

Introduction
Alzheimer's disease (AD) is a progressive and fatal brain disease. According to the Alzheimer's Association, as many as 5.2 million people in the United States are living with AD and 10 million “baby boomers” are expected to develop the disease in their lifetime. AD is the sixth leading cause of death, and the direct and indirect costs of dementia amount to more than $148 billion each year (2008 Alzheimer's Disease Facts and Figures, Alzheimer's Association). Currently, there is no cure. To date, the U.S. Food and Drug Administration (FDA) has approved three cholinesterase inhibitors (donepezil, galantamine, and rivastigmine) and an N‐methylD‐aspartate (NMDA) receptor antagonist (memantine) that offer modest improvement in cognitive function in some individuals, but such treatments are often discontinued within 6 months because of inadequate efficacy and/or tolerability issues. Although treatments with the potential to modify the course of AD by affecting the underlying pathophysiology are rapidly evolving and several of them have entered advanced phases of human clinical trials, none has shown convincing clinical efficacy in a large pivotal trial.
The recent announcement from Myriad Pharmaceuticals, Inc. (Salt Lake City, UT, USA) on June 30, 2008, that Flurizan (Tarenflurbil, MPC‐7869) failed to demonstrate statistically significant efficacy in either of the co‐primary endpoints in the phase III trial represents another high‐profile failure in the pursuit of disease‐modifying therapeutics for AD. It is believed that on the basis of this result, Myriad and partner Lundbeck decided to discontinue the development of Flurizan, including the ongoing international phase III trial, which was due to complete in October. This phase III trial followed 1,684 AD patients over an 18‐month treatment period for a change in the Alzheimer's Disease Assessment Scale‐Cognitive Subscale (ADAS‐Cog) score (a cognitive measure) and Alzheimer's Disease Cooperative Study‐Activities of Daily Living Inventory (ACDS‐ADL) score (a functional measure) relative to placebo as the efficacy endpoints. As this trial was powered to achieve statistical significance in a nonclinically significant change in the ADAS‐Cog score, the failure confirms that Flurizan has no clinical effect beyond the standard of care (and placebo).
Prior to Myriad's announcement, the most recent failure was tramiprosate (Alzhemed), developed by Neurochem (Laval, Quebec, Canada), which was supposed to act by binding soluble amyloid Aβ peptides. It was terminated in August 2007 again after a large phase III trial that included over 1,800 AD patients treated for 18 months showed no statistical significant difference b etween treatment and placebo groups in both primary efficacy endpoints of the ADAS‐Cog and Disability Assessment for Dimentia (DAD). These two large clinical trials with compounds reputed for their antiamyloid mechanism of action were disappointing news to the AD community, especially for the patients and caregivers who are desperately waiting for a disease‐modifying therapy that would offer some hope for disease slowing beyond the short‐term symptomatic relief.
In this commentary, we offer a translational medicine perspective on the choices made to select and develop these compounds for AD treatment. A close examination of the Flurizan case presented below could provide important insights and “lessons learned,” especially regarding the issues of evidence‐based clinical decision making during the drug development program. This case study highlights the importance of appropriate and rigorous use of biomarkers in AD drug development. We take the position, based on our analysis and available literature, that these compounds are inadequate tools to fully investigate the role of amyloid pathways in AD. We further recommend a biomarker‐driven roadmap that could be used at different stages to reduce the risk of development of innovative compounds based on a new mechanism of action in the AD program.
It is widely recognized that today's environment for a new pharmaceutical development is increasingly challenging, particularly since many new targets are not as well validated. In response to this challenge, the pharmaceutical industry is increasingly using biomarkers as rationales and potentially cost‐effective means of predicting potential success of novel therapeutics in drug discovery and development. Biomarkers can be employed to (1) understand the relevance of the drug target to human disease, (2) demonstrate drug‐target interaction, (3) measure the consequences of target modulation (pharmacodynamic [PD] effects), (4) detect modulation of pathophysiological processes, and (5) optimize patient selection to detect medical benefits. The use of such evidence‐based biomarkers can increase confidence during early development, improve the ability to prioritize clinical drug candidates across a broad portfolio, and yield better and more cost‐effective advancement decisions. 1 , 2 , 3 , 4
For convenience and to achieve a uniform lexicon, we group biomarkers into the following categories based on their intended use (Box):
-
A.
Target validation biomarkers provide scientific evidence on the role of the target in human diseases and its potential to be exploited in drug discovery and development campaigns.
-
B.
Target‐compound interaction biomarkers provide evidence on the physical‐chemical interaction of the drug with its intended target.
-
C.
PD biomarkers report on the biological consequences of drug action in the exposed organism or patient. These include biomarkers of efficacy and safety.
-
D.
Disease biomarkers report on disease severity, progression, and regression and could provide guidance as to whether a drug candidate has the potential to fundamentally alter or modify the disease process.
-
E.
Patient selection biomarkers provide information on those patients most likely to respond (or not respond) to the treatment. Such biomarkers provide an opportunity to stratify patients for risk of disease progression and potentially enable shorter trials with higher event rates and earlier outcome assessments.
 Box. Biomarkers: utilitarian classification.
Box. Biomarkers: utilitarian classification.
In the following sections, we examine the case of Flurizan with regard to these five criteria and discuss the potential value of biomarker in decision making.
Target Validation
While the precise etiology and pathogenesis of AD are still unclear, the most convincing hypothesis postulates that the accumulation of Aβ peptide triggers the formation of amyloid plaques and neurofibrillary tangles with subsequent inflammation and brain atrophy 5 ( Figure 1 ). Rare, inherited forms of AD offer strong support for the role of Aβ in the pathogenesis of the disease. Mutations in either the precursor protein or the processing enzymes lead to Aβ accumulation and amyloid plaque and tangle formation. 6 , 7 Despite the fact that the etiology of the disease in sporadic AD remains elusive, Aβ peptides and amyloid plaques also accumulate in these patients.
Figure 1.
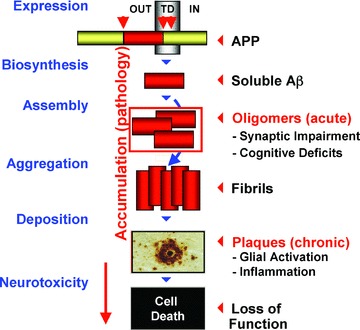
Amyloid cascade hypothesis.
Tremendous progress has been made in understanding the biological role of Aβ, including its production, clearance, and physiological impact on neuronal function and pathology. Multiple agents targeting this pathway, which are expected to further reduce Aβ production or clear amyloid plaques, are in various stages of clinical testing. The results of such ongoing clinical trials are expected to provide proof of concept (POC) for an Aβ‐lowering mechanism as being sufficient to modify the disease course. Although clinical evidence of such amyloid‐targeted therapeutics has not been demonstrated in a pivotal study to date, promising data have been recently emerging. 8 , 9
Gaps in our understanding of Aβ as a drug target remain, and the tools currently available to study Aβ in its various forms are limiting. In vitro and animal studies indicate that soluble forms of Aβ aggregates rather than amyloid deposits are the neurotoxic species and these aggregates are difficult to measure, particularly in a clinical setting. Any measurement of soluble Aβ directly in the human brain is problematic, and although measurements in the cerebrospinal fluid (CSF) or plasma are feasible, their relationship to brain levels is confounded by the differences in the dynamic equilibrium between compartments and the rather ubiquitous expression of the amyloid precursor protein (APP) precursor and processing enzymes. Positron emission tomography (PET) is a promising method that has been used for the in vivo detection of Aβ plaque burden. However, longitudinal studies indicate that amyloid deposition precedes cognitive impairment by decades and correlates with memory loss only in the very mildest stages of AD or in normal elderly. 10 The issues with validating antibody (Ab) as a drug target in AD patients and the inherent limitations of current biomarkers are not unique to Flurizan and relate to all Aβ‐lowering agents.
Target‐Compound Interaction
Flurizan is the pure R‐enantiomer form of flurbiprofen—a nonsteroidal anti‐inflammatory drug (NSAID) with 25 years of clinical experience behind it. Unlike classical NSAIDs, however, Flurizan is not an inhibitor of cyclooxygenase enzymes (COX‐1 and COX‐2). 11 Based on in vitro cell‐based assay results, Flurizan is proposed to be a selective Aβ42‐lowering agent. 12 The data generated to date suggest that Flurizan modulates, rather than inhibits, γ‐secretase to preferentially reduce the generation of the longer toxic Aβ42peptide and favor the production of shorter, less toxic forms. As a selective Aβ42‐lowering agent, Flurizan does this without affecting the processing of other essential γ‐secretase substrates such as Notch, which provides an advantage over direct γ‐secretase inhibitors.
The exact nature of Flurizan's interaction with γ‐secretase complex is not fully understood. One hypothesis is that NSAIDs, including Flurizan, specifically lower Aβ42 through an allosteric binding mechanism that alters the conformation of the presenilin complex. 13 , 14 More recent data indicate that Flurizan does not interact with the enzyme subunits but directly binds to the APP substrate. 12 The authors suggest that drug binding shifts the position of the APP precursor in the plane of the membrane to alter the γ‐secretase cleavage site. Additionally, Flurizan might inhibit Aβ aggregation since the minimal binding site is the same domain that is involved in fibrilization.
In cell‐based systems expressing human APP containing the “Swedish” mutation, Flurizan demonstrated selective Aβ42‐lowering activity. A 50% reduction in Aβ42was observed in human H4 neuroglioma cells at 100 μM using the ELISA method 15 and a 40% reduction was observed at 250 μM in human embryonic kidney cells. 16 Potency was not improved in a broken‐cell model using membrane preparations derived from Chinese Hamster Ovary (CHO) cells to directly measure the inhibition of ‐γ‐secretase in vivo 11 and in vitro. Inhibitory concentration (IC) values for Aβ42‐lowering activity have not been reported. In comparison, several γ‐secretase inhibitors that entered clinical development, including GSI‐953 17 and LY‐450139, 18 have IC50 in the low nanomolar range. Taken together, the ambiguity and controversy regarding the direct target that Flurizan interacts with and the weak in vitro potency in reducing Aβ42 production represent significant knowledge gaps, which increase the risk assumed in further drug development.
PD Response
Assuming that the amyloid cascade is correct and the reduction of Aβ can slow or halt disease progression, it is still unclear how much Aβ42 inhibition is required for clinical efficacy Data from PDAPP transgenic animals that were backcrossed into β‐site of APP cleaving enzyme (BACE) heterozygous knockout mice with a partial reduction of BACE leads to about 12% reduction of soluble total Aβ in brain tissues. 19 , 20 However, this level of reduction in soluble Aβ results in a much more substantial reduction in amyloid plaque load as the animal ages. Similarly, it is possible that in AD patients, a chronic treatment strategy with moderate amyloid reduction might lead to a more substantial impact on amyloid plaque deposition and on the rate of disease progression. One line of evidence comes from the fact that Down's syndrome patients, who process 50% more amyloid precursor protein in brain tissue because of an additional copy of the APP gene on chromosome 21, often develop AD‐like pathology in the third and fourth decades, some 20–30 years earlier than the onset of the sporadic form of the disease. Conversely, one might speculate that reducing Aβ levels for even less than 50% could lead to a substantial delay in disease onset and progression.
When administered orally to transgenic Tg2576 mice, Flurizan lowered Aβ42 in the brain but lacked a dose‐proportional response. A 3‐day subchronic dosing regime at 10, 25, and 50 mg/kg/day produced 26% (p < 0.01), 60% (p < 0.001), and 34% (p < 0.001) reduction of brain Aβ42 levels, respectively. Plasma Aβ40 and Aβ42 levels were decreased by30–50% but lacked direct correlation with brain Aβ levels. The average drug levels for the brain (1.5, 2.6, and 2.5 μM) and plasma (83,117, and 78 μM) at 2 hours after dosing were reported for 10, 25, and 50 mg/kg/day, respectively. 15 Thus, corresponding brain‐to‐plasma ratios were poor (approximately 0.02), perhaps explaining the unpredictable pharmacokinetics (PK)/PD relationship. At best, an exposure multiple of approximately 0.2 was obtained in the brain, in which the concentrations reached approximately 50‐fold lower drug levels than the estimated in vitro IC50 value for Aβ42‐lowering activity. 21 Similar studies performed independently in Tg2576 mice unexpectedly failed to detect significant reductions of Aβ42 levels in the brain. 22 Thus, the reported in vivo PK/PD data did not provide robust evidence for Flurizan's activity as a selective Aβ42‐lowering agent.
Although Flurizan was well tolerated in humans, phase I single‐dose PK data demonstrate that the exposure was not dose‐proportional and plasma drug levels ranged from 131 to 483 μM. 15 , 23 When administered orally twice a day in healthy elderly volunteers, Flurizan demonstrated a half‐life of 6–8 hours and C max of 158 and 185 μM were achieved at T max of 1–2 hours (based on 400 and 800 mg b.i.d.). Th e CSF‐to‐plasma ratio of Flurizan was approximately 0.5%. 24 These two doses were later tested in the larger phase II and phase III trials. Based on the preclinical data described above and assuming a similar brainto‐plasma ratio (approximately 0.2), brain drug levels of 2.6–9.7 μM would achieve exposure multiples of only 0.03–0.1. Although it has been reported that Flurizan displays minimal enantiomeric bioinversion in humans, 23 it is possible that any bioinversion could have further added complexity in understanding the in vivo PK/PD relationship. As a small molecule candidate for a central nervous system (CNS) disease, Flurizan does not have an optimal brain penetration profile and it is possible that drug levels may not have achieved concentrations necessary for efficacy.
More significant early warning signs were raised by a clinical study with healthy elderly subjects: treatment with Flurizan for 21 days did not results in significant Aβ42 reduction measured by ELISA in either the plasma or the CSF compartment. 24 When comparing baseline and posttreatment levels in the four treatment groups of placebo, 200 mg b.i.d., 400 mg b.i.d., and 800 mg b.i.d., reductions in either CSF Aβ42 or plasma Aβ42 were not detected and there was no evidence of selective modulation of Aβ species, as the proposed mechanism would suggest, even though the same surface‐enhanced laser desorption/ionization time‐of‐fiight mass spectrometry (SEDLI‐TOF) method did show a shift in Aβ37/Aβtotal and Aβ42/Aβtotalratios in conditioned media from cells treated with Flurizan in vitro. 24 One caveat of the study results, as the authors argued, is that sampling time in the plasma and CSF may not have been optimal and drug activity might have been missed. In this study, only one time point was used for both predose baseline and on day 21 of the study for both plasma and CSF biomarker measurement. Since it has been reported that Aβ levels, especially in the CSF, can vary quite a bit depending on the time of collection and within hours in the same individual, 25 it would be a more rigorous study design to assess plasma and CSF Aβ levels at multiple time points.
At this point in clinical development, there is no clear evidence demonstrating that Flurizan engaged its target in human subjects. Several sensitive immunoassays have been developed to allow measurement of both plasma and CSF Aβ peptides and continuous sampling of the plasma and CSF from healthy subjects and AD patients is feasible. 26 Moreover, gamma secretase inhibitors (GSIs) have demonstrated a dose‐dependent Aβ lowering in the plasma compartment in both healthy subjects and AD patients. 27 , 28 Reduction of Aβ42 by a variety of means is a widely pursued therapeutic strategy for AD and numerous compounds are under development. In such programs, biomarker evidence that a drug engages its target and can produce a biological response at nontoxic dose range should be considered as an essential criterion for advancing a drug candidate before substantial resources are committed and a larger number of AD patients are exposed for a longer time.
Disease Severity and Progression
Given the variability of clinical efficacy measures, most phase II trials are not powered to detect significant changes in the ADAS‐Cog score or ADL. It is therefore even more important at this phase of AD drug development to incorporate biomarkers to gather supportive evidence of drug activity in the patient population. The most well‐characterized biomarkers that track AD disease progression are imaging measures for brain atrophy (eg, structural magnetic resonance imaging [MRI]) or cerebral glucose metabolism (eg, 2‐fiuoro‐2‐deoxy‐D‐glucose [FDG‐PET]). It is true that although these imaging biomarkers have been shown by multiple groups to correlate with the disease and possibly disease progression, 29 , 30 , 31 precisely how a therapeutic intervention would affect imaging measures is not fully understood. Nevertheless, with the extensive resources required to conduct large, long‐term AD trials to test clinical efficacy and the high risk of failure of disease modifiers, it is prudent to buy down the risk of clinical development by including such imaging biomarkers as early as possible.
The Flurizan phase II POC trial was a double‐blind and placebo‐controlled study over a 12‐month period that did not include any imaging or biochemical biomarker measurements. 32 Flurizan 400 mg b.i.d. or 800 mg b.i.d. dose did not reach statistical significance in reducing cognitive decline in AD, though for patients with milder symptoms, statistical significance was almost reached in the two functional scales (p= 0.059). There were other interesting signals that subsequently influenced the phase III study design. In subgroup analyses, the delayed deterioration in patients with mild disease receiving high doses of the drug and higher drug concentrations were associated with better outcomes. 32 However, such subgroup analyses are known to be unreliable because of the small numbers and the possibility that patients in a particular subgroup had less aggressive disease. The fact that Flurizan showed dose‐related effects on ADL and functional measures but not on cognitive measures was puzzling and should raise concerns, as functional measures are often more variable than the ADAS‐Cog score in AD clinical trials. In addition, in the phase II trials patients with moderate disease had numerically worse scores for Flurizan and placebo patients who then started on the drug did not show benefit. These facts could be interpreted as additional negative signals against Flurizan's efficacy.
As presented at the International Conference on Alzheimer's Disease (ICAD), 33 the phase III study of Flurizan (tarenflurbil), known as Act‐Earli‐AD, was a well‐powered, well‐designed, and well‐conducted trial in subjects with mild AD (mini‐mental state examination [MMSE] 20–26). The 18‐month study was designed to assess the potential effect of Flurizan on cognition, ADL, and global function but did not include any imaging or biochemical biomarker analysis. The treatment groups were well matched at baseline, placebo decline rates were as expected over the 18‐month period, and Apolipoprotein E (ApoE) genotypes were representative of the typical AD population. No difference between placebo and treatment groups in clinical outcome measures left very little question about the Flurizan's lack of efficacy.
Patient Selection Biomarkers
It is recognized that sporadic AD is not a single disease but likely a complex cluster of pathology and syndromes arising from interdependent mechanisms. Therefore, the AD patient population comprises heterogeneous groups with a likely difference in the rate of decline and response to candidate treatment. While numerous putative genetic risk factors have been identified in AD patients, only the presence of the ApoE4 allele has been firmly established and the analysis of the ApoE genotype is increasingly being incorporated into AD drug trials. ApoE protein has been proposed to be involved in trafficking and metabolism of amyloid peptides and ApoE4 carriers have increased risk to develop AD at an earlier onset. A posthoc analysis of ApoE4 carriers versus noncarriers did not reveal a differential effect of Flurizan compared with placebo in the phase III study and further confirmed the lack of clinical activity. 34
AD patients have also been stratified in clinical trials by disease severity, which may influence the drug response. For an Aβ‐lowering therapy, it has been postulated that more mildly affected patients may show a greater clinical benefit. Separate mildly, moderately, or severely affected patient groups have been designated based on cognitive parameters, typically MMSE scores, rather than mechanistic or pathological criteria or the rate of decline. Hints of a response to Flurizan in the mildly affected subgroup from the posthoc analysis of the phase II data led to Myriad's decision to modify its phase III study design and focus exclusively on mild AD patients. In the absence of a successful outcome, whether this was a better strategy remains unanswered.
Biomarker roadmap for developing AD‐modifying therapeutics
We propose a rigorous approach to developing AD treatments in light of the enormous investment and high risk involved in the clinical testing of candidate therapeutics. A robust biomarker data set is needed to mitigate the risk of entering lengthy and expensive phase II and phase III trials. We propose that for agents with proposed Aβ‐lowering mechanisms, a detailed analysis be undertaken in rodent, non‐human primate (NHP), and humans to establish the PK/PD relationship in relevant compartments such as the plasma, CSF, and brain ( Figure 2 ). The direct measurement of Aβ synthesis and clearance rate in and out of human CSF would offer a more direct assessment of central‐acting Aβ‐lowering agents ( Figure 3 ). We also propose that in brain imaging, approaches such as volumetric MRI and cerebral glucose metabolism measured by FDG‐PET be incorporated into POC studies. As the technology and reagents become more mature, patient selection using amyloid imaging might become feasible to select the most appropriate patients with defined amyloid pathology for POC studies in which both plasma‐ and CSF‐soluble amyloid species as well as amyloid deposition in the brain can be monitored for therapeutic activity. Such POC studies will necessitate smaller sample size and maybe a shorter treatment period; therefore, a definitive signal on cognitive and functional endpoints is not expected. Convincing drug activity on such proximal and mechanism‐based biomarkers would increase the confidence in continuing the clinical development program. Conversely, the lack of convincing biomarker results should signal a great risk in further clinical testing.
Figure 2.
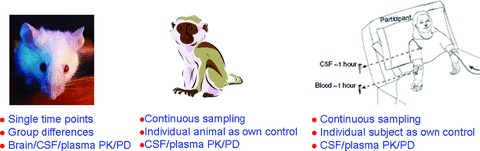
PK/PD analysis can be performed in multiple compartments across species.
Figure 3.
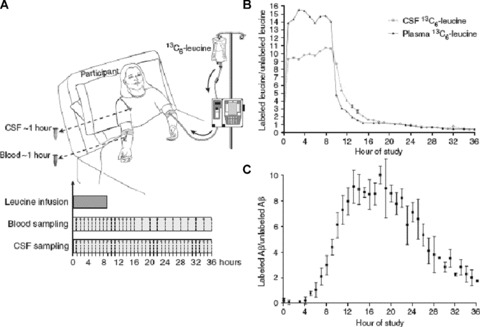
Synthesis and clearance rate of amyloid‐β peptide can be measured in human CSF (adapted from Bateman et al., 25 with permission). (a) Diagram of an individual with an intravenous catheter in antecubital vein and a lumbar catheter in the L3–L4 intrathecal space.13C6‐labeled leucine was infused at a rate of 1.8–2.5 mg/kg/h for 9 or 12 hours after an initial bolus of 2 mg/kg. Plasma samples are collected through the other intravenous line and CSF samples through the lumbar catheter every hour, (b) Labeled leucine in the CSF and blood from an individual during a 36‐hour study. Labeled leucine in the CSF and plasma reaches a near steady‐state level within an hour. There was an exponential decay in labeled leucine levels after the infusion of leucine into the bloodstream was stopped at 9 hours, (c) The average labeled CSF Ab over 36 hours from six individuals. The labeled Ab curves were averaged and the mean for each time point is shown (±SEM). Each participant underwent labeling for 9 or 12 hours, whereas sampling occurred hourly from 0 to 12, 24, or 36 hours. There is no detectable incorporation of the label in the first 4 hours. This is followed by an increase in percent labeled Ab, which plateaus near the steady‐state levels of labeled leucine (approximately 10%), before decreasing over the last 12 hours of the study.
Conclusion
In retrospect, the Flurizan phase III failure should not have been surprising. A careful examination of the accumulated evidence prior to the study start reveals weak potency, poor brain penetration, and a lack of target modulation in human subjects or AD patients. Close to 2,500 AD patients were subsequently exposed to an ineffective treatment in the phase III study, consuming untold resources in the process. The strategy taken with Flurizan certainly did not include sufficient risk mitigation and multiple clinical decision points based on biomarker data could have been incorporated.
References
- 1. Feuerstein GZ. The role of translational medicine and biomarkers research in drug discovery and development. Am Drug Discov. 2007; 2: 23–28. [Google Scholar]
- 2. Feuerstein GZ, Dormer C, Walsh FS, Hurko O, Rutkowski JL. Translational medicine perspectives in drug discovery and development part III: disease biomarkers, disease modifying biomarkers, disease labeling biomarkers and surrogate biomarkers. Am Drug Discov. 2008; 2(4): 36–41. [Google Scholar]
- 3. Feuerstein GZ, Ruffolo RR, Jr , Stiles G, Walsh FS, Rutkowski JL. Translational medicine perspectives of biomarkers in drug discovery and development: part I target selection and validation—biomarkers take center stape. Am Drun Discov. 2007: 2(5): 36–43. [Google Scholar]
- 4. Feuerstein GZ, Dormer C, Ruffolo RR, Jr , Rutkowski JL, Walsh FS, Hurko O. Translational medicine perspectives in drug discovery and development part II: target compound interaction: the vastly neglected biomarkers contributing to early clinical development failure. Am Drug Discov. 2008; 3(2): 48–54. [Google Scholar]
- 5. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physioi Rev. 2001; 81(2): 741–766. [DOI] [PubMed] [Google Scholar]
- 6. Levy‐Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995; 269(5226): 973–977. [DOI] [PubMed] [Google Scholar]
- 7. Sherrington R, Rogaev El, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K. Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature. 1995; 375(6534): 754–760. [DOI] [PubMed] [Google Scholar]
- 8. Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM. Clinical effects of Abeta immunization (AN 1792) in patients with AD in an interrupted trial. Neuroiogy. 2005; 64(9): 1553–1562. [DOI] [PubMed] [Google Scholar]
- 9. Gilman S. Clinical trials of bapineuzumab A‐beta‐amyloid‐targeted immunotherapy in patients with mild‐to‐moderate Alzheimers disease. Paper presented at the International Conference on Alzheimer's Disease and Related Disorders Meeting, July 27–31 , 2008. Chicago , IL , USA .
- 10. Fripp J, Bourgeat P, Acosta O, Raniga P, Modat M, Pike KE, Jones G, O'Keefe G, Masters CL, Ames D, Ellis KA, Muruff P, Currie J, Villemagne VL, Rowe CC, Salvado O, Ourselin S. Appearance modeling of 11C PiB PET images: characterizing amyloid deposition in Alzheimer's disease, mild cognitive impairment and healthy aging. Neuroimage. 2008; 43: 430–439. [DOI] [PubMed] [Google Scholar]
- 11. Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez‐Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001; 414(6860): 212–216. [DOI] [PubMed] [Google Scholar]
- 12. Kukar T, Golde TE. Possible mechanisms of action of NSAIDs and related compounds that modulate gamma‐secretase cleavage. Curr Top Med Chem. 2008; 8(1): 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lleo A, Berezovska O, Herl L, Raju S, Deng A, Bacskai BJ, Frosch MP, Irizany M, Hyman BT. Nonsteroidal anti‐inflammatory drugs lower Abeta42 and change presenilin 1 conformation. Nat Med. 2004; 10(10): 1065–1066. [DOI] [PubMed] [Google Scholar]
- 14. Beher D, Clarke EE, Wrigley JD, Martin AC, Nadin A, Churcher I, Shearman MS. Selected nonsteroidal anti‐inflammatory drugs and their derivatives target gamma‐secretase at a novel site. Evidence for an allosteric mechanism. J Biol Chem. 2004; 279(42): 43419–43426. [DOI] [PubMed] [Google Scholar]
- 15. Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. NSAIDs and enantiomers of flurbiprofen target gamma‐secretase and lower Abeta 42 in vivo. J Gin Invest. 2003; 112(3): 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morihara T, Chu T, Ubeda O, Beech W, Cole GM. Selective inhibition of Abeta42 production byNSAIDR‐enantiomers. J Neurochem. 2002; 83(4): 1009–1012. [DOI] [PubMed] [Google Scholar]
- 17. Martone RL, Zhou H, Atchison K, Comery TA, Xu J, Huang X, Gong X, Jin M, Kreft A, Harrison B. GSI‐953 (begacestat): a novel, selective thiophene sulfonamide inhibitor of APP gamma‐secretase for the treatment of Alzheimer's disease. Manuscript in preparation. 2009. [DOI] [PubMed] [Google Scholar]
- 18. Lanz TA, Karmilowicz MJ, Wood KM, Pozdnyakov N, Du P, Piotrowski MA, Brown TM, Nolan CE, Richter KE, Finley JE, Fei Q, Ebbinghaus CF, Chen YL, Spracklin DK, Tate B, Geoghegan KF, Lau LF, Auperin DD, Schachter JB. Concentration‐dependent modulation of amyloid‐beta in vivo and in vitro using the gamma‐secretase inhibitor, LY‐450139. J Pharmacol Exp Ther. 2006; 319(2): 924–933. [DOI] [PubMed] [Google Scholar]
- 19. Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, Freedman SB, Frigon NL, Games D, Hu K, Johnson‐Wood K, Kappenman KE, Kawabe TT, Kola I, Kuehn R, Lee M, Liu W, Motter R, Nicols NF, Power M, Robertson DW, Schenk D, Schoor M, Shopp GM, Shuch ME, Sinha S, Svensson KA, Tatsuno G, Tintrup H, Wijsman J, Wright S, McConlogue L. BACE knockout mice are healthy despite lacking the primary beta‐secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet. 2001; 10(12): 1317–1324. [DOI] [PubMed] [Google Scholar]
- 20. McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB, Games D, Johnson‐Wood K, Lee M, Zeller M, Liu W, Motter R, Sinha S. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APPtransgenic mice. J Biol Chem. 2007; 282(36): 26326–26334. [DOI] [PubMed] [Google Scholar]
- 21. Kukar T, Prescott S, Eriksen JL, Holloway V, Murphy MP, Koo EH, Golde TE, Nicolle MM. Chronic administration of R‐flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neurosci. 2007; 8: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lanz TA, Fici GJ, Merchant KM. Lack of specific amyloid‐beta(1–42) suppression by nonsteroidal anti‐inflammatory drugs in young, plaque‐free Tg2576 mice and in guinea pig neuronal cultures. J Pharmacol Exp Ther. 2005; 312(1): 399–406. [DOI] [PubMed] [Google Scholar]
- 23. Murray ED, Quiggle D, Gibson K, Leipod D, Loughman B, Wechter W. Phase I single‐dose pharmakinetics and lack of enantiomeric byconversion of E‐7869 (R‐flurbiprofen). Gin Pharmacol Ther. 2000; 67(2): 103. [Google Scholar]
- 24. Galasko DR, Graff‐Radford N, May S, Hendrix S, Cottrell BA, Sagi SA, Mather G, Laughlin M, Zavitz KH, Swabb E, Golde TE, Murphy MP, Koo EH. Safety, tolerability, pharmacokinetics, and Abeta levels after short‐term administration of R‐flurbiprofen in healthy elderly individuals. Alzheimer Dis Assoc Disord. 2007; 21 (4): 292–299. [DOI] [PubMed] [Google Scholar]
- 25. Bateman RJ, Wen G, Morris JC, Holtzman DM. Fluctuations of CSF amyloid‐beta levels: implications for a diagnostic and therapeutic biomarker. Neurology. 2007; 68(9): 666–669. [DOI] [PubMed] [Google Scholar]
- 26. Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid‐beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006; 12(7): 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Siemers ER, Quinn JF, Kaye J, Farlow MR, Porsteinsson A, Tariot P, Zoulnouni P, Galvin JE, Holtzman DM, Knopman DS, Satterwhite J, Gonzales C, Dean RA, May PC. Effects of a gamma‐secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology. 2006; 66(4): 602–604. [DOI] [PubMed] [Google Scholar]
- 28. Wan HI, Bard J, Martone RL, Raje S, Forlow S, Balliet C, Pastore A, Burczynski ME, Atchison K, Zhou H, Mayer S, Adkins K, Gruver M, Rosen B, Harrison B, Magolda R, Reinhart P, Pangalos M, Kreft A, Jacobsen S, Silver P, Paul J, Frick G. GSI‐953, a potent and selective gamma‐secretase inhibitor, modulates Abeta peptides in mice and humans: translating the PK/PD biomarker relationships in different biological compartments between rodent and human. Paper presented at the ICAD, July 27–31 , 2008, Chicago , IL
- 29. Jack CR, Jr , Petersen RC, Xu Y, O'Brien PC, Smith GE, Ivnik RJ, Boeve BF, Tangalos EG, Kokmen E. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000; 55(4): 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET evaluation of cerebral metabolic decline in dementia: a potential outcome measure in Alzheimer's disease treatment studies. Am J Psychiatry. 2002; 159(5):738–745. [DOI] [PubMed] [Google Scholar]
- 31. Mosconi L, Tsui WH, De Santi S, Li J, Rusinek H, Convit A, Li Y, Boppana M, De Leon MJ. Reduced hippocampal metabolism in MCI and AD: automated FDG‐PET image analysis. Neurology. 2005; 64(11): 1860–1867 [DOI] [PubMed] [Google Scholar]
- 32. Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer's disease: a randomised phase II trial. Lancet Neurol. 2008; 7(6): 483–493. [DOI] [PubMed] [Google Scholar]
- 33. Green RCS, LS, Zavitz KH, Amato DA, Beelen AP, Swabb EA. Safety and efficacy of tarenflurbil in subjects with mild Alzheimer's disease: results from an 18‐month multicenter phase 3 trial. Paper presented at the International Conference on Alzheimer's Disease and Related Disorders Meeting, July 27–31 , 2008, Chicago , IL , USA .
- 34. Green RC, Schneider LS, Zavitz KH, Amato DA, Beelen AP, Swabb EA. safety and efficacy of tarenflurbil in subjects with mild Alzheimer's disease: results from an 18‐month multicenter phase 3 trial. Paper presented at the ICAD, July 27–31 , 2008, Chicago , IL , USA .