

Abstract
The pathophysiology of various types of thrombotic microangiopathies is coming progressively into focus. Therapeutic advances are likely to follow at a quickening pace. This discussion focuses on thrombotic thrombocytopenic purpura (TTP), the hemolytic‐uremic syndrome (HUS), thrombotic microangiopathies associated with transplantation‐immunosuppression or anti‐angiogenesis therapy, and the preeclampsia/hemolysis‐elevated liver enzymes and low platelets syndrome (HELLP).
Keywords: TTP, HUS, other thrombotic microangiopathies
Thrombotic Thrombocytopenic Purpura (TTP)
TTP, the most severe microvascular occlusive thrombotic microangiopathy, is characterized by systemic platelet adhesion/aggregation, organ ischemia, profound thrombocytopenia, and fragmentation of erythrocytes. 1 The RBC fragmentation occurs as blood flows through turbulent areas of the microcirculation partially occluded by platelet clumps. Schistocytes, or “split” red cells, appear on the peripheral blood smear (1–4% or more of total RBCs) 1 , 2 as an indication of “microangiopathic hemolytic anemia.” Serum levels of lactate dehydrogenase (LDH) are extremely elevated as a consequence of hemolysis and the leakage of LDH from ischemic or necrotic tissue cells.
The systemic platelet adhesion/aggregation in TTP is often associated with platelet counts below 20,000/uL. Occlusive ischemia of the brain or the GI tract is common, and renal dysfunction may occur. In current clinical practice, the triad of thrombocytopenia, schistocytosis, and an impressively elevated serum LDH are suficient to suggest the diagnosis. 1
Von Willebrand Factor (VWF)
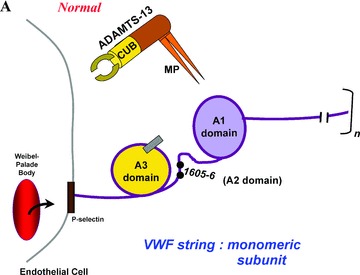
Subunit VWF monomers (approximately 275,000 Daltons) are linked by disulfide bonds into VWF multimers with molecular masses that may reach the millions of Daltons. VWF multimers are constructed predominantly within endothelial cells and stored within Weibel–Palade bodies as immense, coiled, ultralarge (UL) VWF multimers. Upon stimulation, endothelial cells secrete the ULVWF multimers in long strings that remain anchored to the cell membrane. 3 , 4 The long VWF multimeric strings are extremely “sticky” to the glycoprotein Ibα components of platelet glycoprotein Ibα‐IX‐V surface receptors. 5 , 6 The initial adherence of platelets via glycoprotein Ibα to the long VWF strings, and the subsequent coherence of additional platelets to each other (aggregation) via activated glycoprotein IIb–IIIa receptors, produces potentially occlusive platelet thrombi ( Figure 1 ).
Figure 1.
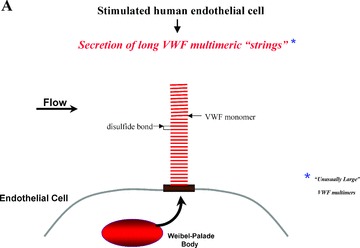

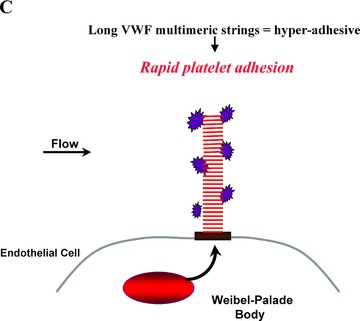
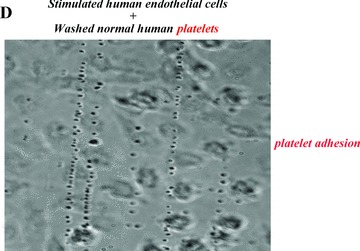
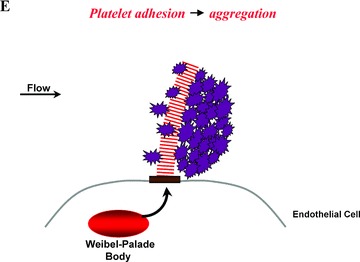
Platelet adhesion and aggregation onto VWF multimeric strings. Stimulation of endothelial cells by a variety of agonists, including inflammatory cytokines and toxins, causes the cells to secrete long, hyperadhesive VWF strings (A, B). Circulating platelets instantly adhere to the cell‐anchored VWF strings (C, D), and then the platelets cohere to each other (aggregate) (E).
In 1982, it was reported that TTP was caused by failure to “process” ULVWF multimers. 7 Subsequent research established that a specific VWF‐cleaving protease 7 , 8 , 9 , 10 , 11 , 12 in normal plasma rapidly cleaves the highly adhesive long VWF strings as they are secreted from endothelial cells. 3 , 13 , 14 Cleavage occurs at tyrosine1605–1606methionine peptide bonds in one or more susceptible VWF monomeric subunits 15 in each multimeric VWF string.
Platelets adhere instantly to the secreted “sticky” VWF strings. Some platelets are likely to circulate with bits of VWF attached after ADAMS‐13 cleavage of the platelet‐VWF strings.
The VWF‐cleaving protease is number 13 in a family of 19 distinct ADAMTS‐type metalloprotease enzymes. ADAMTS‐13 is a disintegrin and metalloprotease with eight thrombospondin‐1‐like domains. It is composed of an aminoterminal metalloprotease domain; a disintegrin domain; a thrombospondin‐1‐like domain; a cysteine‐rich domain and an adjacent spacer portion; 7 additional thrombospondin‐1‐like domains; and 2 similar, nonidentical CUB domains (CUB‐1 and CUB‐2) at the carboxyl‐terminal end of the molecule ( Figure 2 ). 1 , 16 , 17 , 18 , 19 CUB domains, found only in ADAMTS‐13 among the ADAMTS enzymes, contain peptide sequences present in the complement subcomponents, C1r/C1s; the sea Urchin protein, egf; and a bone morphogenic protein. ADAMTS‐13 is a Zn2+‐ and Ca2+‐requiring 190,000 Dalton glycosylated protein that is encoded on chromosome 9q34. 17 , 18 , 19 The enzyme is produced both in hepatic stellate cells 20 and (perhaps predominantly) in endothelial cells for slow, constitutive release into the circulation. 21 , 22 ADAMTS‐13 activity is inhibited in vitro by EDTA and, therefore, functional assays of the enzyme are usually performed using plasma anticoagulated with citrate (a weaker divalent cation binder than EDTA).
Figure 2.
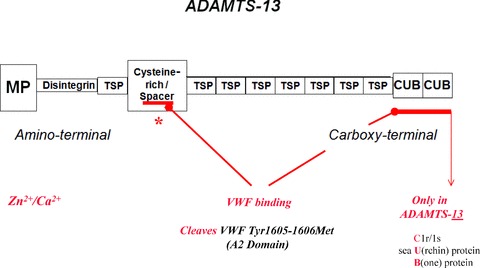
Domain structure of ADAMTS‐13, the VWF‐cleaving metalloprotease. MP = metalloprotease (proteolytic) domain; TSP = thrombospondin‐1‐like domain (a total of eight); CUB = two similar, non‐identical domains (CUB‐1 and CUB‐2) containing peptide sequences found in complement components C1r/C1s, a sea urchin protein, and a bone morphogenic protein. ADAMTS‐13 has a molecular mass of about 190 kD, and is produced in endothelial and hepatic stellate cells. The gene for ADAMTS‐13 is on chromosome #9.
Endothelial cells can be stimulated to secrete long VWF strings by inflammatory cytokines (tumor necrosis factor‐alpha [TNFα], interleukin [IL]‐8 and IL‐6 23 ), Shiga toxins, estrogen, or other agonists. The spacer and CUB‐1 domains of ADAMTS‐13 are involved in binding ADAMTS‐13 to the long VWF strings secreted by endothelial cells, 24 , 25 , 26 , 27 in order to position the enzyme for cleavage of tyr 1605–1606 met peptide bonds.
The ADAMTS‐13‐deficient types of TTP
Severe deficiency of ADAMTS‐13 activity in familial or acquired autoantibody‐mediated TTP patient plasma correlates with a failure to cleave long VWF multimers as they emerge in long strings from the surface of stimulated endothelial cells ( Figure 3 ). 3 , 13 , 14 Familial TTP is rare, usually (but not always) appears initially in infancy or childhood, and may recur as “chronic relapsing TTP” episodes at about 3‐week intervals. 1 , 7 , 8 , 10 Patients with familial, relapsing TTP have less than about 5–10% of normal plasma ADAMTS‐13 (both during and between episodes).
Figure 3.
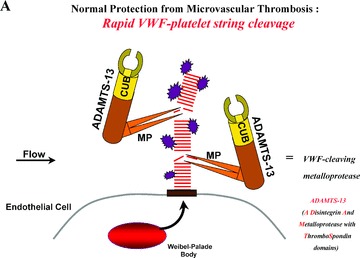
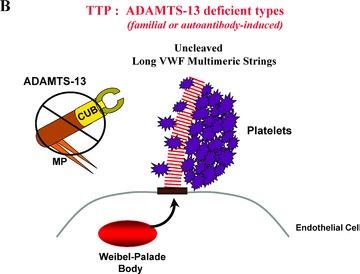
(A) Normal protection from microvascular thrombosis: In normal individuals, ADAMTS‐13 enzyme molecules from the plasma attach to, and then rapidly cleave, long “sticky” VWF multimeric string‐like structures secreted from stimulated endothelial cells. Cleavage occurs at an exposed 1605–6 peptide bond in VWF monomeric subunits. (B) TTP = ADAMTS‐13‐deficient types (familial or autoantibody‐induced). Absent or severely reduced activity of ADAMTS‐13 prevents the timely cleavage of the long VWF multimeric strings secreted by stimulated endothelial cells. Platelets adhere and aggregate onto the uncleaved long VWF strings. Severe familial deficiency of ADAMTS‐13 activity caused by gene mutations, or profound inhibition of ADAMTS‐13 activity caused by acquired autoantibodies, result in a propensity for TTP. Episodes are especially likely to occur in the absence (or severe reduction) of ADAMTS‐13 if there is increased concurrent endothelial cell secretion of long VWF strings (e.g., in the presence of inflammatory cytokines, estrogen, or certain bacterial toxins). CUB represents the two nonidentical C‐terminal CUB domains of ADAMTS‐13; MP represents the N‐terminal metalloproteasse domain.
The absent or severely reduced plasma ADAMTS‐13 activity in familial TTP 1 , 7 , 8 , 10 , 28 , 29 is a consequence of homozygous (or double heterozygous) mutations in both of the ADAMTS‐13 alleles located on chromosome 9q34. 1 , 17 , 18 , 19 The result is defective production or release of ADAMTS‐13 molecules. 1 , 28 , 29 Mutations in familial TTP have been detected all along the gene, in regions encoding different domains. 17 , 18 , 19 In severe familial ADAMTS‐13 deficiency, TTP episodes usually commence in infancy or childhood. In some familial TTP patients with slightly higher plasma ADAMTS‐13 levels, 29 however, overt TTP episodes may not develop until later in life (e.g., during a first pregnancy). 1 , 16 These latter clinical observations suggest that in vivo plasma ADAMTS‐13 activity may sometimes exceed the estimates of activity using in vitro nonphysiologic assays. Additionally, or alternatively, the accentuated secretion of long VWF multimeric strings by endothelial cells stimulated by estrogen (as during menses or pregnancy) or inflammatory cytokines 23 may be required to provoke TTP episodes in some patients with very low, but not absent, plasma ADAMTS‐13 values.
Adults and, sometimes, older children 1 , 16 with acquired ADAMTS‐13 autoantibody‐mediated TTP have transient inhibition of ADAMTS‐13 to less than about 5–10% of normal during acute episodes or later recurrences. 1 , 11 , 12 , 16 Plasma ADAMTS‐13 levels then increase toward normal during recovery. 1 , 16 Recurrences occur at irregular intervals in about one‐third of patients. Polyclonal IgG autoantibodies that inhibit plasma ADAMTS‐13 activity during episodes are detected in most of these patients, 1 , 11 , 12 indicating transiently defective immune regulation. TTP induced by this mechanism may occur occasionally late in pregnancy or immediately after delivery, or in patients with HIV/AIDS. 1 , 16
The IgG autoantibodies against ADAMTS‐13 are of restricted clonality, containing VH1–69 heavy chain variable region gene‐encoded components. 30 Their preferred, but not exclusive, epitope target is the spacer portion of the cysteine‐rich/spacer domain that is important (along with the CUB‐1 domain), in docking ADAMTS‐13 to the long VWF strings. 24 , 25 , 26 , 27
A small fraction of patients treated for arterial thrombosis with the platelet P2Y12 adenosine diphosphate receptor‐inhibiting thienopyridine drugs, ticlopidine (Ticlid) or clopidogrel (Plavix) develop TTP within a few weeks after the initiation of therapy. 31 , 32 , 33 , 34 Antibodies that inhibit plasma ADAMTS‐13 have been demonstrated in a few of these patients. 31 , 33 , 34 It has been suggested that the immune dysregulation induced by these similar thienopyridine compounds may be analogous to the anti‐RBC antibody “escape” associated with the once‐popular antihypertensive drug, α‐methyldopa (Aldomet). 34 An alternative possibility is that the binding of thienopyridines to P2Y12 molecules on endothelial cell (and other cell) types may, in a fraction of exposed individuals, initiate anomalous intracellular signaling patterns or provoke antibody production against thienopyridine (as hapten)‐bound P2Y12 protein complexes on cell surfaces. Malfunction or injury to endothelial and other cells with surface P2Y12 receptors (lymphocytes, CD34 stem cells) may result. 34
Plasma ADAMTS‐13 activity in healthy adults ranges from about 50–180%. Activity is often reduced below normal in liver disease, disseminated malignancies, chronic metabolic and inf ammatory conditions, pregnancy, and newborns. 35 With the exception of occasional peri‐partum women who develop overt TTP, the ADAMTS‐13 activity in these conditions is not reduced to the extremely low values (i.e., < 5–10% of normal) found in patients with familial or acquired autoantibodymediated TTP.
Therapy for ADAMTS‐13‐deficient types of TTP
During the more than 50 years between the initial description of TTP and the first use of plasma therapy, almost all patients died of the disease. It is now recognized that normal fresh‐frozen plasma, the cryoprecipitate‐poor fraction of plasma (cryosupernatant), and solvent/detergent‐treated plasma all contain active ADAMTS‐13. 1 , 12 The infusion about every 2–3 weeks of normal plasma into familial TTP patients lacking effective enzyme production/release prevents, or reduces the frequency of, TTP episodes. The plasma t1/2 of infused ADAMTS‐13 activity is about 2–4 days. Some ADAMTS‐13 molecules may continue to dock and cleave one secreted long VWF string after another over an even longer time period. 1 , 13 , 14
Adults and some older children with acquired ADAMTS‐13 autoantibody‐mediated TTP require daily plasma exchange until remission. Plasma exchange combines the infusion of fresh‐frozen plasma or cryosupernatant (containing uninhibited ADAMTS‐13) with plasmapheresis (which may remove autoantibodies against ADAMTS‐13 and cytokines that stimulate endothelial cells to secrete long VWF strings). Although both solvent‐detergent‐treated plasma 1 , 12 and methylene blue/light‐treated plasma (for inactivation of lipid envelope viruses) also contain active ADAMTS‐13, the lower‐than‐normal protein S quantity in solvent‐detergent plasma has restricted its use.
Plasma exchange allows about 80–90% of acquired ADAMTS‐13 autoantibody‐mediated TTP patients to survive an episode, usually with minimal organ damage. 1 , 36 Lower titers of autoantibodies are associated with better responses to plasma exchange procedures. Production of ADAMTS‐13 autoantibodies may be suppressed by high‐doses of glucocorticoids, 4–8 weekly injections of rituximab (monoclonal antibody against CD20 on B‐lymphocytes), or removal of autoantibody‐producing cells by splenectomy. 1 , 16 There is a small risk of progressive multi‐focal leukoencephalopathy (PML) associated with rituximab therapy, as with several other immunosuppressive agents. 37
ADAMTS‐13 has been partially purified 17 and produced in active recombinant form 38 for possible eventual therapeutic use. Because a plasma ADAMTS‐13 level of only a little more than 5–10% is suficient to prevent or truncate TTP episodes, 1 , 16 , 28 gene therapy may eventually be used to extend remissions in familial TTP patients.
In 1988, 39 Phillips et al. discovered that a triphenyl‐methyl compound, aurin tricarboxylic acid (ATA), binds to large and ultra‐large VWF multimers and prevents VWF attachment to GPIbα. It was subsequently found that polymeric forms of ATA could inhibit coronary thrombosis in dogs without causing excessive bleeding. 40 , 41 This was the first demonstration that inhibiting VWF‐platelet adhesion might be an effective approach to antiarterial thrombotic therapy ( Figure 4 ). 39 , 40 , 41 ATA has not been formulated for testing in humans, however, and so there is no information available on possible efficacy or toxicity of the parent compound (or any chemical derivative).
Figure 4.
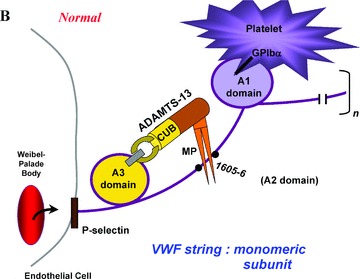
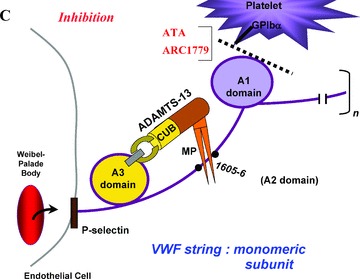
(A) A “close‐up” of one of the many monomeric subunits that comprise a long VWF multimeric string. The VWF strings are secreted from endothelial cell Weibel–Palade bodies, and remain anchored in the endothelial cell membrane. The A1, A2 (containing the tyr1605–1606met ADAMTS‐13 proteolytic cleavage site), and A3 domains are shown. (B) Adequate quantities of ADAMTS‐13 enzymes are present in the plasma of normal individuals. The two carboxy‐terminal CUB domains are indicated, and the metalloprotease domain (MP) is drawn as a pincer‐like stricture on the amino‐terminal portion of the enzyme. Platelets from flowing blood adhere to the long VWF strings immediately after string secretion. Platelet adherence is via platelet GPIbα receptors to the A1 domain of VWF monomeric subunits. ADAMTS‐13 attaches via its spacer and CUB‐1 domain to the A3 domain of VWF monomeric subunits, and cleaves the adjacent tyr1605–1606met bond. ADAMTS‐13 cleavage occurs in various monomers along the length of VWF multimeric strings that have had their A2 domains unfolded. Unfolding occurs as a result of the propulsive force of VWF secretion from Weibel–Palade bodies, and is augmented by the shear stresses associated with fluid flow. (C) Both ATA and a pegylated oligonucleotide, ARC1779, block the interaction of the A1 domain of VWF monomers and GPIbα on platelets. ARC1779 is under development commercially for therapeutic use in TTP and arterial thrombosis.
Recently, a similar concept for inhibiting arterial thrombosis has been used as the basis for constructing an anti‐VWF aptamer. This polymer inhibits, as does the polymetic form of ATA, VWF‐platelet adhesion. The newly designed anti‐VWF aptamer, so far designated “ARC‐1779,” is an oligonucleotide that binds to the VWF A1 domain and blocks A1 domain binding to the platelet GPIbα component of GPIbα‐IX‐V complexes ( Figure 4 ). 42
ARC has been approved by the FDA for use in TTP as an “orphan drug” because, in preliminary trials, it was effective in the treatment of a few patients with relapsing and refractory types of TTP. 43 , 44
Other Thrombotic Microangiopathies
Systemic or predominantly renal microvascular platelet clumping (+/− fibrin polymer formation), thrombocytopenia, erythrocyte fragmentation, and increased serum levels of LDH are also characteristic of the other thrombotic microangiopathies discussed below. Unlike ADAMTS‐13‐deficient types of TTP, the entities to be summarized are not usually associated with absent or severely reduced plasma ADAMTS‐13 activity. 1 , 16 , 45 This latter f nding may explain the poor response of patients with these disorders to plasma infusion or exchange.
Hemolytic‐Uremic Syndrome (HUS)
Acquired HUS is frequently preceded by hemorrhagic enterocolitis that is caused by the inadvertent ingestion of cytotoxin‐producing serotypes of E. coli (e.g., 0157:H7) or Shigella species. 1 , 46 Shiga toxin (Stx)‐1 and Stx‐2 produced by enterohemorrhagic E. coli are relatively common causes of bloody diarrhea‐associated HUS ( Figure 5 ). This disorder is the result of obstruction of the glomerular microvasculature by platelet‐f brin thrombi. Stx‐1 and Stx‐2 stimulate the rapid and profuse secretion of long VWF multimeric strings from endothelial cells, including glomerular microvascular endothelial cells. 47 Platelets immediately adhere to the secreted long VWF strings, and the rate of platelet‐VWF string cleavage by ADAMTS‐13 is delayed in the presence of Stx‐1 or Stx‐2. 47 (The toxins may interfere with ADAMTS‐13 binding to the VWF strings.) Over the course of about 1 week, progressive platelet adhesion to long VWF strings atop Stx‐stimulated glomerular endothelial cells in diarrhea‐associated HUS may explain the evolving glomerular microvascular occlusions and renal failure. 47 The “cytokine storm” that accompanies diarrhea‐associated HUS 1 , 47 potentiates these processes: TNF‐α, IL‐8, and IL‐6 (bound to its soluble receptor) stimulate endothelial cell secretion of long VWF strings; and IL‐6 also interacts with the long VWF strings to inhibit the rate of VWF string cleavage ( Figure 5 ). 23 The incorporation of Shiga toxins into glomerular endothelial cells, and the resultant interference with peptide synthesis over the course of hours to days, may cause additional renal endothelial cell injury and sub‐endothelial exposure. 47
Figure 5.
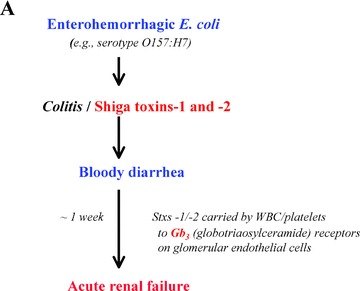
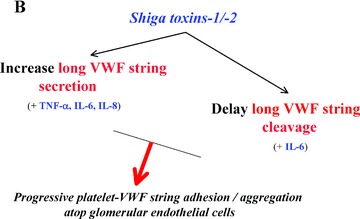
Proposed pathophysiology of the diarrhea‐associated hemolytic‐uremic syndrome (HUS) that is most common in North America. (A) Enterohemorrhagic E. coli injure and efface colonic mucosa and induce bloody diarrhea, and produce Shiga toxins‐1 and ‐2. These toxins are transported by white cells and platelets through the bloodstream to globotriaosylceramide (Gb3) receptors that are present in high concentrations on glomerular endothelial and other renal cells. (B) In response to the Shiga toxins, and elevated levels of circulating inflammatory cytokines (TNF‐α, IL‐8, IL‐6), glomerular endothelial cells profusely secrete long “sticky” VWF strings. The Shiga toxins, in conjunction with one of the inflammatory cytokines (IL‐6), also slow the rate of ADAMTS‐13‐mediated cleavage of cell‐bound VWF strings. The result is excessive platelet adhesion/aggregation on VWF strings atop glomerular microvascular endothelial cells over the course of about 1 week, progressive loss of nephron function, and increasing renal failure.
Therapy for hemorrhagic diarrhea‐associated HUS is, thus far, limited to supportive care for the renal and hematological complications.
HUS usually occurs as a single episode, except in rare individuals who have a familial, recurrent type of the disease. 16 In these latter patients (often children) with “atypical” HUS, there may be deficient quantity or defective function of the plasma alternative complement pathway regulatory protein, factor H 48 ( Figure 6 ). The result is over‐activation of complement component C3 to C3b whenever the alternative complement component pathway is activated. The generation of (C3b)2Bb complexes activates C5 to C5b, and C5b initiates the formation of C5b678(9)n membrane attack complexes that form destructive pores in target cell membranes. These latter may include the membranes of renal glomerular endothelial cells.
Figure 6.
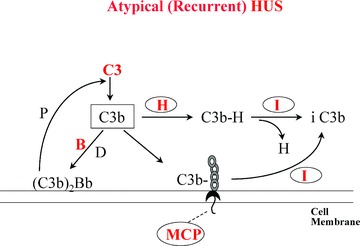
The alternative complement pathway and atypical (recurrent) HUS. C3b = activated complement factor C3. B = complement factor B, which is activated to Bb by complement factor D. (C3b)2Bb = the alternative complement pathway active C3 convertase that is stabilized by properdin (P). H = plasma factor H, which displaces Bb from C3b and allows its inactivation to iC3b by plasma complement factor I (C3b‐cleaving protease). MCP = membrane cofactor protein, which functions on cell membranes as factor H functions (predominantly) in plasma. Heterozygous mutations in factor H, less commonly MCP, or least commonly factor I cause increased susceptibility to a familial, recurrent type of HUS. Rare gain‐of‐function mutations in C3 or factor B may also cause the syndrome. The mutations known to be associated with atypical, recurrent HUS are shown in red. The rare production of autoantibodies to factor H also causes aytpical HUS.
A similar atypical HUS clinical syndrome results from familial deficiencies or abnormalities in the following other alternative complement pathway control substances or pathway components: membrane cofactor protein (MCP or CD46); 49 C3b‐cleaving protease (factor I); 50 factor B; 51 or C3 itself. 52 (Factor B and C3 abnormalities are gain‐of‐function mutations.) ( Figure 6 ) Acquired factor H autoantibodies have also been found in a few patients with atypical HUS. 53 , 54 , 55
It is not yet known whether or not eculizumab, the monoclonal antibody to C5 that prevents the formation of the C5b678(9)n membrane attack complex, may be useful therapy in “atypical,” complement‐mediated HUS. (Eculizumab is effective treatment for paroxysmal nocturnal hemoglobinuria.)
Transplantation‐Immunosuppression, Chemotherapy, and Antiangiogenesis Therapy
A thrombotic microangiopathy that clinically more often resembles HUS than TTP sometimes occurs weeks to months after exposure to one of the following: cyclosporine or tacrolimus (FK 506), inhibitors of the protein phosphatase 2B (calcineurin) that are often used as immunosuppressants with allogeneic bone marrow or solid organ transplantation; mitomycin; gemcitabine; bevacizumab (Avastin) or sunitinib (Sutent); combinations of chemotherapeutic agents; and/or total‐body irradiation. 56 , 57 , 58 Clues to the cause of several of these entities have emerged recently.
Cyclsporin and tacrolimus
Cyclsporin, a cyclic nonapeptide, and tacrolimus, a macrolide, inhibit protein phosphatase 2B (calcineurin) in immune and endothelial cells. The biochemical effect is to maintain some proteins in a phosphorylated state for a prolonged time. Cyclosporin‐ or tacrolimus‐treated endothelial cells profusely secrete long VWF multimeric strings. 59 This vigorous secretion may slowly overwhelm the capacity of ADAMTS‐13 to defend the microvasculature against platelet thrombotic occlusion and thrombotic microangiopathy.
There may be some analogy between the pathophysiology of cyclosporin/tacrolimus‐associated thrombotic microangiopathy and both diarrhea‐associated HUS (described previously) and sepsis/DIC/renal failure. In sepsis/DIC/renal failure, cytokine‐stimulated VWF string secretion from stimulated endothelial cells occurs (along with progressive consumption of plasma ADAMTS‐13). 60
Treatment for transplantation‐immunosuppression‐induced or chemotherapy‐radiotherapy‐associated thrombotic microangiopathy is, to date, limited to supportive care and the discontinuation of any putative of ending drug.
Bevacizumab and sunitinib
Bevacizumab (Avastin) is a humanized monoclonal antibody that binds/inactivates vascular endothelial growth factor (VEGF). It has been used as adjunctive antiangiogenesis therapy for various malignant solid tumors. Unfortunately, in some patients the antibody may also bind so much of the VEGF produced naturally by the patients’ renal epithelial podocytes that it results in glomerular endothelial cell dysfunction and a HUS‐like disorder. 56 Inhibition of the VEGF receptor‐1 tyrosine kinase signaling mechanism by the anti‐neoplastic agent, sunitinib (Sutent), may also cause thrombotic microangiopathy in occasional recipients of the drug. 57
Discontinuation of the of ending agent is the therapy for drug‐associated thrombotic microangiopathy.
Preeclampsia/HELLP Syndrome
The bevacizumab effect has a pathophysiologic analogy to the preeclampsia/HELLP syndrome (hemolysis‐elevated liver enzymes‐low platelets), a serious microangiopathic complication of pregnancy. 61 In preeclampsia/HELLP, the hypoxic placenta releases soluble extra‐vascular domains of at least two receptors for angiogenic factors: soluble VEGF receptor‐1 (also known as soluble fms‐like tyrosine kinase receptor‐1, or sflt‐1), which binds/inactivates VEGF; and soluble endoglin (Eng), which binds/inactivates transforming growth factor (TGF) β‐1 and TGF β‐3. These circulating soluble portions of pro‐angiogenic receptors prevent VEGF and TGFβ‐1/‐3 from binding to their physiologic receptors in cell membranes and, therefore, contribute to (or primarily cause) the progressive renal dysfunction/ hypertension and hepatic necrosis in preeclampsia and the HELLP syndrome. 61
Delivery of the fetus and placenta is usually effective therapy in preeclampsia/HELLP syndrome. It is not yet known if some VEGF formulation may be useful adjunctive treatment for these entities.
Chemotherapy–Radiotherapy
It is also not known if an excessive release of one or more soluble angiogenic receptors from neoplastic cells during chemotherapy/radiotherapy might cause thrombotic microangiopathy in a fraction of patients under treatment for malignant disorders.
Conclusion
Considerable progress in elucidating the pathogenesis of several types of thrombotic microangiopathies has been made in recent years. Table 1 summarizes these advances in knowledge. Perhaps the emerging clues will eventually help unravel the causes of the still‐mysterious types of thrombotic microangiopathies and lead to overdue therapeutic breakthroughs.
Table 1.
Pathophysiology of thrombotic microangiopathies
| Deficient/inhibited ADAMTS‐13 |
| TTP |
| Excessive secretion/persistence of long VWF strings |
| HUS |
| Cyclosporin/tacrolimus (?) |
| Inactivation of pro‐angiogenic factor(s) (or signaling mechanism) |
| Bevacizumab/sunitinib |
| Preeclampsia/HELLP syndrome |
References
- 1. Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002; 347: 589–600. [DOI] [PubMed] [Google Scholar]
- 2. Ruutu T, Barosi G, Benjamin RJ, Clark RE, George JN, Gratwohl A, Holler E, Iacobelli M, Kentouche K, Lämmle B, Moake JL, Richardson P, Socié G, Zeigler Z, Niederwieser D, Barbui T. Diagnostic criteria for hematopoietic stem cell transplant‐associated microangiopathy: results of a consensus process by an International Working Group. Haematologica. 2007; 92: 95–100. [DOI] [PubMed] [Google Scholar]
- 3. Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, López JA. ADAMTS‐13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002; 100: 4033–4039. [DOI] [PubMed] [Google Scholar]
- 4. Padilla A, Moake JL, Bernardo A, Ball C, Wang Y, Arya M, Nolasco L, Turner N, Berndt MC, Anvari B, López JA, Dong JF. P‐selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood. 2004; 103: 2150–2156. [DOI] [PubMed] [Google Scholar]
- 5. Moake JL, Turner NA, Stathopoulos NA, Nolasco LH, Hellums JD. Involvement of large plasma von Willebrand factor (vWF) multimers and unusually large vWF forms derived from endothelial cells in shear stress‐induced platelet aggregation. J Clin Invest. 1986; 78: 1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arya M, Anvari B, Romo GM, Cruz MA, Dong JF, McIntire LV, Moake JL, López JA. Ultralarge multimers of von Willebrand factor form spontaneous high‐strength bonds with the platelet glycoprotein Ib‐IX complex: studies using optical tweezers. Blood. 2002; 99: 3971–3977. [DOI] [PubMed] [Google Scholar]
- 7. Moake JL, Rudy CK, Troll JH, Weinstein MJ, Colannino NM, Azocar J, Seder RH, Hong SL, Deykin D. Unusually large plasma factor VIII: von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med. 1982; 307: 1432–1435. [DOI] [PubMed] [Google Scholar]
- 8. Moake JL, Byrnes JJ, Troll JH, Rudy CK, Hong SL, Weinstein MJ, Colannino NM. Effects of fresh‐frozen plasma and its cryosupernatant fraction on von Willebrand factor multimeric forms in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1985; 65: 1232–1236. [PubMed] [Google Scholar]
- 9. Frangos JA, Moake JL, Nolasco L, Phillips MD, McIntire LV. Cryosupernatant regulates accumulation of unusually large vWF multimers from endothelial cells. Am J Physiol. 1989; 256: H1635–H1644. [DOI] [PubMed] [Google Scholar]
- 10. Furlan M, Robles R, Solenthaler M, Wassmer M, Sandoz P, Lämmle B. Deficient activity of von Willebrand factor‐cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1997; 89: 3097–3103. [PubMed] [Google Scholar]
- 11. Furlan M, Robles R, Galbusera M, Remuzzi G, Kyrle PA, Brenner B, Krause M, Scharrer I, Aumann V, Mittler U, Solenthaler M, Lämmle B. von Willebrand factor‐cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic‐uremic syndrome. N Engl J Med. 1998; 339: 1578–1584. [DOI] [PubMed] [Google Scholar]
- 12. Tsai HM, Lian EC. Antibodies to von Willebrand factor‐cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998; 339: 1585–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bernardo A, Ball C, Nolasco L, Choi H, Moake JL, Dong JF. Platelets adhered to endothelial cell‐bound ultra‐large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005; 3: 562–570. [DOI] [PubMed] [Google Scholar]
- 14. Dong JF, Moake JL, Bernardo A, Fujikawa K, Ball C, Nolasco L, López JA, Cruz MA. ADAMTS‐13 metalloprotease interacts with the endothelial cell‐derived ultra‐large von Willebrand factor. J Biol Chem. 2003; 278: 29633–29639. [DOI] [PubMed] [Google Scholar]
- 15. Dent JA, Galbusera M, Ruggeri ZM. Heterogeneity of plasma von Willebrand factor multimers resulting from p;roteolysis of the constituent subunit. J Clin Invest. 1991; 88: 774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moake JL. Thrombotic thrombocytopenic purpura and the hemolytic uremic syndrome In Michelson AD, ed. Platelets, 2nd ed, Burlington , MA : Academic Press, 2006, 903–923. [Google Scholar]
- 17. Fujikawa K, Suzuki H, McMullen B, Chung D. Purification of human von Willebrand factor‐cleaving protease and its identifi cation as a new member of the metalloproteinase family. Blood. 2001; 98: 1662–1666. [DOI] [PubMed] [Google Scholar]
- 18. Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD Jr, Ginsburg D, Tsai HM. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001; 413: 488–494. [DOI] [PubMed] [Google Scholar]
- 19. Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Wille‐brand factor‐cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombo‐cytopenic purpura. J Biol Chem. 2001; 276: 41059–41063. [DOI] [PubMed] [Google Scholar]
- 20. Zhou W, Inada M, Lee TP, Benten D, Lyubsky S, Bouhassira EE, Gupta S, Tsai HM. ADAMTS13 is expressed in hepatic stellate cells. Lab Invest. 2005; 85: 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Turner N, Nolasco L, Dong JF, Moake J. ADAMTS‐13 cleaves long von Willebrand factor multimeric strings anchored to endothelial cells in the absence of flow, platelets or conformation‐altering chemicals. J Thromb Haemost. 2009; 7: 229–232. [DOI] [PubMed] [Google Scholar]
- 22. Turner N, Nolasco L, Tao Z, Dong JF, Moake J. Human endothelial cells synthesize and release ADAMTS‐13. J Thromb Haemost. 2006; 4: 1396–1404. [DOI] [PubMed] [Google Scholar]
- 23. Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell‐derived ultralarge von Willebrand factor multimers under flow. Blood. 2004; 104: 100–106. [DOI] [PubMed] [Google Scholar]
- 24. Luken BM, Turenhout EA, Hulstein JJ, Van Mourik JA, Fijnheer R, Voorberg J. The spacer domain of ADAMTS13 contains a major binding site for antibodies in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 2005; 93: 267–274. [DOI] [PubMed] [Google Scholar]
- 25. Tao Z, Peng Y, Nolasco L, Cal S, Lopez‐Otin C, Li R, Moake JL, López JA, Dong JF. Recombinant CUB‐1 domain polypeptide inhibits the cleavage of ULVWF strings by ADAMTS13 under flow conditions. Blood. 2005; 106: 4139–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Banno F, Chauhan AK, Kokame K, Yang J, Miyata S, Wagner DD, Miyata T. The distal carboxyl‐terminal domains of ADAMTS‐13 are required for regulation of in vivo thrombus formation. Blood. 2009; 113: 5323–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klaus C, Plaimauer B, Studt JD, Dorner F, Lämmle B, Mannucci PM, Scheiflinger F. Epitope mapping of ADAMTS13 autoantibodies in acquired thrombotic thrombocytopenic purpura. Blood. 2004; 103: 4514–4519. [DOI] [PubMed] [Google Scholar]
- 28. Bianchi V, Robles R, Alberio L, Furlan M, Lämmle B. Von Willebrand factor‐cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood. 2002; 100: 710–713. [DOI] [PubMed] [Google Scholar]
- 29. Tao Z, Anthony K, Peng Y, Choi H, Nolasco L, Rice L, Moake JL, Dong JF. Novel ADAMTS‐13 mutations in an adult with delayed onset thrombotic thrombocytopenic purpura. J Thromb Haemost. 2006; 4: 1931–1935. [DOI] [PubMed] [Google Scholar]
- 30. Pos W, Luken BM, Hovinga JA, Turenhout EA, Scheiflinger F, Dong JF, Fijnheer R, Voorberg J. VH1–69 germline encoded antibodies directed towards ADAMTS13 in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2009; 7: 421–428. [DOI] [PubMed] [Google Scholar]
- 31. Tsai HM, Rice L, Sarode R, Chow T W, Moake JL. Antibody inhibitors to von Willebrand factor metalloproteinase and increased binding of von Willebrand factor to platelets in ticlopidine‐associated thrombotic thrombocytopenic purpura. Ann Intern Med. 2000; 132: 794–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bennett CL, Weinberg PD, Rozenberg‐Ben‐Dror K, Yarnold PR, Kwaan HC, Green D. Thrombotic thrombocytopenic purpura associated with ticlopidine: a review of 60 cases. Ann Intern Med. 1998; 128: 541–544. [DOI] [PubMed] [Google Scholar]
- 33. Bennett CL, Connors JM, Carwile JM, Moake JL, Bell WR, Tarantolo SR, McCarthy LJ, Sarode R, Hatfield AJ, Feldman MD, Davidson CJ, Tsai HM. Thrombotic thrombocytopenic purpura associated with clopidogrel. N Engl J Med. 2000; 342: 1773–1777. [DOI] [PubMed] [Google Scholar]
- 34. Zakarija A, Kwaan HC, Moake JL, Bandarenko N, Pandey DK, McKoy JM, Yarnold PR, Raisch DW, Winters JL, Raife TJ, Cursio JF, Luu TH, Richey EA, Fisher MJ, Ortel TL, Tallman MS, Zheng XL, Matsumoto M, Fujimura Y, Bennett CL. Ticlopidine‐ and clopidogrel‐associated thrombotic thrombocytopenic purpura (TTP): review of clinical, laboratory, epidemiological, and pharmacovigilance fi ndings (1989–2008). Kidney Int Suppl. 2009; S20–S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mannucci PM, Canciani MT, Forza I, Lussana F, Lattuada A, Rossi E. Changes in health and disease of the metalloprotease that cleaves von Willebrand factor. Blood. 2001; 98: 2730–2735. [DOI] [PubMed] [Google Scholar]
- 36. Kennedy AS, Lewis QF, Scott JG, Kremer Hovinga JA, Lämmle B, Terrell DR, Vesely SK, George JN. Cognitive deficits after recovery from thrombotic thrombocytopenic purpura. Transfusion. 2009; 49: 1092–1101. [DOI] [PubMed] [Google Scholar]
- 37. Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, Laubach J, Bawn SD, Gordon LI, Winter JN, Furman RR, Vose JM, Zelenetz AD, Mamtani R, Raisch DW, Dorshimer GW, Rosen ST, Muro K, Gottardi‐Littell NR, Talley RL, Sartor O, Green D, Major EO, Bennett CL. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV‐negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports Project. Blood. 2009; 113: 4834–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Plaimauer B, Zimmermann K, Völkel D, Antoine G, Kerschbaumer R, Jenab P, Furlan M, Gerritsen H, Lämmle B, Schwarz HP, Scheiflinger F. Cloning, expression, and functional characterization of the von Willebrand factor‐cleaving protease (ADAMTS13). Blood. 2002; 100: 3626–3632. [DOI] [PubMed] [Google Scholar]
- 39. Phillips MD, Moake JL, Nolasco L, Turner N. Aurin tricarboxylic acid: a novel inhibitor of the association of von Willebrand factor and platelets. Blood. 1988; 72: 1898–1903. [PubMed] [Google Scholar]
- 40. Strony J, Phillips M, Brands D, Moake J, Adelman B. Aurin tricarboxylic acid in a canine model of coronary artery thrombosis. Circulation. 1990; 81: 1106–1114. [DOI] [PubMed] [Google Scholar]
- 41. Weinstein M, Vosburgh E, Phillips M, Turner N, Chute‐Rose L, Moake J. Isolation from commercial aurintricarboxylic acid of the most effective polymeric inhibitors of von Willebrand factor interaction with platelet glycoprotein Ib. Comparison with other polyanionic and polyaromatic polymers. Blood. 1991; 78: 2291–2298. [PubMed] [Google Scholar]
- 42. Gilbert JC, DeFeo‐Fraulini T, Hutabarat RM, Horvath CJ, Merlino PG, Marsh HN, Healy JM, Boufakhreddine S, Holohan TV, Schaub RG. First‐in‐human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation. 2007; 116: 2678–2686. [DOI] [PubMed] [Google Scholar]
- 43. Jilma B, Jilma P, Paulinska P, Gilbert JC, Hutabarat R, Wagner T, Knoebl P. Proof of concept for the anti von Willebrand factor aptamer in patients with relapsing thrombotic thrombocytopenic purpura (TTP). Am Soc Hematol Annual Meeting. 2008; abstract no. 2290.
- 44. Firbas C, Jilma B, Wagner PG, Hutabarat R, Schaub RG, Gilbert JC, Knoebl P. Anti von Wille‐brand factor aptamer ARC1779 for refractory thrombotic thrombocytopenic purpura. Am Soc Hematol Annual Meeting. 2008; abstract no. 2298.
- 45. Van Der Plas RM, Schiphorst ME, Huizinga EG, Hené RJ, Verdonck LF, Sixma JJ, Fijnheer R. von Willebrand factor proteolysis is deficient in classic, but not in bone marrow transplantation‐associated, thrombotic thrombocytopenic purpura. Blood. 1999; 93: 3798–3802. [PubMed] [Google Scholar]
- 46. Karmali MA, Petric M, Lim C, Fleming PC, Arbus GS, Lior H. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin‐producing Escherichia coli. J Infect Dis. 1985; 151: 775–782. [DOI] [PubMed] [Google Scholar]
- 47. Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, Dong JF, Moake JL. Hemolytic ure‐mic syndrome‐associated Shiga toxins promote endothelial‐cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. 2005; 106: 4199–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Warwicker P, Goodship TH, Donne RL, Pirson Y, Nicholls A, Ward RM, Turnpenny P, Goodship JA. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998; 53: 836–844. [DOI] [PubMed] [Google Scholar]
- 49. Bonnardeaux A, Pichette V. Complement dysregulation in haemolytic uraemic syndrome. Lancet. 2003; 362: 1514–1515. [DOI] [PubMed] [Google Scholar]
- 50. Fremeaux‐Bacchi V, Dragon‐Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, Loirat C, Rondeau E, Fridman WH. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. 2004; 41: e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goicoechea de Jorge E, Harris CL, Esparza‐Gordillo J, Carreras L, Arranz EA, Garrido CA, López‐Trascasa M, Sánchez‐Corral P, Morgan BP, Rodríguez de Córdoba S. Gain‐of‐function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Nat Acad Sci USA. 2007; 104: 240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fremeaux‐Bacchi V, Miller EC, Liszewski K, Strain L, Blouin J, Brown AL, Moghal N, Kaplan BS, Weiss RA, Lhotta K, Kapur G, Mattoo T, Nivet H, Wong W, Gie S, Hurault de Ligny B, Fischbach M, Gupta R, Hauhart R, Meunier V, Loirat C, Dragon‐Durey MA, Fridman WH, Janssen BJ, Goodship TH, Atkinson JP. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008; 112: 4948–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dragon‐Durey M‐A, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, Weiss L, Fridman WH, Frémeaux‐Bacchi V. Anti‐factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005; 16: 555–563. [DOI] [PubMed] [Google Scholar]
- 54. Józsi M, Strobel S, Dahse HM, Liu WS, Hoyer PF, Oppermann M, Skerka C, Zipfel PF. Anti‐factor H autoantibodies block C‐terminal recognition function of factor H in hemolytic uremic syndrome. Blood. 2007; 110: 1516–1518. [DOI] [PubMed] [Google Scholar]
- 55. Józsi M, Licht C, Strobel S, Zipfel SL, Richter H, Heinen S, Zipfel PF, Skerka C. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood. 2008; 111: 1512–1514. [DOI] [PubMed] [Google Scholar]
- 56. Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008; 358: 1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bollée G, Patey N, Cazajous G, Robert C, Goujon JM, Fakhouri F, Bruneval P, Noël LH, Knebelmann B. Thrombotic microangiopathy secondary to VEGF pathway inhibition by sunitinib. Nephrol Dial Transplant. 2009; 24: 682–685. [DOI] [PubMed] [Google Scholar]
- 58. Moake JL, Byrnes JJ. Thrombotic microangiopathies associated with drugs and bone marrow transplantation. Hematol Oncol Clin North Am. 1996; 10: 485–497. [DOI] [PubMed] [Google Scholar]
- 59. Nolasco LH, Gushiken FC, Turner NA, Khatlani TS, Pradhan S, Dong JF, Moake JL, Vijayan KV. Protein phosphatase 2B inhibition promotes the secretion of von Willebrand factor from endothelial cells. J Thromb Haemost. 2009; 7: 1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ono T, Mimuro J, Madoiwa S, Soejima K, Kashiwakura Y, Ishiwata A, Takano K, Ohmori T, Sakata Y. Severe secondary deficiency of von Willebrand factor‐cleaving protease (ADAMTS13) in patients with sepsis‐induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood. 2006; 107: 528–534. [DOI] [PubMed] [Google Scholar]
- 61. Mutter WP, Karumanchi SA. Molecular mechanisms of preeclampsia. Microvasc Res. 2008; 75: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]