

Abstract
In the failing human heart (FHH) the induction of a fetal contractile protein gene program is directly and selectively associated with the dilated cardiomyopathy (DCM) phenotype and involves multiple signaling pathways. In response to cardiac stress signals, class II HDACs are subject to phosphorylation dependent nuclear export, which allows for activation of fetal cardiac genes via the transcription factor MEF2. The current study tests the hypothesis that MEF2 activation produced by class II HDAC de‐repression is present in the FHH. In this study, human left ventricular tissue from nonfailing and failing adult hearts was analyzed for the presence of MEF2, HDACs 4 and 5. CaMK and HDAC kinase activities were measured in tissue homogenates. In nuclear fractions from failing ventricles, HDAC4 and HDAC5 protein was decreased versus nonfailing controls. MEF2 was not reduced in failing nuclear fractions. CaMK and HDAC kinase activities were increased in failing versus nonfailing hearts. pkcμ (pkd1) activity was increased in nuclear fractions from failing human LVs. These data provide support for decreased nuclear compartment class II HDACs in the FHH, associated with increased activities of kinases known to phosphorylate class II HDACs.
Keywords: heart failure, histone deacetylases, protein phosphorylation, transcription factors
Introduction
Myocyte enhancer factor 2 (MEF2) is a family of transcription factors involved in muscle‐specific gene expression and in the regulation of cardiac development. 1 In mammalian species, there are four members (MEF2 A‐D) that are widely expressed in skeletal and cardiac muscle, and in neural and angiogenic tissue. MEF2 proteins homo‐ and hetero‐dimerize and bind to DNA through an amino‐terminal DNA‐binding domain. MEF2 can be regulated by phosphorylation, through a number of serine/threonine kinases of the mitogen‐activated protein (MAP) kinase family. 2
In eukaryote chromatin, structure is regulated by remodeling, covalent modification, and histone replacement. 3 Histones, which form the nucleosomal structure, are modified by acetylation, phosphorylation, and other processes. Histone deacetylases (HDACs) are the enzymes responsible for terminal lysine deacetylation, while the reverse process is effected by histone acetylases (HATs). 4 A total of 19 HDACs have been identified in humans. 3 HDACs have been grouped into three distinct classes, with corresponding homology to the yeast S. Cerevisiae. 5 Class I HDACs are represented by HDAC1, 2, 3, and 8, with homology to the yeast Rpd3, where the catalytic domain constitutes the majority of the protein. HDAC1 and 2 form multiprotein complexes as Sin3/HDAC and NuRD/MI2/NRD/HDAC, while HDAC3 binds to other transcriptional compressors. Class III members are related to the yeast SIR2 and exhibit ADP‐ribosylation activity in addition to deacetylase activity. Class II HDACs comprise HDAC4, 5, 6, 7, 9, and 10. Class II HDACs are most prominently represented in striated muscle and brain, in contrast to the ubiquitously expressed Classes I and III. Class IIa HDACs (HDAC4, 5, 7, 9) share nuclear localization signals flanked by two conserved serines, which when phosphorylated are targets for binding of the chaperone protein 14–3‐3, which escorts the HDAC from the nucleus. 6 , 7 , 8 Class IIb HDACs are thus far limited to HDAC6, remarkable for its double catalytic domain, and HDAC10, a newly discovered member that possesses a leucine rich domain. Class II HDACs possess N‐terminal MEF2 docking domains and have been shown to function as transcriptional repressors of MEF2 target gene expression. 9 , 10 , 11
Under pathophysiological conditions, hypertrophic signals reaching the damaged heart stimulate multiple receptor pathways that raise intracellular Ca2+, which activates a variety of kinases capable of transducing a hypertrophic response. Ca2+/calmodulin‐dependent protein kinases (CaMK) as well as novel HDAC kinases, such as Protein Kinase Dl (PKD1), phosphorylate class II HDACs. 12 , 13 The ensuing conformational change and dissociation from MEF2 leads to class II HDAC nuclear export. MEF2 freed of its repression by class II HDACs may then activate muscle‐specific gene expression, in particular by acting combinatorially with other muscle specific transcription factors such as GATA4 and NFAT3. 1 , 14 This regulatory mechanism is summarized in Figure 1 .
Figure 1.
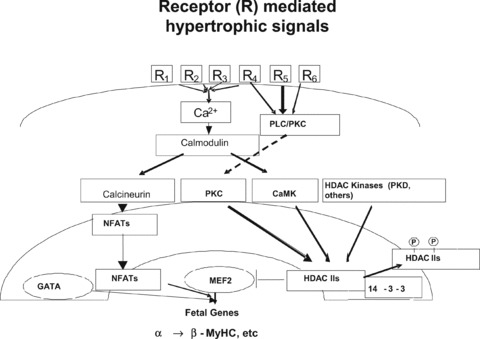
MEF2‐HDAC model, schematic representation of class II HDAC repression of MEF2 transcriptional activation. In the stressed or damaged heart multiple receptor‐activated pathways increase intracellular calcium, which among other processes, activates calcium dependent CaMKII. Downstream from CaMK, another kinase activated by hypertrophic signals, PKD1, phosphorylates class II HDACs, leading to a conformational change and dissociation from MEF2. The class II HDACs then bind to the chaperone 14–3‐3 and are exported to the cytosol.
As in rodent myocardium, the failing human heart (FHH) exhibits evidence of induction of a “fetal” contractile protein gene program, consisting of increased expression of the fetal genes encoding β‐myosin heavy chain and natriuretic peptides, and decreased expression of the adult genes α‐myosin heavy chain and Sarcoplasmic reticulum‐Ca2+ ATPase (SERCA). 15 , 16 Moreover, reversal of this pattern is associated with improvement of the dilated cardiomyopathy (DCM) phenotype, 17 , 18 suggesting that induction of a fetal gene program is an important component of the molecular pathophysiology of human systolic heart failure. However, in human myocardium there is little information available on potential molecular mechanisms responsible for fetal gene induction. In the current work, we test the hypothesis that the nuclear export of class II HDACs is associated with increased activities of multiple kinases that phosphorylate class II HDACs in the FHH.
Materials and Methods
Tissue procurement
Nonfailing (n= 24) human hearts were obtained from unused organ donors with no history of cardiac dysfunction (left ventricular ejection fractions 0.65 ± 0.09 by echocardiograms performed as part of the organ recovery process) or coronary artery disease. Failing hearts (n= 24) were obtained from end stage cardiac transplant recipients with advanced nonischemic dilated cardiomyopathies (left ventricular ejection fractions 0.16 ± 0.07, p= < 0.001 vs. nonfailing). The two groups of hearts exhibited no difference in age (nonfailing, 48.3 ± 8 years; failing, 50.4 ± 11 years). The gender distribution was 11 males, 13 females in nonfailing; 13 males, 11 females in failing. The CaMK activity was measured in a subset of 20 hearts (10 nonfailing and 10 failing). Phosphorylated and nonphosphorylated HDAC5, CaMKII, and PKD were measured in an additional subset of eight nonfailing and eight failing hearts.
Tissue processing
For protein abundance measurements human left ventricular free wall full thickness 1 g aliquots frozen at −80°C were pulverized in liquid nitrogen and resuspended in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris‐HCl, pH 7.5; 150 mM NaCl; 1% Nonidet P‐40; 0.5% sodium deoxycholate; 0.1% SDS; 1 mM EDTA; 1 mM NaF; 2 μg/mL of aprotinin, pepstatin A, and leupeptin; 6 μg TPCK; 6 μg TLCK). The tissues were homogenized at 4°C for 30 seconds × 3 with a Polytron homogenizer. The homogenates were centrifuged at 3000 ×g for 10 minutes at 4°C. The supernatants were then centrifuged 48000 ×g for 30 minutes at 4°C. Aliquots of these preparations were frozen at −20°C for short‐term use. For nuclear‐cytosolic localization studies human cardiac tissue was prepared using the NE‐PER system from Pierce Chemicals (Rockford, IL, USA). All the mentioned tissue preparation steps and performed assays were done in the presence of complete protease and phosphatase inhibition.
Western blot analysis
Total cardiac tissue homogenates (50–60 μg loaded in equal protein amounts in a volume of 20 μL) were electrophoretically resolved on a 7.5% or 10% SDS‐polyacrylamide gel. The separated proteins were transferred to nitrocellulose or PVDF membranes and processed for western blot analysis. Complete details will be found in the accompanying supplemental material file.
Tissue fractionation into nuclear and cytosolic fractions
The tissue fractionation was performed according to the manufacturer's (Pierce, Rockford, IL, USA) protocol with minor modifications. A brief description of the previously mentioned modifications is given in the supplemental material file. Results of western blot data from nuclear and cytosolic fractions were expressed as a fraction of the relative abundance of calnexin in the respective nonfailing compartments.
CaMK isoform identification and protein abundance
The amount and isoform pattern of CaMK were assessed in total protein homogenates as well as in nuclear and cytoplasmic fractions with total CaMKII and CaMKII δ antibodies.
CaMK enzyme activity
Measurement of CaM kinase enzyme activity was performed in total protein homogenates by previously described techniques 19 with some modifications. Additional information on the CaMK enzyme activity measurement can be found in the supplemental file.
PKCμ (PKD1) activity assay
Nuclear and cytoplasmic fractions of the additional subset of eight nonfailing and eight failing hearts were fractionated on 8% SDS‐PAGE and the proteins were transferred to nitrocellulose and processed as described previously. PKD activity was measured by probing the membrane with an antibody that recognizes the human autophosphorylation site pS910 (homologous site with the murine pS916), which indicates activation of PKCμ. ( Figure 8 ).
Figure 8.
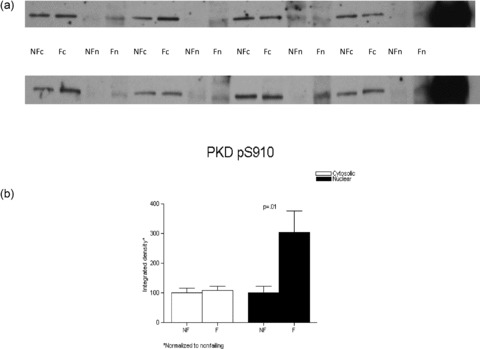
PKD activity as assessed by the detection of phosphorylatyed serine 910 signal is highly increased in the nuclear fraction in the failing versus nonfailing LVs. Nuclear and cytosolic fractions from eight nonfailing and eight failing LVs were loaded on an 8% SDS‐PAGE gel. (A) Detection of PKD pS910 was obtained with a rabbit antibody and the proteins were visualized with enhanced chemiluminescence. (B) The results normalized to their corresponding nonfailing compartment values were graphed.
HDAC kinase enzyme activity
Global HDAC kinase activity was assessed by the measurement of radioactive ATP incorporation into an HDAC5 substrate fused to glutathione S‐transferase when incubated with homogenates of normal and failing human left ventricles (LVs). Additional description of the methods employed is given in the supplemental file.
Data handling and statistical methods
Variance of mean values was expressed as standard deviations for baseline data, and standard error of the mean for changes from baseline. Differences between cytosolic and nuclear expression (normalized to calnexin as a loading control) were determined by comparing the difference between each ventricle's cytosolic and nuclear densitometry value to the null hypothesis value of zero difference, within nonfailing and failing groups. Differences between failing and nonfailing groups (for nuclear and cytosolic fractions) were determined by comparing each densitometry value to the average of the respective nonfailing cytosolic and nuclear fraction values, which were set at 1 for each protein on each western blot. All comparisons were made by the Wilcoxon Signed Rank test. A p value of <0.05 in a two‐tailed distribution was considered statistically significant.
Results
Tissue fractionation validation
The integrity of the cytosolic and nuclear fractionation was assessed by western blotting with antibodies raised against the cytosolic protein LDH‐H4, and the nuclear transcription factor SP‐1. The results are shown in Figure SI in the accompanying supporting information.
Equivalent levels of MEF2 in homogenates and tissue fractions of nonfailing and failing hearts
Nonfailing and failing left ventricular homogenates were probed for the presence of MEF2 proteins, using antibodies raised to MEF2. Immunodetection of the abundance of MEF2 in the cytosolic versus nuclear fractions in nonfailing and failing hearts revealed that, when normalized to the corresponding calnexin values and to each fractions average nonfailing value, there was no difference in protein expression in the cytoplasmic (failing 0.96 ± 0.06 vs. nonfailing 1 ± 0.12; p= 0.58) or nuclear (failing 0.85 ± 0.08, nonfailing 1 ± 0.09; p= 0.23) fractions ( Figure 2 ).
Figure 2.
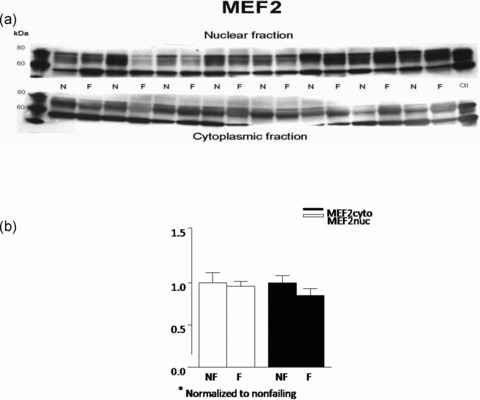
MEF2 protein abundance is unchanged in FHH. Nuclear and cytoplasmic lysates of eight nonfailing and eight failing LV were fractionated on 8% SDS‐PAGE gel and the proteins were transferred to nitrocellulose. (A) MEF2 protein abundance was assessed with a polyclonal rabbit antibody that recognizes mainly MEF2A. Similar results were obtained in an additional 16 nonfailing and 16 failing hearts. (B) The above results were also quantified and are rendered in graph form.
HDAC4 and HDAC5 protein abundance in cytosolic and nuclear fractions of nonfailing and failing hearts
Since MEF2 protein levels were unchanged in failing versus nonfailing hearts, we next investigated nuclear versus cytosolic localization of its transcriptional repressors HDAC4 and HDAC5. Nuclear export of either HDAC can result in increased MEF2 activity with no increase in protein levels. 11 As shown in Figure 3, immunodetectable HDAC4 was unchanged in failing versus nonfailing cytosolic fractions (normalized to nonfailing, failing = 0.84 ± 0.06, p= 0.31). Similarly, the immunodetectable HDAC5 ( Figure 4 ) in cytosolic fractions of failing versus nonfailing LVs exhibited no difference in abundance (0.91 ± 0.06, p= 0.45).
Figure 3.
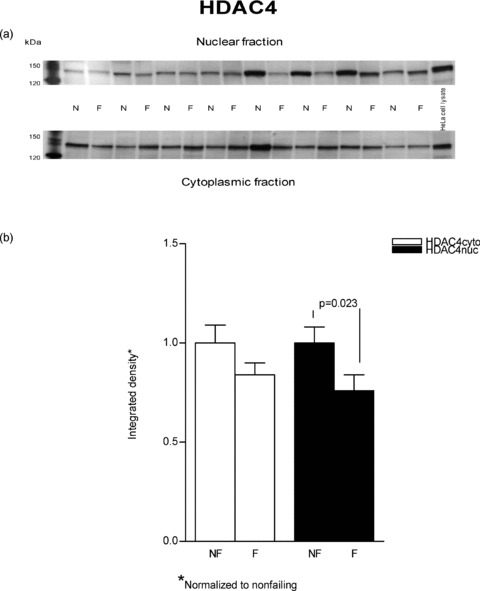
HDAC4 protein levels are marginally increased in cytosolic fractions from failing versus nonfailing LVs. Proteins (50 μg) from nuclear and cytoplasmic lysates of eight nonfailing and eight failing LVs were separated on an 8% SDS‐PAGE gel and transferred to PVDF membrane. A rabbit polyclonal antibody to HDAC4 was used to determine the protein abundance by using enhanced chemiluminescence visualization. (A) This figure is a typical representative of the immunodetection of HDAC4 in the examined samples. (B) The chemiluminescent signals were quantified and the results are reported in graph form.
Figure 4.
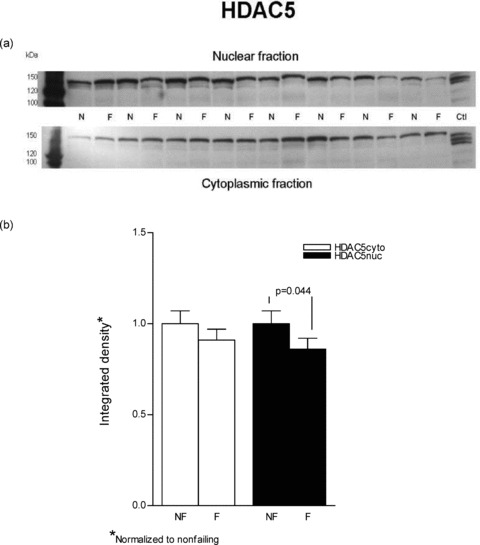
HDAC5 is decreased in nuclear fractions from failing versus nonfailing LVs. Lysates from eight nonfailing and eight failing LVs were loaded on an 8% SDS‐PAGE gel. (A) The proteins were separated and transferred to a PVDF membrane. Immunoblotting for HDAC5 was done with a rabbit polyclonal antibody and the protein was visualized with enhanced chemiluminescence. Similar results were obtained in an additional 16 nonfailing and 16 failing hearts. (B) The results from the above representative blot are further expressed in graph form.
In contrast to cytosolic fractions, in nuclear preparations immunodetectable HDAC4 and HDAC5 were both decreased in failing hearts. As shown in Figure 3 HDAC4 was substantially decreased in the failing nuclear extracts (0.76 ± 0.08 vs. 1 ± 0.08, p= 0.023). In Figure 4 it can be seen that HDAC5 abundance was also decreased in nuclear fractions prepared from failing hearts compared to nonfailing (0.86 ± 0.06 vs. 1 ± 0.07, p= 0.044.). In an alternate analysis that compares each sample's cytosolicmuclear distribution, the ratios of cytosolic to nuclear integrated values (C/N) were compared between nonfailing and failing hearts. In this instance, the differences noted in the previous statistical analysis were not significant. For HDAC4 the failing C/N value, 1.41 ± 0.23, was not different from the nonfailing value, 1.19 ± 0.38 (p= 0.3). Similarly, the failing HDAC5 C/N, 1.42 ± 0.44, was not statistically significant compared to the nonfailing value, 0.64 ± 0.07 (p= 0.06). It should be noted that the variance expressed as the coefficient of variation is 3–4‐fold higher in the C/N ratios than within nuclear or cytosolic measurements, which contributes to the HDAC5 increase in failing of 2.2‐fold not being statistically significant.
HDAC kinase activity in failing and nonfailing hearts
Since HDAC4/5 nuclear localization is decreased in the failing hearts and HDAC kinases are known to phosphorylate class II HDACs we next asked whether global HDAC kinase activity is changed in the failing heart. 12 , 13 As shown in Figure 5, total HDAC kinase activity, as assessed by phosphorylation of an HDAC5 substrate, was uniformly increased in homogenates of failing human LVs, by 3 ± 0.3 fold (p= 0.04).
Figure 5.
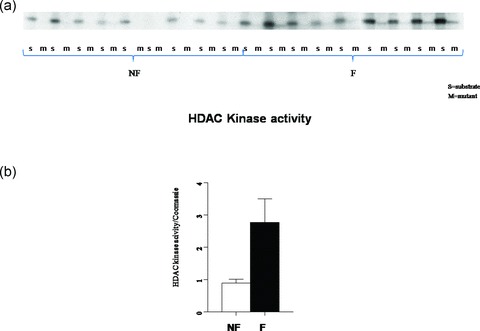
HDAC kinase activity is increased three fold in the FHH. Equal amounts of GST‐HDAC5 substrate and GST‐HDACmutant were incubated with nonfailing and failing hearts in the presence of 5 μCi [γ‐32P]‐ATP. (A) Phosphoproteins were resolved on SDS‐PAGE. There was equal loading by corresponding Coomassie staining of the gel (not shown). (B) The signals normalized to the corresponding Coomassie loading are presented in graph form.
CaMK activity and isoform abundance in failing and nonfailing hearts
Among the kinases that phosphorylate class II HDACs, CaMK occupies a prominent role. The activity of CaMK was assessed in failing and nonfailing LVs ( Figure 6a ). Homogenized tissue was incubated under a broad range of calcium conditions (10 pM to 3mM) in the presence of calmodulin, Syntide II substrate, and hot ATP. The results are expressed as incorporated phosphate per minute per milligram of protein. The increase in CaMK activity in failing hearts (by 2.5 ± 0.3 fold, p= 0.05) is in concordance with previous published results. 20 To determine whether the increased activity correlated with increased amounts of CaMK protein 30 μg of total protein homogenates were separated on 10% SDS‐PAGE and the proteins blotted onto nitrocellulose. The blot was probed for CaMKII δ protein, which was normalized to total GAPDH levels on the same blot ( Figure 6b ). The signal normalized to the corresponding GAPDH levels obtained on the same blot shows no difference between failing and nonfailing hearts.
Figure 6.
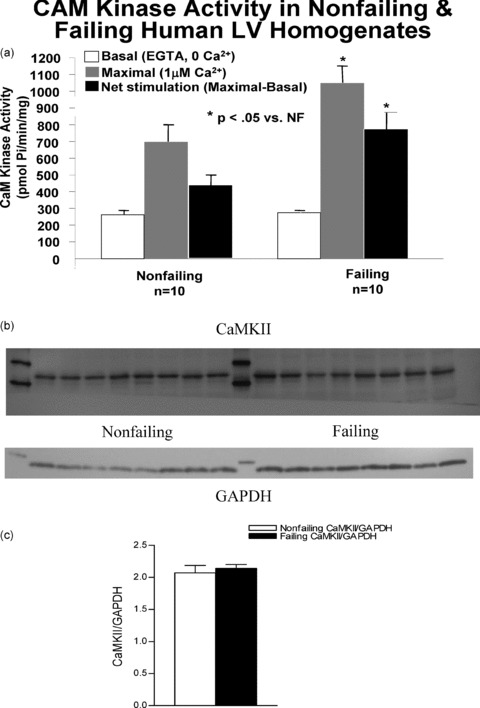
CaMK activity in human nonfailing versus failing LV. (A) The activity of CaMK was assessed in failing and nonfailing LVs. CaM kinase activity is approximately double that in the normal heart. CaMKII protein levels are unchanged in nonfailing versus FHHs. Protein levels in ten nonfailing and eight failing human LV total lysates were measured. Homogenized tissue was fractionated on a 10% SDS‐PAGE gel, transferred to nitrocellulose and immunoblotted with an antibody to CaMKII. (B) The upper band shows the presence of CaMKII at around 56kDa. On the same blot, the GAPDH signal shows equal loading in all lanes. (C) The accompanying graph shows that the ratio of CaMKII/ GAPDH integrated intensity values in not different in nonfailing and failing hearts.
In tissue cytosolic and nuclear fractions, we also measured the immunodetection of CaMKII phosphorylated on threonine 286 as a measure of total CaMKII activity ( Figure 7 ). CaMKII pT286 abundance was higher in failing versus nonfailing in the nuclear fraction (1.65 ± 0.15 vs. 1 ± 0.12, respectively, p= 0.003) but nonsignificantly different in the cysolic fraction (1.37 ± 0.15 vs. 1 ± 0.16, p= 0.06).
Figure 7.
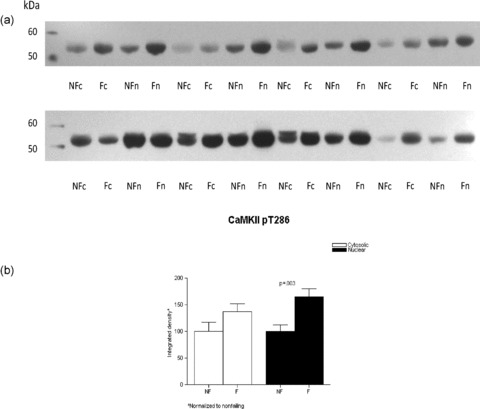
CaMKII phosphorylation is increased in the FHH. Lysates from eight nonfailing and eight failing LVs were loaded on a 10% SDS‐PAGE gel, the proteins fractionated and transferred to nitrocellulose membrane. The membranes were immunoblotted with a rabbit polyclonal antibody to CaMKII pT286, which has been shown to mirror total CaMKII activity. (A) The proteins were then visualized with enhanced chemiluminescence. Panel B illustrates the above results in graph form.
PKCμ activity in nuclear and cytosolic fractions of nonfailing and failing left ventricles
We next examined whether PKCμ (PKD1) activity was increased in the failing heart. As shown in Figure 8, PKCμ activity as assessed by a phospho Ab that recognizes the human autophosphorylation site was increased in nuclear fractions from failing human LVs (3.04 ± 0.76 vs. 1 ± 0.22, failing vs. nonfailing, respectively, p= 0.01) and unchanged in cytosolic fractions.
Discussion
In adult cardiac myocytes, MEF2 is associated with class II HDACs, resulting in repression of downstream MEF2 target genes. In model systems of pathologic cardiac hypertrophy, class II HDACs have been shown to undergo phosphorylation‐dependent nuclear export in response to stress signals, thereby freeing MEF2 to stimulate expression of genes that drive cardiac growth. To begin to assess whether this paradigm is relevant to the failing/hypertrophied human heart, we performed multiple experiments designed to test the hypothesis that de‐repression of MEF2 by nuclear export of class II HDACs is present in the failing, hypertrophied human LV.
In humans or animal models, chronic hemodynamic overload results in a hypertrophic, remodeling process that may ultimately result in decreased contractile function and overt clinical heart failure. In both established rodent models and the FHH 15 , 16 , 17 , 18 the remodeling process is characterized by changes in the gene expression of regulators of cell growth and contractile function. The pattern of gene expression in the normal adult is changed to a “fetal” gene pattern in the pathologically hypertrophied heart, where adult genes such as α‐MyHC exhibit decreased expression, and fetal isoforms such as β‐MyHC and natriuretic peptides are increased.
On a transcriptional level, recent studies have provided information about the network of signal transduction pathways mediating myogenic regulation. The hypertrophic stimuli that elicit a pathologic change in the heart have been shown to act at least in part through an increase of intracellular calcium and activation of CaMK. 21 , 22 , 23 In turn, CaMK stimulates MEF2 activity, which leads to the activation of myogenic genes. 24 , 25 , 26 At least part of the action of CaMK in this regard is mediated by phosphorylation of class II HDACs. 11
In the present report, we show that CaMK enzyme activity is increased in left ventricular homogenates of patients with DCM, in agreement with previous reports. 19 , 20 In addition, in nuclear fractions from failing hearts, CaMKII pT286 abundance was increased by 65%, indicating a greater degree of enzyme activation by autophosphorylation in failing hearts. However, protein abundance of CaMKIδ, which is the predominant member of the multifunctional group of CaMK in cardiac muscle, was not different in nonfailing versus failing hearts. These data imply that a posttranslational modification of CaMKIδ or up‐regulation of another, undetected isoform of CaMKII, was responsible for the increased activity in failing preparations.
Recently, a new kinase, protein kinase D1 (protein kinase Cμ) was shown to phosphorylate class II HDACs in the same position as CaMK. 27 In failing hearts we found a 3‐fold increase in HDAC kinase activity, as assessed by phosphorylation of S259 of HDAC5. This could be due to increased PKD activity 13 , 28 as demonstrated in nuclear fractions of failing versus nonfailing LVs, but could also be related to the increased CaMKII activity or to increased activity of both kinases. Regardless of its origin, global HDAC kinase functional activity is clearly increased in the FHH, and presumably this would lead to nuclear export of class II HDACs.
A test for the class II HDAC‐MEF2 activation hypothesis in the failing heart would logically include an assessment of their relative abundance in nuclear and cytosolic fractions, in nonfailing versus failing left ventricular preparations. HDAC4, whose abundance was greater in nuclear versus cytosolic fractions in nonfailing hearts only, exhibited a 24% decrease in failing versus nonfailing nuclear fractions. HDAC5 also exhibited a significant, lesser decrease 14% in that location without a change in the cytosol. The increase in CaMK and HDAC kinase activity suggests that nuclear export of HDAC4 and 5 is the mechanism responsible for decreased HDAC4 and 5 levels in the nuclear fraction. However, since no increase in HDAC4 and 5 in the cytosolic fraction of failing heart preparations was observed, it is possible that the decrease in nuclear HDAC4 and 5 is due to other factors, such as compartment‐specific protein degradation.
Recently Bossuyt et al. 28 also tested the class II nuclear export hypothesis in FHHs, and several of our findings are in agreement with their data. That study 28 also examined left ventricular homogenates from nonfailing and failing human LVs, and our data on increased CaMK enzyme activity are in agreement with their data on increased autophosphorylated CaMKII. In addition, we demonstrated that the nuclear fraction contained an increase in autophosphorylated CAMKII activity, but the cytosolic fraction only a nonsignificant trend. However, unlike what was shown by Bossuyt et al. 28 our data for CaMKII protein abundance did not demonstrate an increase in the failing heart, suggesting that a posttranslational modification of CaMKII, such as phosphorylation, accounted for the increase in enzyme activity. Evidence for a decrease in HDAC5 in nuclear fractions of failing human LV was also found in both studies, indirectly assessed by Bossuyt et al. 28 as an increase in HDAC5 phosphorylation in failing LV homogenates and in our study directly by Western blotting in failing LV nulear fractions. Evidence for increased PKD1 activity was found in failing LV homogenates by the Bossuyt study 28 and in failing LV nuclear fractions in our study. In addition, our study demonstrated that immunodetectable MEF2 protein abundance, likely predominately MEF2A, 29 is unchanged in homogenates and nuclear fractions of failing human LVs. Our data also demonstrated directly and unequivocally that global HDAC kinase activity is increased in failing human ventricles, presumably related to the increase in CaMKII and PKD1 activities. Our data also indicate that decreased HDAC4 in nuclear fractions may be more important than decreases in HDAC5, which would correlate with increased CaMKII activity 30 Finally, while our data are strongly suggestive that nuclear export of class II HDACs contributes to the transcriptional de‐repression on MEF2, such an effect in the form of increased MEF2 binding to target genes or increased target gene transcriptional activity has not been directly demonstrated in this study.
In summary, these data provide support for the hypothesis that nuclear to cytoplasm export of class II HDACs is activated in the FHH, under the control of up‐regulated HDAC kinases.
Supporting information
Figure SI. Nucleo‐cytoplasmic compartmentalization was assessed by measuring the protein abundance of the nuclear protein Spl and the cytoplasmic protein LDH‐H4.
Please note: Wiley‐Blackwell Publishing is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
Some of these data were first presented in abstract form at the 2002 AHA meeting.
Sources of support: 2R01 HL48013, R01 HL616401, unrestricted grant from Gilead Inc.
We also would like to acknowledge the expertise of Joseph Cleveland, M.D., and Brian D. Lowes, M.D., in the obtention of the cardiac tissue used in this study.
References
- 1. Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor‐2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998; 14: 167–196. [DOI] [PubMed] [Google Scholar]
- 2. Zhao M, New L, Kravchenko VV, Kato Y, Gram H, Di Padova F, Olson EN, Ulevitch RJ, Han J. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999; 19: 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang XJ, Seto E. Histones, which form the nucleosomal structure, are modified by acetylation, phosphorylation and other processes. Curr Opin Genet Dev. 2003; 13: 143–153. [DOI] [PubMed] [Google Scholar]
- 4. Khochbin S, Verdel A, Lemercier C, Seigneurin‐Berny D. Functional significance of histone deacetylase diversity. Curr Opin Genet Dev. 2001; 11: 162–166. [DOI] [PubMed] [Google Scholar]
- 5. De Ruijter AJ, Van Gennip AH, Caron HN, Kemp S, Van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family–review. Biochem J. 2003; 370(Pt 3): 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McKinsey TA, Zhang CL, Olson EN. Identification of a signal‐responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol. 2001; 21(18): 6312–6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kao HY, Verdel A, Tsai CC, Simon C, Juguilon H, Khochbin S. Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J Biol Chem. 2001; 276(50): 47496–47507. [DOI] [PubMed] [Google Scholar]
- 8. Ellis JJ, Valencia TG, Zeng H, Roberts LD, Deaton RA, Grant SR. CaM kinase NdeltaC phosphorylation of 14–3‐3beta in vascular smooth muscle cells: activation of class II HDAC repression. Mol Cell Biochem. 2003; 242(1–2): 153–161. [PubMed] [Google Scholar]
- 9. Lu J, McKinsey TA, Nicol RL, Olson EN. Signal‐dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA. 2000; 97: 4070–4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000; 6: 233–244. [DOI] [PubMed] [Google Scholar]
- 11. McKinsey TA, Zhang CL, Lu J, Olson EN. Signal‐dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature (London). 2000; 408: 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal‐responsive repressers of cardiac hypertrophy. Cell. 2002; 110(4): 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist‐dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004; 24(19): 8374–8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morin S, Charron F, Robitaille L, Nemer M. GATA‐dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000; 19(9): 2046–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lowes BD, Minobe WA, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, Badesch DB, Groves BM, Gilbert EM, Bristow MR. Changes in gene expression in the intact human heart: down‐regulation of α‐myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest 1997; 100:2315–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997; 100: 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lowes BD, Gilbert EM, Abraham WT, Minobe WA, Larrabee P, Ferguson D, Wolfel EE, Lindenfeld J, Tsvetkova T, Robertson AD, Quaife RA, Bristow MR. Myocardial gene expression in dilated cardiomyopathy treated with beta‐blocking agents. N Engl J Med. 2002; 346(18): 1357–1365. [DOI] [PubMed] [Google Scholar]
- 18. Abraham WT, Gilbert EM, Lowes BD, Minobe WA, Larrabee P, Roden RL, Dutcher D, Sederberg J, Lindenfeld JA, Wolfel EE, Shakar SF, Ferguson D, Volkman K, Linseman JV, Quaife RA, Robertson AD, Bristow MR. Coordinate changes in Myosin heavy chain isoform gene expression are selectively associated with alterations in dilated cardiomyopathy phenotype. Mol Med. 2002; 11: 750–760. [PMC free article] [PubMed] [Google Scholar]
- 19. Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta‐isoforms of the multifunctional Ca2+/calmodulin‐dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999; 84(6): 713–721. [DOI] [PubMed] [Google Scholar]
- 20. Kirchhefer U, Schmitz W, Scholz H, Neumann J. Activity of cAMP‐dependent protein kinase and Ca2+/calmodulin‐dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res. 1999; 42: 254–261. [DOI] [PubMed] [Google Scholar]
- 21. Zhang T, Brown JH. Role of Ca2+/calmodulin‐dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res. 2004; 63: 476–486. [DOI] [PubMed] [Google Scholar]
- 22. Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J Jr, Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003; 92: 912–919. [DOI] [PubMed] [Google Scholar]
- 23. Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, Ross J Jr, Brown JH. The cardiac‐specific nuclear delta(B) isoform of Ca2+/calmodulin‐dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002; 277 1261–1267. [DOI] [PubMed] [Google Scholar]
- 24. Youn HD, Grozinger CM, Liu JO. Calcium regulates transcriptional repression of myocyte enhancer factor 2 by histone deacetylase 4. J Biol Chem. 2000; 275: 22563–22567 [DOI] [PubMed] [Google Scholar]
- 25. McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor‐2 transcription factor by calcium/calmodulin‐dependent protein kinase‐stimulated binding of 14–3‐3 to histone deacetylase 5. Proc NatlAcad Sci USA. 2000; 97(26): 14400–14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, Overbeek P, Richardson JA, Grant SR, Olson EN. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000; 105(10): 1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Valencia TG, Roberts LD, Zeng H, Grant SR. Tetracycline‐inducible CaM kinase II silences hypertrophy‐sensitive gene expression in rat neonate cardiomyocytes. Biochem Biophys Res Commun. 2000; 274(3): 803–810. [DOI] [PubMed] [Google Scholar]
- 28. Bossuyt J, Helmstadter K, Wu X, Clements‐Jewery H, Haworth RS, Avkiran M, Martin JL, Pogwizd SM, Bers DM. Ca2+/calmodulin‐dependent protein kinase II{delta} and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Cite Res 2008; 102(6): 695–702. [DOI] [PubMed] [Google Scholar]
- 29. Mora S, Pessin JE. The MEF2A isoform is required for striated muscle‐specific expression of the insulin‐responsive GLUT4 glucose transporter. J Biol Chem. 2000; 275(21): 16323–16328. [DOI] [PubMed] [Google Scholar]
- 30. Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel‐Duby R, Maier LS, Olson EN. The {delta) isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA. 2009; 106(7): 2342–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure SI. Nucleo‐cytoplasmic compartmentalization was assessed by measuring the protein abundance of the nuclear protein Spl and the cytoplasmic protein LDH‐H4.
Please note: Wiley‐Blackwell Publishing is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item