]
- 24. Ingenuity® Systems . Available at: http://www.ingenuity.com. Accessed March 10, 2009.
- 25. Thomas EP, Dewhirst B, Todd J, Anim T, Downey‐Jones M, Tilley D, Servenius B, Pawlowski K, Westcott A, Woodwark M, Firth M, Dix I. AstraZeneca Gene Catalogue—a novel view of the human transcriptome and proteome. ISMB ECCB; 2004; Glasgow , UK . (Presentation)
- 26. Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita M, Chan Z, Baron A, Franklin W, Drabkin HA, Girard L, Gazdar AF, Minna JD, Bunn PA, Jr . Restoring E‐cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006; 66: 944–950. [DOI] [PubMed] [Google Scholar]
- 27. Frederick BA, Helfrich BA, Coldren CD, Zheng D, Chan D, Bunn PA Jr, Raben D. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non‐small cell lung carcinoma. Mol Cancer Ther. 2007; 6: 1683–1691. [DOI] [PubMed] [Google Scholar]
- 28. Charoenrat P, Rhys‐Evans PH, Archer DJ, Eccles SA. C‐erbB receptors in squamous cell carcinomas of the head and neck: clinical significance and correlation with matrix metalloproteinases and vascular endothelial growth factors. Oral Oncol. 2002; 38: 73–80. [DOI] [PubMed] [Google Scholar]
- 29. Shintani S, Funayama T, Yoshihama Y, Alcalde RE, Matsumura T. Prognostic significance of ERBB3 overexpression in oral squamous cell carcinoma. Cancer Lett. 1995; 95: 79–83. [DOI] [PubMed] [Google Scholar]
- 30. Wu JY, Wu SG, Yang CH, Gow CH, Chang YL, Yu CJ, Shih JY, Yang PC. Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clin Cancer Res. 2008; 14: 4877–4882. [DOI] [PubMed] [Google Scholar]
- 31. Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel‐Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, Da Cunha Santos G, Lagarde A, Richardson F, Seymour L, Whitehead M, Ding K, Pater J, Shepherd FA. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med. 2005; 14: 133–144. [DOI] [PubMed] [Google Scholar]
- 32. Hirsch FR, Varella‐Garcia M, Bunn PA Jr, Franklin WA, Dziadziuszko R, Thatcher N, Chang A, Parikh P, Pereira JR, Ciuleanu T, Von Pawel J, Watkins C, Flannery A, Ellison G, Donald E, Knight L, Parums D, Botwood N, Holloway B. Molecular predictors of outcome with gefitinib in a phase III placebo‐controlled study in advanced non‐small‐cell lung cancer. J Clin Oncol. 2006; 24: 5034–5042. [DOI] [PubMed] [Google Scholar]
- 33. Parissenti AM, Hembruff SL, Villeneuve DJ, Veitch Z, Guo B, Eng J. Gene expression profiles as biomarkers for the prediction of chemotherapy drug response in human tumour cells. Anticancer Drugs. 2007; 18: 499–523. [DOI] [PubMed] [Google Scholar]