

Abstract
Acute lung injury (ALI) occurs in up to 37% of patients following trauma/hemorrhagic shock (T/HS) and, in other settings, is due to alveolar epithelial cell (AEC) apoptosis. To determine if AEC apoptosis is a key contributor to ALI following T/HS and whether or not signal transducer and activator of translation (Stat)3 activation can prevent it, rats were pretreated with a Stat3 inhibitor or placebo and subjected to T/HS or sham protocol and resuscitated without or with interleukin (IL)‐6. T/HS induced apoptosis in up to 15% of lung cells, 82% of which were AEC. Apoptosis increased with increasing duration of shock and required resuscitation. IL‐6 treatment stimulated lung Stat3 activation and prevented AEC apoptosis. Pretreatment of rats with a Stat3 inhibitor blocked the antiapoptotic effect of IL‐6. Mice deficient in Stat3β, a naturally occurring dominant negative isoform of Stat3, were resistant to T/HS‐induced lung apoptosis. T/HS altered the expression of 87% of apoptosis‐related genes. IL‐6 treatment normalized expression of 75% of the genes altered by T/HS; Stat3 inhibition prevented normalization of 65% of the gene whose expression was normalized by IL‐6. Thus, T/HS‐induced AEC apoptosis, which depended on the duration of hypotension, required resuscitation and was prevented by IL‐6‐mediated activation of Stat3, which acted to normalize the apoptosis transcriptome.
Keywords: alveolar epithelial cell, apoptosis, IL‐6, Stat3
Introduction
Trauma is the leading cause of death for those under 45 years of age in the United States. 1 , 2 The leading cause of death beyond 48 hours is multiple organ failure (MOF), which is preceded by acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) in 83% of cases. 3 , 4 , 5
ALI/ARDS from all causes results in over 70,000 deaths each year in the United States, with an overall mortality that exceeds 40%. 6 , 7 Increasing evidence attributes an important role for alveolar epithelial cell (AEC) apoptosis in the pathogenesis of ALI/ARDS; 7 , 8 , 9 however, the cellular and molecular events contributing to AEC apoptosis are not completely understood, especially in the setting of trauma complicated by hemorrhagic shock (T/HS).
We developed a model of T/HS in rats that results in AEC apoptosis as a function of increased duration of hypotension. AEC apoptosis also required resuscitation, which provided an opportunity for intervention. Interleukin (IL)‐6 administration at the start of resuscitation completely reversed AEC apoptosis and was associated with increased Stat3 activity within the lungs. Mice deficient in Stat3β, a naturally occurring dominant negative isoform of Stat3, were resistant to T/HS‐induced lung apoptosis. Microarray analysis of the lungs showed that the main effect of IL‐6 was to normalize the T/HS‐induced apoptosis transcriptome. Pharmacological inhibition of Stat3 activity within the lungs blocked the ability of IL‐6 to prevent AEC apoptosis and normalize the T/HS‐induced lung apoptosis transcriptome.
Methods
Rat and mouse protocols for trauma plus hemorrhagic shock
These studies were approved by the Baylor College of Medicine Institutional Review Board for animal experimentation and conform to the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals. Adult male Sprague–Dawley rats were obtained from Harlan (Indianapolis, IN, USA).
Stat3β homozygous‐deficient (Stat3βΔ/Δ) mice were generated as described 10 and re‐derived at Jackson Labs (Bar Harbor, ME, USA). Pups from heterozygous matings were tailed and genotyped by polymerase chain reaction (PCR), as described, with minor modifications. 10
The rats were subjected to the sham or trauma/hemorrhagic shock (T/HS) protocols, as described. 11 , 12 Blood was withdrawn into a heparinized syringe episodically to maintain the target mean arterial blood pressure (MAP) at 35 mmHg until blood pressure compensation failed. Blood was then returned as needed to maintain the target MAP. The amount of shed blood returned (SBR) defined five different levels of shock severity reflected in the duration of hypotension: 0% SBR (SBR0) represented the lowest level of shock severity (duration of hypotension 78.0 ± 2.5 minutes), 10% SBR (SBR10; duration of hypotension 149.0 ± 41.4 minutes), 20% SBR (SBR20; duration of hypotension 165.0 ± 32.7 minutes), 35% SBR (SBR35; duration of hypotension 211.0 ± 7.6 minutes), and 50% SBR (SBR50; duration of hypotension 273.0 ± 24.9 minutes). At the end of the hypotensive period, the rats were resuscitated, as described, 11 , 12 and humanely sacrificed 60 minutes after the start of resuscitation. Where indicated, the rats received an intraarterial bolus of 10 μg/kg of recombinant human IL‐6 in 0.1 mL PBS at the initiation of the resuscitation or PBS alone. Sham rats were anesthetized and cannulated for 250 minutes but were not subjected to hemorrhage or resuscitation. One group of rats unresuscitated hemorrhagic shock (UHS) was subjected to the most severe hemorrhagic shock protocol (SBR50), but not resuscitated, and kept at the target MAP (35 mmHg) for an additional 60 minutes (duration of hypotension 336.0 ± 10.3 minutes) before sacrifice.
Stat3βΔ/Δ mice and wild‐type littermate mice were subjected to the T/HS protocol, as described. 13 Sham mice were anesthetized and immobilized in a pairwise fashion with T/HS mice and sacrificed at the same time as their T/HS companion.
Rats and mice lungs were harvested 1 hour after the start of resuscitation, flash‐frozen in liquid nitrogen (left lung), and fixed in 2% paraformaldehyde (right lung).
In vivo pharmacological inhibition of Stat3
To achieve pharmacological inhibition of Stat3 activity within the lungs, the rats were randomized to receive by tail vein injection the G‐rich, quartet‐forming oligodeoxynucleotide (GQ‐ODN) T40214 or non‐specific (NS)‐ODN (2.5 mg ODN/kg) complexed in polyethyleneimine, as described, 14 24 hours prior to subjecting them to the most severe T/HS protocol (SBR50 protocol) with IL‐6 treatment. The half‐life of T40214 in tissues is ≥48 hour. 14
Nucleosome ELISA
Determination of cytoplasmic histone‐associated DNA fragments (nucleosomes) was performed using the Cell Death Detection ELISA Plus Kit (Roche Applied Science, Indianapolis, IN, USA), following the manufacturer's instructions, and modified for the detection of nucleosomes within tissues, as described before. 11 , 12
Terminal deoxynucleotidyl transferase (TdT)‐mediated nick end labeling (TUNEL) staining
TUNEL was performed using the ApopTag® Plus Peroxidase In Situ Apoptosis Detection Kit on lung tissue sections, as previously described. 11 , 12 Data are presented as TUNEL‐positive nuclei in ×1,000 high‐power field (hpf)/total nuclei number ×100%.
Immunoblotting
Levels of Stat3 activation within high‐salt protein extracts of frozen lungs were assessed by immunoblotting with mouse monoclonal antibody to Tyr705 phosphorylated (p)Stat3, as described. 11 , 12 Immunoblotting for the detection of serine 473‐phosphorylated (p)Akt sing mouse monoclonal antibody (Cell Signaling Technology, Inc., Danvers, MA, USA; 1:1,000 dilution) was performed, as previously described. 15
RNA isolation and microarray hybridization and analysis procedures
Total RNA was isolated from 4‐ to 5‐μm cryotome sections of the lungs using TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA) single‐step RNA isolation protocol followed by purification with RNeasy® Mini Kit (Qiagen, Hilden, Germany), as instructed by the manufacturer. Gene expression profiling was performed with the Affymetrix Rat Array RAE 230A following Affymetrix protocols used within the Baylor College of Medicine Microarray Core Facility, as previously described. 11 , 12
Microarray analysis
We used Affymetrix GCOS (Santa Clara, CA, USA), dChip (http://www.dchip.org), and Array Analyzer (Insightful Corporation, TIBCO, Palo Alto, CA, USA) software packages for quality assessment and statistical analysis and annotation. Expression estimation and group comparisons were done with the Array Analyzer. Low‐level analyses included background correction, quartile normalization, and expression estimation using GC robust multiarray average (GCRMA). 16 One‐way analysis of variance (ANOVA) with contrasts 15 was used for group comparisons on all genes and on the list of apoptosis‐related genes only. p values were adjusted for multiple comparisons using the Benjamini–Hockberg method. 17 The adjusted p values represent false discovery rates (FDR) and are estimates of the proportion of “significant” genes that are false or spurious “discoveries.” We used an FDR = 10% as cutoff. 11 , 12
Quantitative reverse transcription‐polymerase chain reaction (Q‐RT‐PCR)
To validate gene expression patterns of microarray hybridization, we performed Q‐RT‐PCR for four genes: Pik3r1, Lgals1, MT1A, and Ccl2, as described. 11 , 12
Statistical analysis
Data throughout the manuscript are presented as mean ± standard error of the mean (SEM). Multiple group comparisons of the means were done by one‐way ANOVA and the Student–Newman–Keuls test. A linear regression analysis demonstrated a linear association between the duration of hypotension and nucleosome levels or%TUNEL‐positive nuclei. Accordingly, the Pearson correlation coefficient was used to evaluate the strength of the association between the duration of hypotension and apoptosis.
Results
T/HS‐induced lung apoptosis depends on the severity of shock and requires resuscitation
To determine whether lung apoptosis occurs in our T/HS model and the contribution of the severity of shock to apoptosis, we measured histone‐associated DNA fragments (nucleosomes) in extracts of the lungs from rats subjected to increasing duration of shock ( Figure 1A ). Nucleosome levels increased with increasing duration of shock (Pearson correlation coefficient 0.764, p < 0.001), with the level of nucleosomes in the SBR50 group (2,580.5 ± 412.3 units/mL) achieving a level 14 times higher than the sham group (146.9 ± 55.6 units/mL, p < 0.0001; 1, 2).
Figure 1.
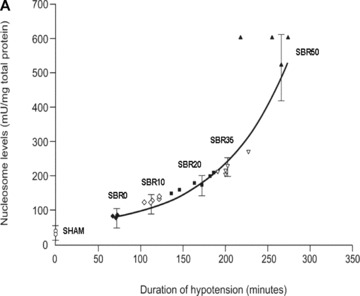
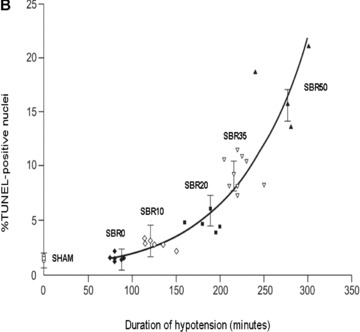
Effect of shock severity on lung apoptosis. Rats were subjected to sham protocol (S) or to T/HS protocol with increasing duration of shock as indicated followed by resuscitation. The lungs were harvested 60 minutes after the start of resuscitation. (A) Nucleosome levels were measured in protein extracts of frozen sections of each lung and the results were plotted after correction for total protein as a function of the duration of the hypotensive period for each animal. Curve fitting was performed and the best‐fitting curve is shown; nucleosome levels increased with the duration of hypotension (Pearson correlation coefficient = 0.764, p < 0.0001). (B) Sections of paraformaldehyde‐fixed lung were stained using the TUNEL assay and TUNEL‐positive nuclei were counted. Data shown represent the percentage of TUNEL‐positive nuclei in 20 random 1,000× fields. Curve fitting was performed and the best‐fitting curve is shown; the percentage of TUNEL‐positive nuclei increased with the duration of hypotension (Pearson correlation coefficient = 0.866, p < 0.0001).
Figure 2.
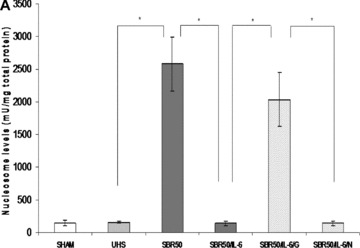
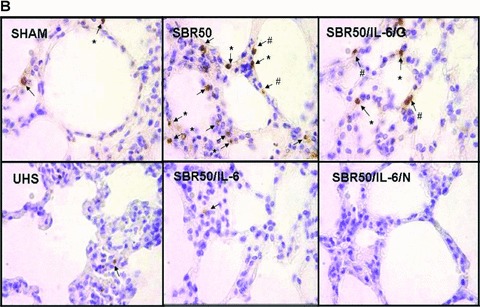
Effect of resuscitation, IL‐6 treatment, and GQ‐ODN pretreatment on T/HS‐induced lung apoptosis. Rats were subjected to sham protocol (Sham, n= 4), unresuscitated T/HS (UHS, n= 3), T/HS treated with placebo at the beginning of resuscitation (SBR50, n= 4), T/HS treated with IL‐6 at the beginning of resuscitation (SBR50/IL‐6, n= 5), T/HS preceded by treatment with GQ oligodeoxynucleotide (GQ‐ODN) 24 hours prior to resuscitation with IL‐6 (SBR50/IL‐6/G, n= 5), or T/HS preceded by treatment with nonspecific ODN (NS‐ODN) 24 hours prior to resuscitation with IL‐6 (SBR50/I‐6/N, n= 5). The lungs were harvested 60 minutes after the start of resuscitation. (A) Nucleosome levels were measured in protein extracts of frozen sections of each lung. Data presented are mean ± SEM of nucleosome level corrected for total protein for each group. Bars marked with an asterisk (*) differ significantly within the pair (p < 0.05, one‐way ANOVA). (B) Sections of paraformaldehyde‐fixed lung were stained using the TUNEL assay. Representative photomicrographs of 1,000× fields of lung specimens from each experimental group are shown. Apoptotic nuclei are indicated by arrows. Type I alveolar epithelial cells (AEC) are indicated by the number symbol (#). Type II AECs are indicated by an asterisk (*). (C) TUNEL‐positive nuclei were counted. Data shown are the mean ± SEM percentage of TUNEL‐positive nuclei per 1,000× fields (20 fields counted). Bars marked with an asterisk (*) differ significantly within the pair (p < 0.05).
To confirm the nucleosome ELISA findings, we performed TUNEL staining of lung sections from T/HS and sham rats. Similar to the nucleosome ELISA results, the percentage of TUNEL‐positive nuclei increased with increasing severity of shock ( Figure 1B ; Pearson correlation coefficient 0.866, p < 0.0001), with the percentage of TUNEL positive nuclei in the SBR50 group (15.38%± 2.84) increasing 7 times over the sham levels (1.93%± 0.44, p < 0.001; 1, 2).
To determine which cells within the lungs were undergoing apoptosis, 10 representative 1,000× fields in each TUNEL‐stained slide were reviewed by an experienced histologist. Alveolar epithelial cells (AEC) type I and II (AEC‐I and AEC‐II) were classified according to morphological criteria, as previously described. 18 TUNEL‐positive nuclei included AEC‐I, AEC‐II, neutrophils, and alveolar macrophages ( Figure 2B ); however, AEC‐II were the predominant TUNEL‐positive cell (59.2% of total TUNEL‐positive cells, p < 0.05), followed by AEC‐I (23.2% of total TUNEL‐positive cells). Thus, AEC accounted for 82% of all TUNEL‐positive cells in the lungs.
To determine the time course of progression to apoptotic cell death in the lungs, TUNEL‐positive nuclei were assessed in the lungs of rats subjected to SBR35 T/HS protocol and sacrificed 4 hours after the start of resuscitation and the results were compared with those sacrificed at 1 hour. The percentage of TUNEL‐positive nuclei in the lungs harvested 4 hours after resuscitation (8.07%± 1.27) was lower than that in the lungs harvested 1 hour after resuscitation (9.79%± 0.69, p > 0.05). Thus, lung apoptosis assessed by TUNEL staining was maximal within 1 hour of resuscitation.
To determine the contribution of resuscitation to lung apoptosis, we assessed nucleosome levels in the lungs of rats subjected to T/HS without resuscitation (UHS group; Figure 2A ). The level of nucleosomes in the UHS group (158.1 ± 15.95 units/mg total protein) was similar to that of the sham group (146.5 ± 45.05 units/mg total protein, p > 0.05). Similar results were obtained when lung apoptosis was assessed by TUNEL staining ( Figure 2C ). The percentage of TUNEL‐positive nuclei in the UHS group (9.43%± 2.34) was not statistically different from that of the sham group (1.93%± 0.22, p > 0.05). Thus, lung apoptosis in the setting of T/HS requires resuscitation, which provided an opportunity to intervene.
IL‐6 administration at the beginning of resuscitation prevented T/HS‐induced lung apoptosis
We previously demonstrated that IL‐6 administration at the start of resuscitation decreased T/HS‐induced lung inflammation.To evaluate the effect of IL‐6 administration on T/HS‐induced lung apoptosis, we measured apoptotic cell death in SBR50 rats randomly assigned to receive either PBS (SBR50) or IL‐6 (10 μg/kg, SBR50/IL‐6) at the beginning of resuscitation. Nucleosome levels in the SBR50/IL‐6 group (142.1 ± 35.4 units/mL) were decreased by 95% compared with those of the SBR50 group (2,580.5 ± 412.3 units/mL, p < 0.001) and were similar to those of the sham group (146.9 ± 55.6 units/mL,p > 0.05; Figure 2A ). In addition, the percentage of TUNEL‐positive nuclei in the SBR50/IL‐6 group (2.65%± 0.43) was decreased by 83% compared with that of the SBR50 group (15.28%± 2.75, p < 0.001) and to levels statistically indistinguishable from those of the sham group (1.93%± 0.22, p > 0.05; Figure 2C ). In particular, the number of TUNEL‐positive AEC‐II per high‐power field in the SBR50/IL‐6 group (2.0 ± 0.2) was reduced by 75% compared with the SBR50 group (8.1 ± 1.75, p < 0.001) and was similar to that of the sham group (3.1 ± 1.9, p > 0.05). Similarly, the number of TUNEL‐positive AEC‐I per high‐power field in the SBR50/IL‐6 group (1.8 ± 1.13) was 44% lower than that of the SBR50 group (3.2 ± 1.03, p < 0.01), and similar to that of the sham group (2.1 ± 0.74, p > 0.05).
The ability of IL‐6 to prevent T/HS‐induced lung apoptosis is Stat3‐dependent
Binding of IL‐6 to its receptor on the surface of cells activates intracellular Stat3, which has previously been demonstrated to activate the transcription of several antiapoptotic genes and contribute to apoptosis resistance in cancer cells. 20 To assess if the antiapoptotic effect of IL‐6 is mediated by Stat3, we first determined if Stat3 is activated in the lungs of rats resuscitated with IL‐6. Extracts of cryotome sections of the lungs harvested 1 hour after IL‐6 treatment were examined by immunoblotting with mouse monoclonal antibody to Tyr705 phosphorylated (p)Stat3 ( Figure 3A ). Analysis of the signal of the Stat3 bands intensity indicated that Stat3 activity is increased 1.8‐fold in the lungs of IL‐6‐treated rats compared with placebo‐treated rats (p < 0.0001; Figure 3B ).
Figure 3.
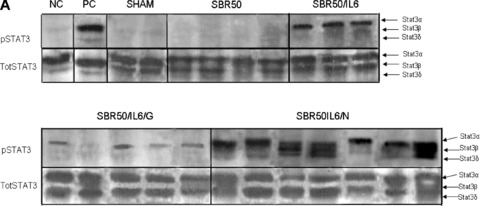
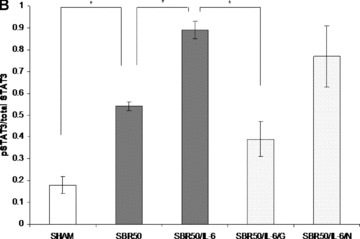
Effect of IL‐6 treatment and GQ‐ODN pretreatment on lung Stat3 activation within the lungs. Rats were subjected to sham (S) protocol, T/HS treated with placebo at the beginning of resuscitation (SBR50), T/HS treated with IL‐6 at the beginning of resuscitation (SBR50/IL‐6), T/HS preceded by treatment with GQ oligodeoxynucleotide (GQ‐ODN) 24 hours prior to resuscitation with IL‐6 (SBR50/IL‐6/G), or T/HS preceded by treatment with nonspecific ODN (NS‐ODN) 24 hours prior to resuscitation with IL‐6 (SBR50/IL‐6/N). The lungs were harvested 60 minutes after the start of resuscitation. (A) Protein extracts of whole lung were immunoblotted for phosphorylated (p)Stat3 and total Stat3. Bands representing Stat3α, Stat3β, and Stat3δ are indicated on the right. 51 , 52 (B) The pStat3 and total Stat3 bands were quantitated by densitometry and data are presented as mean ± SEM of pStat3 signal corrected for total Stat3 signal for each group. Bars marked with an asterisk (*) differ significantly within the pair (p < 0.0001).
To assess if Stat3 activated in the lungs of IL‐6‐treated rats contributes to apoptosis protection, we pretreated rats with the G‐rich, quartet‐forming oligodeoxynucleotide (GQ‐ODN) T40214, a novel Stat3 inhibitor, 14 , 21 24 hours prior to subjecting them to the SBR50 protocol and resuscitation with IL‐6 (SBR50/IL‐6/G rats). Stat3 activity within the lungs of SBR50/IL‐6/G rats was reduced by 78% compared with rats pretreated with a nonspecific ODN (SBR50/IL‐6/N; p < 0.0001; Figure 3B ). Importantly, nucleosome levels within the lungs of SBR50/IL‐6/G rats (2,034.8 ± 409.5 units/mL) were 14.3‐fold higher than those of the SBR50/IL‐6 rats (142.1 ± 35.4 units/mL, p < 0.001; Figure 2A ) and were statistically indistinguishable from those of the SBR50 group (2,580.5 ± 412.3 units/mL, p > 0.05). Similarly, the percentage of TUNEL‐positive nuclei in the lungs of SBR50/IL‐6/G rats (15.83%± 3.16) was 6‐fold higher than that of SBR/IL‐6 rats (2.65%± 0.43, p < 0.0001) and was statistically indistinguishable from the percentage of TUNEL‐positive nuclei in SBR50 rats (15.28%± 2.75, p > 0.05; Figure 2B and C ). In contrast, pretreatment of rats with the nonspecific control ODN (NS‐ODN) did not alter the IL‐6 effect on Stat3 activity ( Figure 3 ), nucleosome levels ( Figure 2A ) or percentage of TUNEL‐positive cells ( Figure 2B and C ). Thus, administration of a Stat3 inhibitor blocked IL‐6‐mediated Stat3 activation and prevention of T/HS‐mediated lung apoptosis.
Two isoforms of Stat3 are expressed in all cells—α(p92) and β (p83)—both derived from a single gene by alternative mRNA splicing, with Stat3αpredominating. Stat3αfunctions as an oncogene, 22 in part, through inhibiting apoptosis, whereas Statβ antagonizes the oncogenic function of Stat3α. 23 Although mice deficient in both isoforms of Stat3 are embryonically lethal at day 6.5 to 7, 24 and mice deficient in Stat3αdie within 24 hours of birth, mice deficient in Stat3β have normal survival and fertility. 10 , 25 To further support the hypothesis that Stat3, in particular Stat3α, contributes to resistance to apoptosis within the lungs in the setting of T/HS, we subjected Stat3β homozygous‐deficient (Stat3βΔ/Δ) mice and their littermate control wild‐type mice to our T/HS protocol (target MAP 30 mmHg for 5 hours) and examined their lungs for nucleosome levels 1 hour after the start of resuscitation ( Figure 4 ). Nucleosome levels in wild‐type T/HS mice were increased compared with wild‐type sham mice (p < 0.001). However, nucleosome levels in the lungs of Stat3βΔ/Δ T/HS mice were reduced by 80% compared with wild‐type T/HS mice (p < 0.001) and were similar to sham mice. These findings indicate that Stat3, in particular Stat3α, protects the lungs from apoptosis in the setting of T/HS.
Figure 4.
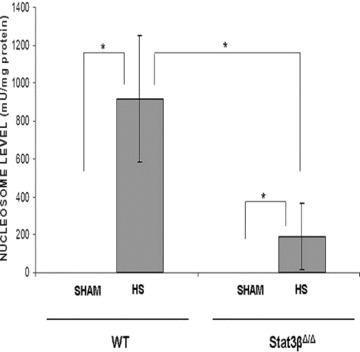
Effect of Stat3β ablation on T/HS‐induced lung apoptosis. Stat3β homozygous‐deficient (Stat3βΔ/Δ) mice and their littermate control wild‐type mice were subjected to the murine T/HS protocol or sham protocol and their lungs were harvested 1 hour after the start of resuscitation. Nucleosome levels were measured in protein extracts of frozen sections of the lungs and the results were corrected for total protein. Data presented are the mean ± SEM of each group (n≥ 3). Significant differences (p < 0.05, one‐way ANOVA) are indicated by an asterisk (*).
Binding of IL‐6 to its receptor results in recruitment of the protein tyrosine phosphatase nonreceptor 11/growth factor receptor‐bound protein2/extracellular signal‐regulated kinases (SHP‐2/Grb‐2/ERK) signaling pathway and in activation of phosphoinositol‐3 kinase (PI‐3K), which promote apoptosis resistance through activation of protein kinase B (Akt). 26 Akt promotes survival by inhibiting the proapoptotic protein Bcl‐associated death promoter (Bad) 27 and caspase (Casp) 9. 28 , 29 To evaluate the role of activated Akt in the antiapoptotic effect of IL‐6 in our model of T/HS, we measured serine 473 phosphorylated in extracts of cryotome sections of the lungs of rats subjected to T/HS and resuscitated without and with IL‐6. pAkt band intensity quantification showed no difference between rats subjected to sham protocol and to T/HS and rats resuscitated without IL‐6 and with IL‐6 (p > 0.05, ANOVA; data not shown). These results underscore the findings outlined in the previous paragraph and indicate that Stat3 is the critical signaling intermediate of the antiapoptotic effects of IL‐6 in our model of T/HS.
Microarray analysis of the lung transcriptome focusing on differential expression of apoptosis‐related genes
In addition to increasing the transcription of antiapoptotic genes (Bcl‐xL, Bcl‐2, and Mcl‐1), 22 , 30 , 31 , 32 , 33 Stat3 has been shown to decrease the transcription of proapoptotic genes (Bad, Bnip3l, and Casp3). To evaluate the role of Stat3 downstream of IL‐6 at the transcriptome level and to identify genes altered within the lungs of animals subjected to T/HS, especially those involved in apoptosis in a global and unbiased manner, we performed Affymetrix oligonucleotide microarray analysis with RAE 230A chips. Sixteen chips were hybridized using mRNA isolated from four lungs, each from the sham, SBR50, SBR50/IL‐6, and SBR50/IL‐6/G groups. All sixteen chips were included in the normalization and expression estimation steps of the analysis and in the statistical analysis and differential expression comparison. The 15,866 probe sets on the RAE 230A chip represent 9,818 annotated genes or expressed sequence tags, including 859 apoptosis‐related genes. The list of 859 apoptosis‐related genes present on the RAE 230A was created by combining gene lists obtained by querying annotation databases provided in GeneSpring (Agilent Technologies, Santa Clara, CA, USA) and dChip, which were derived from the Gene Ontology (GO) Consortium.
To identify genes differentially expressed among the experimental groups, the data were filtered to remove genes with nearly uniformly low expression (absent on ≥80% of chips). Of the 859 apoptosis‐related genes represented on the chips, 630 genes met the requirement of this filtering process and were included in the analysis. One‐way ANOVA (see Methods) was then performedthat identified 611 apoptosis genes with differential expression among four experimental groups—sham, SBR50, SBR50/IL‐6, and SBR50/IL‐6/G—at an FDR = 10%. Of the 611 apoptosis pathway genes whose expression was altered among the four groups, 536 were altered in the SBR50 versus sham comparison (Table S1, Supporting Information). The differential expression of 191 of these genes was 2‐fold or higher (Table S1, Supporting Information, and Figure 5A ). Among the genes whose differential expression was altered by 2‐fold or more in the SBR50 versus sham comparison, the transcripts of the majority of these genes (112 genes) were increased in the SBR50 versus sham comparison by 6.8 ± 3.7‐fold (range = 2‐ to 96.3‐fold), whereas transcripts of 79 genes were decreased in the SBR50 versus sham comparison by 2.2 ± 1.0‐fold (range = 2‐ to 12.3‐fold; Figure 5B ). Importantly, 84 of the 112 genes that were increased in the SBR50 versus sham group were decreased significantly in the SBR50/IL‐6 versus SBR50 group by 2.1 ± 0.9‐fold (range = 1.1‐ to 15.2‐fold), and 60 of the 79 genes that were decreased in SBR50 group were increased significantly in the SBR50/IL‐6 group by 2.0 ± 1.4‐fold (range = 1.1‐ to 9.7‐fold; Figure 5B ). Thus, of the 191 genes whose transcript levels were altered by 2‐fold or higher in the SBR50 versus sham group, 144 or 75% returned to the sham level or were “normalized” in the SBR50/IL‐6 group (Table S1, Supporting Information).
Figure 5.
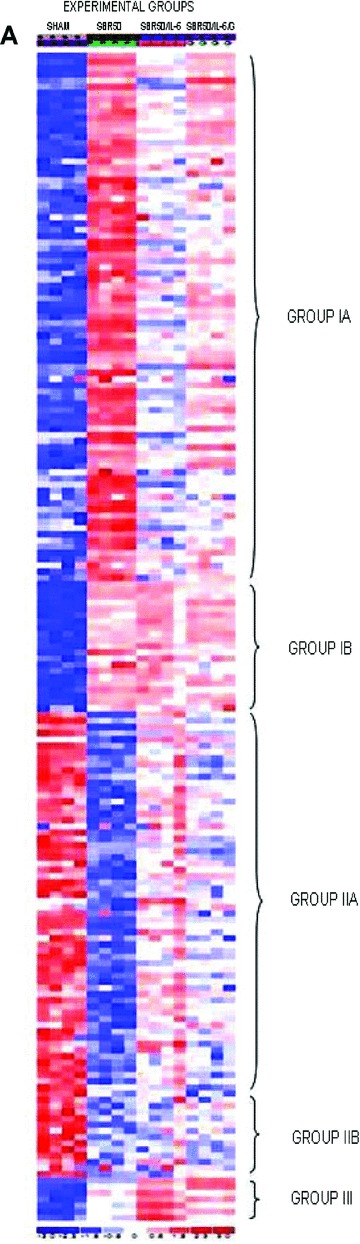
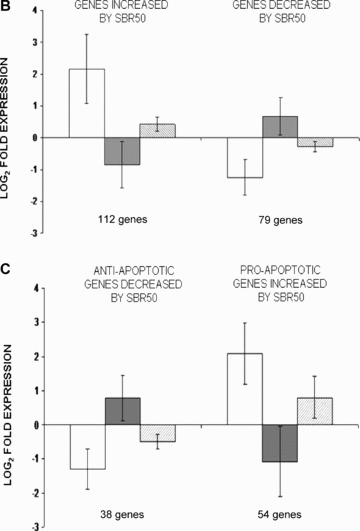
Effect of T/HS without or with IL‐6 treatment on lung apoptosis‐related gene expression and impact of Stat3 inhibition on the IL‐6 effect. (A) A heat map of apoptosis pathway genes is shown containing those genes whose expression is altered 2‐fold or more within the four groups. Columns represent samples from the four groups examined as indicated (S = Sham; P = placebo‐treated SBR50; I = IL‐6‐treated SBR50/IL‐6; and G = animals pretreated with G‐quartet ODN prior to T/HS and IL‐6 treatment, SBR50/IL‐6/G). Rows represent genes as listed in Table S1 (Supporting Information). Red indicates a level of expression above the mean expression of a gene within the experimental group. White indicates a level of expression at the mean within the experimental group, whereas blue indicates a level of expression below the mean within the experimental groups. Logarithm in base 2 of fold expression levels (Log2‐fold) changes in expression levels of subsets of apoptosis‐related genes are shown in (B) and (C) comparing SBR50 versus sham (open bars), SBR50/IL‐6 versus SBR50 (gray bars), and SBR50/IL‐6/G versus SBR50/IL‐6/N (stippled bars). (B) The 191apoptosis‐related genes whose expression levels were changed by 2‐fold or more in SBR50 versus sham were separated into those genes whose transcript levels were increased in SBR50 versus sham (112 genes; left side of the panel) and those whose transcript levels were decreased in SBR50 versus sham (79 genes; right side of the panel). Bars shown represent mean ± SD of the log2‐fold change in gene expression levels for each comparison. (C) The overall effect of T/HS on transcript levels of anti‐ and proapoptotic genes is shown. In the left side of the panel, the mean ± SD of the log2‐fold change in gene expression levels of 38 antiapoptotic genes whose expression was decreased in the SBR50 versus sham comparison is shown (open bar). The expression of all of these genes was increased in the SBR50/IL‐6 versus SBR50 comparison (gray bar). In the right side of the panel, the mean ± SD of the log2‐fold change in gene expression levels of 54 proapoptotic genes whose expression was increased in the SBR50 versus sham comparison is shown (open bar). The expression of 51 of 54 of these genes was decreased in the SBR50/IL‐6 versus SBR50 comparison (gray bar).
Ninety‐one of the 144 genes altered in the SBR50 versus sham comparison and normalized in the SBR50/IL‐6 versus SBR50 comparison were also altered in the SBR50/IL‐6 versus SBR50/IL‐6/G comparison. Remarkably, 86 of these 91 genes (95%) were altered in the opposite direction as the SBR50/IL‐6 versus SBR50 comparison, consistent with the hypothesis that IL‐6 normalized the T/HS‐induced lung apoptosis transcriptome in large part through activation of Stat3 (Table S1, Supporting Information).
Apoptosis‐related genes consist of those encoding proteins that prevent apoptosis (antiapoptotic genes) and those encoding proteins that induce apoptosis (proapoptotic genes). To identify candidate apoptosis‐related genes whose altered expression caused T/HS‐induced AEC apoptosis, we focused on two sets of genes: (1) antiapoptotic genes whose transcript levels were decreased by T/HS and (2) proapoptotic genes whose transcript levels were increased by T/HS. Among the genes with a 2‐fold or higher differential expression in the SBR50 versus sham comparison, 38 antiapoptotic genes were decreased and 54 proapoptotic genes were increased ( Figure 5C ). Expression levels of all 38 of the antiapoptotic genes that were decreased by T/HS were increased by IL‐6 treatment; 94% of the proapoptotic genes that were increased by T/HS were decreased by IL‐6 treatment. Finally, the expression levels of 67% of antiapoptotic genes increased by IL‐6 treatment were decreased by pretreatment with T40214, and conversely, the expression levels of 79% of proapoptotic genes decreased by IL‐6 treatment were increased by T40214 pretreatment ( Figure 5C ), indicating that the normalizing effect of IL‐6 on both sets of genes was mediated largely through activation of Stat3.
The antiapoptotic genes whose expression was decreased by at least 4‐fold during T/HS were glucagon‐like peptide‐1 receptor (Glp1r; 12.5‐fold), phosphatidylinositol‐3‐kinase regulatory subunit‐1 (Pik3r1; 5.5‐fold), lectin galactose binding soluble‐1 (Lgals1; 5.3‐fold), and procollagen, type1, alpha1 (Col1a1; 4.0‐fold; Table S1, Supporting Information). The expression of each was increased in the IL‐6‐treated group by 1.3‐ to 9.7‐fold. Proapoptotic genes whose expression was increased ≥4‐fold by T/HS(≥4.0‐fold upregulated) were branched chain aminotransferase 1 (Bcat1; 17.7‐fold), growth arrest and DNA damage‐inducible 45‐gamma (Gadd45g; 16.9‐fold), oxidized low‐density lipoprotein (ox‐LDL) receptor 1 (Oldlr1; 15.8‐fold), granzyme B (Gzmb; 12.9‐fold), Schaflen3 (Slfn3; 11.8‐fold), Casp4 (7.1‐fold), complement component 3a‐receptor1 (C3ar1; 6.8‐fold), tumor necrosis factor (TNF; 6.3‐fold), protein kinase interferon‐inducible, double‐stranded RNA‐dependent (Prkr; 6.3‐fold), receptor‐interacting serine‐threonine kinase 3 (Ripk3; 6.1‐fold), lipocalin 2 (Lcn2; 5.6‐fold), GTP cyclohydrolase (Gch; 4.7‐fold), IL‐1b (4.2‐fold) and perforin 1 (Prf1; 4.1‐fold). The expression of each was decreased in the IL‐6‐treated group by 1.8‐ to 15.2‐fold (Table S1, Supporting Information).
Q‐RT‐PCR was performed on a subset of the 191 genes whose expression was altered by 2‐fold or higher by T/HS to establish the concordance rate between alteration in gene expression determined by microarray and by Q‐RT‐PCR, an alternative method for assessing the levels of gene expression. The genes evaluated were Pik3r1, Lgals1, MT1A, and Ccl2. The rate of agreement between microarrays and Q‐RT‐PCR in the direction of change in gene expression was 75%, which is similar to or better than that reported by others. 34
Discussion
We demonstrated that T/HS in rats results in lung apoptosis, especially of AECs. Apoptosis increased with increasing duration of hypotension and required resuscitation, which provided an opportunity for therapeutic intervention. IL‐6 administered at the start of resuscitation completely prevented T/HS‐induced AEC apoptosis and was accompanied by increased Stat3 activity. Pharmacological inhibition of Stat3 using GQ‐ODN T40214 developed by our group completely blocked IL‐6‐mediated Stat3 activation and prevention of AEC apoptosis. Mice deficient in the naturally occurring dominant‐negative isoform of Stat3, Stat3β, were completely resistant to T/HS‐induced lung apoptosis, confirming a role for Stat3, particularly Stat3α, in the antiapoptotic effect of IL‐6‐activated Stat3. Lung microarray analysis showed that 87% of known apoptosis‐related genes were altered in T/HS. IL‐6 “normalized” the expression of 75% of these genes; Stat3 was responsible for this normalization in the majority of cases 65.2%). Further examination of the microarray results indicated that the effect of IL‐6‐activated Stat3 was 2‐fold; IL‐6‐activated Stat3 increased the levels of antiapoptotic gene transcripts whose levels were decreased by T/HS and also decreased the transcript levels of proapoptotic genes whose levels were increased by T/HS.
ALI progression to ARDS occurs in 83% of T/HS patients who develop MOF and is associated with significant morbidity and mortality. 3 , 4 , 5 However, the cellular and molecular bases for ALI/ARDS in the setting of T/HS are poorly defined. AEC apoptosis has been described in other settings of lung insult such hypoxia, hyperoxia, adenoviral infection, and bleomycin. 35 , 36 , 37 , 38 Buccellato et al. demonstrated that apoptosis of respiratory epithelial cells due to hyperoxia resulted from generation of reactive oxygen species (ROS), which in turn promoted the activation of Bax at the mitochondrial membrane. 35 AEC apoptosis in adenovirus infections was shown to be mediated by cytotoxic T lymphocytes through either a granzyme or a Fas ligand/receptor pathway. 37 Bleomycin‐induced AEC apoptosis was accompanied by upregulation of Fas and Fas ligand. 38 We describe for the first time that T/HS‐induced AEC apoptosis increased with increasing duration of hypotension and required resuscitation, which strongly suggests a role for reactive oxygen species (ROS).
We previously demonstrated that exogenous administration IL‐6 decreased cardiomyocyte and hepatocyte apoptosis in rodents following T/HS. 11 , 12 , 39 Also, studies by Ward et al. using transgenic mice in which IL‐6 was overexpressed in the lungs indicated that IL‐6 is cytoprotective for Clara cells and alveolar epithelial cells in the setting of hyperoxia. 40 The mechanism(s) of these IL‐6‐mediated effects, however, were not examined. IL‐6 is an antiapoptotic cytokine that activates Stat3 upon interaction with its receptor. 41 We observed that prevention of T/HS‐induced AEC apoptosis by IL‐6 administration was accompanied by increased lung Stat3 activity. Furthermore, pharmacological inhibition of Stat3, by pretreatment with T40214, decreased Stat3 activity and completely prevented the antiapoptotic effect of IL‐6. Thus, activation of Stat3 is critical for the IL‐6‐mediated prevention of AEC apoptosis following T/HS and may also be critical for mediating the antiapoptotic effects of IL‐6 demonstrated in other organs in the setting of T/HS, 11 , 12 as well in other settings. The finding that the lungs of mice deficient the naturally occurring dominant‐negative isoform of Stat3, Stat3β, were completely resistant to T/HS‐induced apoptosis further supports this conclusion ( Figure 4 ).
Activation of Stat3 has been shown to increase the transcription antiapoptotic genes such as Bcl‐xL, Bcl‐2, and Mcl‐1, as well as decrease the transcription of proapoptotic genes, most notably Bad, Bnip3, and Casp3. 22 , 30 , 31 , 32 , 33 A global and unbiased assessment the apoptosis transcriptome using oligonucleotide microarray analysis determined that T/HS altered the expression of 87% of apoptosis‐related genes, of which 55% were antiapoptotic genes and 45% were proapoptotic genes ( Figure 5B ). The predominant effect of IL‐6 administration on the T/HS‐induced apoptosis transcriptome was to increase the expression of antiapoptotic genes whose expression was decreased by T/HS and decrease the expression of proapoptotic genes whose expression was increased by T/HS ( Figure 5C ). Stat3 inhibition, by GQ‐ODN administration, reversed the IL‐6 effect on gene expression in 67% of antiapoptotic genes and 79% of proapoptotic genes ( Figure 5C ). These results suggest that IL‐6‐induced activation of Stat3 results in protection from AEC apoptosis by shifting the balance of anti‐ and proapoptotic transcript levels away from one favoring apoptosis toward one promoting cell survival.
Antiapoptotic gene transcripts that were decreased the greatest during shock and normalized by IL‐6 administration include Glp1r, Pik3r1, and Lgals1. Glp1r is expressed in the intestinal epithelium, cholangiocytes, and neurons of the mammalian hindbrain. Overexpression of Glp1r has been shown to protect against pancreatic islet cell apoptosis. 42 Pik3r1 encodes the p85α regulatory subunit of PI‐3K. Pik3r1 is expressed in several cell types including non‐small cell lung cancer cells, mammary epithelium, fibroblasts, neurons, and muscle. Complete depletion of Pik3r1 in fibroblasts increased apoptosis by preventing Akt‐mediated phosphorylation of Bad. 43 Lgals1 is expressed by neonatal smooth muscle cells in mice embryo, as well as in adult endothelial cells, and has previously been shown to increase cell division in cultured vascular endothelial cells. 44
Proapoptotic transcripts most upregulated following T/HS and downregulated in the IL‐6 treatment group were Bcat1, Gadd45g, Oldlr1, Gzmb, Prf1, Ripk3, and Lcn2. Bcat1 is a cytosolic enzyme that catalyzes transamination of branched‐chain amino acids producing glutamate and branched‐chain alpha‐ketoacids. It is expressed by neurons throughout the nervous system. During states of increased catabolism and decreased energy, Bcat1 converts branched‐chain amino acids to branched‐chain ketoacids, which in turn induce DNA damage and apoptosis. 45 Gadd45g belongs to a family of proteins involved in DNA damage response and cell growth arrest. It is ubiquitously expressed in all normal adult and fetal tissues. Gadd45g activates MTK1 kinase activity in response to environmental stresses, leading to apoptosis through the p38/c‐Jun kinase pathway. Downregulation of Gadd45g prevents apoptosis of cancer cells. 46 Oldlr1 is expressed in highly vascularized organs such as the lungs and placenta and in endothelial cells, smooth muscle cells, cardiomyocytes, and activated macrophages. Oldlr1 is a type II glycoprotein and acts as a receptor for ox‐LDL. Interaction with ox‐LDL induces ROS, reduces NO, and activates nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NFκB). It also increases expression of Bax and decreases expression of Bcl‐2. Oldlr1 is known to induce apoptosis of vascular endothelial cells and vascular smooth muscle cells in a model of cardiac ischemia‐reperfusion. 47 Gzmb (a serine protease) and Prf1 (a pore‐forming protein) have been implicated in the pathogenesis of lung injury in the setting of sepsis 48 and bleomycin‐induced ALI. 38 The cells predominantly expressing Gzmb and Prf1 in these settings were lymphocytes 48 and macrophages. 38 RIP3 is a serine/threonine kinase that localizes to the mitochondria and contributes to TNF‐α‐mediated apoptosis. 49 Lcn2 is a protease that cleaves Casp3; Lcn2 is upregulated during mammary gland involution and is induced by lipopolysaccharide (LPS), TNF‐α, and retinoic acid. 50
The list of anti‐ and proapoptotic genes whose expression was altered the greatest by T/HS and normalized by IL‐administration contains genes that are known to be expressed by epithelial cells—Glp1r, Pik3r1, and Lcn2—as well as genes—Oldlr1, Gzmb, and Prf1—that are expressed by neutrophils, macrophages, and cytotoxic T lymphocytes that infiltrate the lungs during systemic inflammatory states such as T/HS. These observations raise the possibility that IL‐6‐mediated activation of Stat3 confers protection against T/HS‐induced AEC apoptosis through two distinct pathways, one “intrinsic” and the other “extrinsic.” Intrinsic protection by Stat3 is mediated directly within AECs by increasing the levels of antiapoptosis gene transcripts such as Glp1r and Pik3r1 and by decreasing the transcript level of the proapoptotis gene Lcn2. Extrinsic protection by Stat3 is mediated indirectly by decreasing the transcript levels of proapoptotic genes Oldlr1, Gzmb, and Prf1 within cells such as lymphocytes, macrophages, and neutrophils that infiltrate the lungs in T/HS. Further studies are needed to establish whether either or both of these pathways are involved in T/HS‐induced AEC apoptosis and determine which transcripts, in particular, are most important for apoptosis prevention.
In addition to providing new insight into T/HS‐induced ALI by identifying candidate genes important for AEC apoptosis, our results suggest that IL‐6 administration deserves further study to determine its usefulness as an adjuvant for resuscitation of patients suffering from severe T/HS to prevent ALI/ARDS.
Supporting information
Supporting info item
Supporting info item
Acknowledgments
This work was supported, in part, by grants HL076169 (David J. Tweardy) and T32‐HL66991 (Ana Moran) from the National Heart, Lung and Blood Institute of the NIH and grant H48839 (Ana Moran) from the American Lung Association.
References
- 1. Minino AM, Fingerhut LA, Boudreault MA, Warner M. Deaths: Injuries 2002. Natl Vital Stat Rep. 2006; 54: 1–124. [PubMed] [Google Scholar]
- 2. McGee K, Peden M, Waxweiler R, Sleet D. Injury Surveillance. Inj Control Safe Promot. 2003; 10:105–108. [DOI] [PubMed] [Google Scholar]
- 3. Carrico CJ, Holcomb JB, Chaudry IH. Scientific priorities and strategic planning for resuscitation research and life saving therapy following traumatic injury: report of the PULSE Trauma Work Group. Acad Emerg Med. 2002; 9(6): 621–626. [DOI] [PubMed] [Google Scholar]
- 4. Ciesla DJ, Johnson JL, Burch JM, Cothren CC, Sauaia A. The role of the lung in postinjury multiple organ failure. Surgery. 2005; 138: 749–758 [DOI] [PubMed] [Google Scholar]
- 5. Moore FASA, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury multiple organ failure: a bimodal phenomenon. J Trauma. 1996; 40: 501–512. [DOI] [PubMed] [Google Scholar]
- 6. Rubenfeld GD, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005; 353: 1685–1693. [DOI] [PubMed] [Google Scholar]
- 7. Martin TR, Nakamura M, Matute‐Bello G. Apoptosis and epithelial injury in the lungs. Proc Am Thorac Soc. 2005; 2: 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martin TR, Matute‐Bello G. The role of apoptosis in acute lung injury. Crit Care Med. 2003; 31: S184–S188. [DOI] [PubMed] [Google Scholar]
- 9. Jernigan TW, Fabian TC. Apoptosis and necrosis in the development of acute lung injury after hemorrhagic shock. Am Surg. 2004; 12: 1094–1098. [PubMed] [Google Scholar]
- 10. Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X, Murray‐Tait T, Chiarle R, Poli V. The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol. 2004; 5(4): 401–409. [DOI] [PubMed] [Google Scholar]
- 11. Alten JA, Moran A, Tsimeltzon AI, Mastrangelo MA, Hilsenbeck SG, Tweardy DJ. Prevention of hypovolemic circulatory collapse by IL‐6 activated Stat3. PLoS ONE. 2008; 3(2): e1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moran A, Arikan‐Ackan A, Mastrangelo MA, Wu Y, Yu B, Tweardy DJ. Prevention of trauma and hemorrhagic shock‐mediated liver apoptosis by activation of Stat3alpha. Int J Clin Exp Med. 2008; 1(3): 213–247. [PMC free article] [PubMed] [Google Scholar]
- 13. Hierholzer C, Harbrecht B, Menezes JM, Kane J, MacMicking J, Nathan CF, Peitzman AB, Billar TR, Tweardy DJ. Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. J Exp Med. 1998; 187(6): 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jing N, Li Y, Xiong W, Sha W, Jing L, Tweardy DJ. G‐quartet oligonucleotides: a new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res. 2004; 64(18): 6603–6609. [DOI] [PubMed] [Google Scholar]
- 15. Wang Q, Li L, Xu E, Wong V, Rhodes C, Brubaker PL. Glucagon‐like peptide‐1 regulates proliferation and apoptosis via activation of protein kinase B in pancreatic INS‐1 beta cells. Diabetologia. 2004; 47(3): 478–487. [DOI] [PubMed] [Google Scholar]
- 16. Wu JY, Feng L, Park HT, Havlioglu N, Wen L, Tang H, Bacon KB, Jiang ZH, Zhang XC, Rao Y. The neuronal repellent Slit inhibits leukocyte chemotaxis induced by chemotactic factors. Nature. 2001; 410(6831): 948–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001; 125(1–2): 279–284. [DOI] [PubMed] [Google Scholar]
- 18. Parent R. Comparative Biology of the Normal Lung. CRC Press: Boca Raton , FL : 1992. [Google Scholar]
- 19. Meng ZH, Billiar TR, Tweardy DJ. Distinct effects of systemic infusion of G‐CSF versus IL‐6 on lung and liver inflammation and injury in hemorrhagic shock. Shock. 2000; 14: 41–48. [DOI] [PubMed] [Google Scholar]
- 20. Darnell JE. Validating Stat3 in cancer therapy. Nat Med. 2005; 11(6): 595–596. [DOI] [PubMed] [Google Scholar]
- 21. Jing N, Li Y, Xu X, Sha W, Li P, Feng L, Tweardy DJ. Targeting Stat3 with G‐quartet oligodeoxynucleotides in human cancer cells. DNA Cell Biol. 2003; 22(11): 685–696. [DOI] [PubMed] [Google Scholar]
- 22. Bromberg JF, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE. Stat3 as an oncogene. Cell. 1999; 98: 295–303. [DOI] [PubMed] [Google Scholar]
- 23. Turkson J, Bowman T, Garcia R, Caldenhoven R, DeGroot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol. 1998; 18: 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takeda KN, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA 1997; 94: 3801–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dewilde S, Vercelli A, Chiarle R, Poli V. Of alphas and betas: distinct and overlapping functions of STAT3 isoforms. Front Biosci. 2008; 13: 6501–6514. [DOI] [PubMed] [Google Scholar]
- 26. Ishihara K, Hirano T. Molecular basis of the cell specificity of cytokine action. Biochim Biophys Acta. 2002; 1592(3): 281–296. [DOI] [PubMed] [Google Scholar]
- 27. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell‐intrinsic death machinery. Cell. 1997; 91(2): 231–241. [DOI] [PubMed] [Google Scholar]
- 28. Cardone MH, Roy N, Stennicke HR, Salvensen GS, Franke TF, Stanbridge E, Frisch S, Reed J. Regulation of cell death protease caspase‐9 by phosphorylation. Science. 1998; 282(5392): 1318–1321. [DOI] [PubMed] [Google Scholar]
- 29. Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002; 14(5): 381–395. [DOI] [PubMed] [Google Scholar]
- 30. Catlett‐Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez‐Luna JL, Nunez G, Dalton WS, Jove R. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999; 10(1): 105–115. [DOI] [PubMed] [Google Scholar]
- 31. Chen J, Lopez JA. Interactions of platelets with subendothelium and endothelium. Microcirculation. 2005; 12(3): 235–246. [DOI] [PubMed] [Google Scholar]
- 32. Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002; 109(9): 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Taub R. Hepatoprotection via the IL‐6/Stat3 pathway. J Clin Invest. 2003; 112(7): 978–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao M, Chow A, Powers J, Fajardo G, Bernstein D. Microarray analysis of gene expression after transverse aortic constriction in mice. Physiol Genomics. 2004; 19(1): 93–105. [DOI] [PubMed] [Google Scholar]
- 35. Buccellato LJ, Akinci OI, Chandel NS, Budinger GR. Reactive oxygen species are required for hyperoxia‐induced Bax activation and cell death in alveolar epithelial cells. J Biol Chem. 2004; 279: 6753–6760. [DOI] [PubMed] [Google Scholar]
- 36. Lian X, Qin Y, Hossain SA, Yan L, White A, Xu H, Shipley JM, Li T, Senior AM, Du H, Yan C. Overexpression of Stat3C in pulmonary epithelium protects against hyperoxic lung injury. J Immunol. 2005; 174(11): 7250–7256. [DOI] [PubMed] [Google Scholar]
- 37. Matsuzaki Y, Xu Y, Ikegami M, Besnard V, Park KS, Hull WM, Wert SE, Whitsett JA. Stat3 is required for cytoprotection of the respiratory epithelium during adenoviral infection. J Immunol. 2006; 177(1): 527–537. [DOI] [PubMed] [Google Scholar]
- 38. Miyazaki H, Kuwano K, Yoshida K, Maeyama T, Yoshimi M, Fujita M, Hagimoto N, Yoshida R, Nakanishi Y. The perforin mediated apoptotic pathway in lung injury and fibrosis. J Clin Pathol. 2004; 57(12): 1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arikan AA, Mastrangelo MA, Tweardy DJ. Interleukin‐6 treatment reverses apoptosis and blunts susceptibility to intraperitoneal bacterial challenge following hemorrhagic shock. Crit Care Med. 2006; 34: 771–777. [DOI] [PubMed] [Google Scholar]
- 40. Ward NS, Homer RJ, Mantell LL, Einarsson O, Du YF. Interleukin‐6‐induced protection in hyperoxic acute lung injury. Am J Respir Cell Mol Biol. 2000; 22: 535–542. [DOI] [PubMed] [Google Scholar]
- 41. Hirano T. Interleukin 6 and its receptor: ten years later. Int Rev Immunol. 1998; 16(3–4): 249–284. [DOI] [PubMed] [Google Scholar]
- 42. Li L, El‐Kholy W, Rhodes CJ, Brubaker PL. Glucagon‐like peptide‐1 protects beta cells from cytokine‐induced apoptosis and necrosis: role of protein kinase B. Diabetologia. 2005; 48(7): 1339–1349. [DOI] [PubMed] [Google Scholar]
- 43. Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3‐kinase regulates cell signaling and survival. Mol Cell Biol. 2002; 22(3): 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Foster JJ, Goss KL, George CL, Bangsund PJ, Snyder JM. Galectin‐1 in secondary alveolar septae of neonatal mouse lung. Am J Physiol Lung Cell Mol Physiol. 2006; 291(6): L1142–L1149. [DOI] [PubMed] [Google Scholar]
- 45. Eden A, Benvenisty N. Involvement of branched‐chain amino acid aminotransferase (Bcat1/Eca39) in apoptosis. FEBS Lett. 1999; 457(2): 255–261. [DOI] [PubMed] [Google Scholar]
- 46. Zerbini LF, Libermann TA. Life and death in cancer. GADD45 alpha and gamma are critical regulators of NF‐kappaB mediated escape from programmed cell death. Cell Cycle. 2005; 4(1): 18–20. [DOI] [PubMed] [Google Scholar]
- 47. Kataoka K, Hasegawa K, Sawamura T, Fujita M, Yanazume T, Iwai‐Kanai T, Kawamura T, Hirai T, Kita T, Nohara R. LOX‐1 pathway affects the extent of myocardial ischemia‐reperfusion injury. Biochem Biophys Res Commun. 2003; 300(3): 656–660. [DOI] [PubMed] [Google Scholar]
- 48. Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000; 161(1): 237–243. [DOI] [PubMed] [Google Scholar]
- 49. Kasof GM, Prosser JC, Liu D, Lorenzi MV, Gomes BC. The RIP‐like kinase, RIP3, induces apoptosis and NF‐kappaB nuclear translocation and localizes to mitochondria. FEBS Lett. 2000; 473(3): 285–291. [DOI] [PubMed] [Google Scholar]
- 50. Playford RJ, Belo A, Poulsom R, Fitzgerald AJ, Harris K, Pawluczyc I, Ryon J, Darby T, Nilsen‐Hamilton M, Ghosh S, Marchbank T. Effects of mouse and human lipocalin homologues 24p3/lcn2 and neutrophil gelatinase‐associated lipocalin on gastrointestinal mucosal integrity and repair. Gastroenterology. 2006; 131(3): 809–817. [DOI] [PubMed] [Google Scholar]
- 51. Chakraborty A, Tweardy DJ. Granulocyte colony‐stimulating factor activates a 72‐kDa isoform of STAT3 in human neutrophils. J Leukoc Biol. 1998; 64(5): 675–680. [DOI] [PubMed] [Google Scholar]
- 52. Chakraborty A, White SM, Schaefer TS, Ball ED, Dyer KF, Tweardy DJ. Granulocyte colony‐stimulating factor activation of Stat3 alpha and Stat3 beta in immature normal and leukemic human myeloid cells. Blood. 1996; 88(7): 2442–2449. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item