

Abstract
To model a clinical trial of dendritic cell (DC) therapy of a poorly immunogenic mammary tumor, we treated BALB/c mice bearing an established TS/A mammary tumor with lysate‐pulsed DCs and CpG DNA. We observed that the dose of CpG DNA required to activate DCs in vitro was insufficient to mediate tumor rejection in vivo. We therefore undertook in vivo studies to identify an optimized dose of CpG DNA for tumor therapy, defined as the lowest and least frequently administered dose of CpG DNA that mediated complete tumor rejection. We show that one priming dose of 15 nanomoles and one booster dose of 10 nanomoles of CpG DNA given 7 days apart, respectively, with lysate‐loaded DCs were sufficient to mediate complete tumor rejection in vivo. This dose of CpG DNA was 42‐fold higher than that required to activate DCs in vitro but was not associated with any toxicity in mice. Also, the cured mice rejected a subsequent challenge with fresh TS/A tumor, and both CD4+ and CD8+ T cells were required for tumor rejection. We conclude that effective DC‐based therapy of a poorly immunogenic TS/A tumor is enhanced by optimized dosing of CpG DNA. Our data have important implications for DC‐based clinical trials of breast cancer immunotherapy.
Keywords: immunotherapy, vaccines, cells
Introduction
Antigen‐presenting cells (APC) such as dendritic cells (DCs) are required to prime cytotoxic T lymphocytes (CTL) against tumor‐associated antigens (TAA) to generate effective antitumor immunity (reviewed in Ref. 1). DCs maybe induced to synthesize important immunostimulatory cytokines such as interferon‐alpha (IFN‐α) and interleukin‐12 (IL‐12). 2 , 3 In turn, IL‐12 stimulates T‐helper 1 (Th1) immunity 3 important for the enhancement of CTL activity. Studies have demonstrated that TAA‐loaded DCs enhance the rejection of murine tumors, 4 , 5 , 6 , 7 , 8 and such preclinical studies have formed the basis for DC‐based therapies of patients with cancer. 9 , 10 , 11 , 12 Of note, a recent study 13 demonstrated that a therapeutic DC‐tumor fusion vaccine generated two partial responses and one instance of stabilization of disease in 10 treated patients with metastatic breast cancer. Therefore, these encouraging results bode well for DC‐based therapies of patients with advanced cancer.
Although DCs are potent APCs, efforts have nevertheless been undertaken to further amplify their immunostimulatory capacity, and bacterial DNA oligodeoxynucleotides containing unmethylated CpG sequences (CpG DNA) provide such a stimulus (reviewed in Ref. 14). Thus, CpG DNA upregulates the expression of major histocompatibility complex (MHC) and costimulatory molecules such as CD40 and CD86 by DCs. 15 , 16 , 17 It is also a potent stimulator of DC‐mediated CTL responses in vivo, 18 , 19 , 20 and stimulates DCs to secrete potent immunomodulatory cytokines such as IL‐12, TNF‐α, and IFN‐γ 21 , 22 and augments the activities of natural killer cells and B cells. 23 , 24 , 25 , 26 Therefore, CpG DNA has considerable promise as a potent adjuvant for DC‐based cancer therapy.
We wished to model a clinical trial of DC‐based therapy of a poorly immunogenic mammary tumor in immunosuppressed patients. Therefore, we undertook a therapeutic study of tumor lysate‐pulsed DCs plus CpG DNA within the Th2‐biased and Tregrich BALB/c mouse 27 bearing an established poorly immunogenic and rapidly growing TS/A mammary tumor. 28 However, we discovered that the dose of CpG DNA required to maximally activate DCs in vitro was insufficient to stimulate DC‐mediated tumor regression in vivo. We therefore hypothesized that there was a requirement beyond DC activation by CpG DNA to enhance antitumor immunity, and that optimized dosing of CpG DNA would require both activation of DCs as well as stimulation of systemic Thl immunity to enhance tumor rejection in vivo. To test this hypothesis, we undertook a dose‐finding study to identify an optimal dose of CpG DNA that augmented DC‐mediated tumor rejection of an established TS/A tumor in BALB/c mice.
Materials and Methods
Mice
Female BALB/c mice (6–8 weeks of age) were purchased from Taconic (Hudson, NY, USA) and housed in the Animal Facility of the Hillman Cancer Center, University of Pittsburgh Cancer Institute. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh and conducted according to its published guidelines.
TS/A tumor cell
The BALB/c‐derived mammary adenocarcinoma TS/A, a poorly immunogenic and rapidly growing tumor cell line, was generously provided by Dr. Guido Forni, Turin, Italy. The tumor cells were maintained in complete tumor cell medium (RPMI 1640, 10% heat‐inactivated fetal bovine serum, 2 mM L‐glutamine, 100 U/mL penicillin, 100 μ/mL streptomycin, 1 mM sodium pyruvate, 0.1 mM non‐essential amino acids, and 5.5 × 10−5M 2‐mercaptoethanol) and used in experiments when the cells were in logarithmic growth phase.
Generation of bone marrow‐derived DCs
Bone marrow cells were obtained from the femurs of the BALB/c mice. After treatment with ACKhypotonic buffer (0.15 M NH4C1, 0.02 M KHC03, and 0.1 mM EDTA, pH 7.4) to lyse erythrocytes, the bone marrow cells were put in 6‐well plates in 4 mL of normal medium (RPMI 1640,10% heat‐inactivated fetal bovine serum, 2 mM L‐glutamine, 100 U/mL penicillin, 100 μ/mL streptomycin, 1 mM sodium pyruvate, 0.1 mM non‐essential amino acids, and 10 mM HEPES) at a concentration of 106 cells/mL, and incubated overnight in 5% CO2 at 37°C. On the next day, 1,000 U/mL each of murine (m) granulocyte macrophage colony stimulating factor (GM‐CSF) and m IL‐4 (R&D System, Minneapolis, MN, USA) was added to the normal medium (“DC medium”) and cultured for 7 days. Nonadherent cells were harvested and purified by CDllc microbeads (Miltenyi Biotec, Auburn, CA, USA) based on the manufacturer s recommended protocol. Thus, the cells were incubated with anti‐CDllc magnetic beads and passed through a positive selection column (type LS+) in a magnetic field. After rinsing, the column was removed from the magnetic field and CD1 lc+ cells were eluted from the column and washed with PBS before use.
Cytokine production by CpG DNA‐stimulated DCs
The commercially synthesized CpG DNA (5′‐TCCATGA‐CGTTCCTGATGCT‐3′) 26 (Invitrogen, Carlsbad, CA, USA) was phosphorothioate‐modified to resist degradation by intracellular nucleases. A “non‐CpG” DNA (5′‐TCCATGAGC‐TTCCTGATGCT‐3′) was used as a negative DNA control, where the CG dinucleotide from the test DNA was inverted to a GC sequence. DNAs were negative for endotoxin. DCs were generated as described above and 1 × 106 DCs were plated in a 96‐well plate in 200 μL of DC medium/well and 0, 0.1, 1.0, or 3.0 μM (i.e., 0, 0.13,1.3, or 3.9 μg) of CpG or non‐CpG DNA was added to the DC culture. DCs and CpG DNA were cocultured for 24 hours at 37°C, and supernatants were collected and assessed for p70 IL‐12, IFN‐γ, and tumor necrosis factor (TNF‐α) by ELISA (R&D Systems, Minneapolis, MN, USA).
In vivo tumor therapy
Ear‐tagged BALB/c mice were inoculated subcutaneously in their right flanks with 5 × 104 TS/A tumor cells suspended in PBS to a total volume of 100 μL. The tumors were allowed to grow for an average of 7 days until they were palpable (approximately 1–3 mm2). One day prior to therapy, TS/A antigen was generated by five freeze–thaw (−80°C → 37°C) cycles of TS/A tumor cells. Next, the antigen was cocultured overnight at a 0.5–1.0:1 tumor:DC ratio in 5% CO2 at 37°C, with DCs cultured for 7 days in the DC medium. On the following day, tumor‐antigen loaded DCs were washed three times with PBS to remove excess tumor material. TS/A tumor‐bearing mice were then treated twice, 7 days apart, by subcutaneous injections in the left flank with 100 μL of PBS, TS/A antigen‐loaded DCs alone (5 × 105 DCs), (c) CpG DNA alone at the test dose (nanomoles, nmol), or TS/A antigen‐loaded DCs + CpG DNA, where the DCs and CpG DNA were mixed just prior to injection. The tumors were measured with calipers, twice a week, in a blinded fashion. Tumor area (mm2) was determined by calculating the product of the two longest perpendicular diameters.
In vivo T‐cell depletion
The tumor‐bearing mice were depleted of CD4+ and CD8+ T cells in vivo using anti‐CD4 and anti‐CD8 antibodies (GK1.5 and 2.43, respectively; National Cell Culture Center, Minneapolis, MN, USA). The antibodies were administered by intraperitoneal injection on days −2, +2 or +3, +5, +8, or +9 and +12 with respect to the first DC vaccination. The first dose of anti‐CD4 antibody was 0.5 mg and of anti‐CD8 was 1 mg per mouse, followed by subsequent same doses to maintain T‐cell depletion. In preliminary experiments, this antibody dosing schema depleted the mice of CD4 T cells by 64% and CD8 T cells by 50%, as determined by fluorescence activated cell sorting (FACS) (data not shown).
Statistics
Survival data were analyzed using the log‐rank test and the two‐tailed p values are presented. The antibody‐blocking studies are presented as the proportion of tumor‐free animals, and p values were calculated using an exact two‐tailed chi‐squared test for the equality of proportions.
Results
CpG DNA is a potent stimulator of DC function
Prior to our in vivo tumor therapy studies, we evaluated the stimulatory effect of CpG DNA on BALB/c DCs using the in vitro production of immunomodulatory cytokines by DCs as our surrogate for DC activation. As shown in Figure 1 , we determined that CpG DNA is a potent stimulator of DCs, as evidenced by high levels of production of IL‐12, IFN‐γ, and TNF‐α. The results revealed that peak IL‐12 production (2,400 picograms (pg)/106 DC/24 hours) and IFN‐γ production (21,600 pg/106 DC/24 hours) by DCs were observed using 0.1 μM (0.13 μg/200 μL) CpG DNA. PeakTNF‐α production (1,600 pg/106 DC/24 hours) was observed using 3 μM (3.9 μg/200 μl) CpG DNA.
Figure 1.
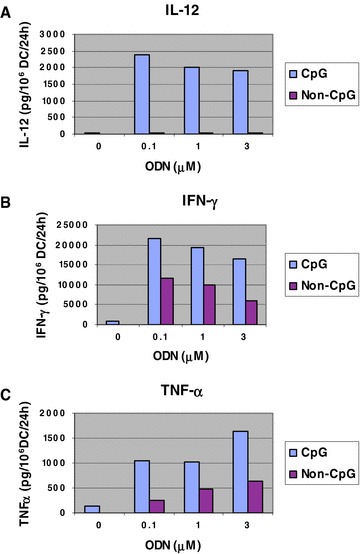
CpG DNA dose response of cytokine production by DCs. One million BALB/c DCs were plated in a 96‐well plate in 200 μL of DC medium/well and 0, 0.1, 1.0, or 3.0 μM (i.e., 0, 0.13, 1.3, or 3.9 μg) of CpG DNA or non‐CpG DNA was added to the wells by gentle mixing. DCs were cocultured with the DNAs for 24 hours at 37°C. Supernatants were then collected and assessed by ELISA for (A) p70 IL‐12, (B) IFN‐γ, and (C) TNF‐α.
Therapy of TS/A tumor‐bearing BALB/c mice with DC + CpG DNA
In a preliminary in vivo study of TS/A tumor therapy in BALB/c mice using D C + CpG DNA, we found that the dose of CpG DNA required to maximally activate DCs in vitro (i.e., 3.9 μg/200 μL of medium) was insufficient to mediate the rej ection of an established TS/A tumor in vivo when combined with tumor antigen‐loaded DCs (data not shown). Therefore, to determine an effective dose of CpG DNA with no toxicity for in vivo use as an immune adjuvant, we conducted a series of adaptive in vivo dose‐response studies using varying doses of CpG DNA combined with a fixed number of TS/A tumor lysate‐loaded DCs (5 × 105 DCs) against an established TS/A tumor in individual groups of three BALB/c mice per group. As shown in our first dose tier study ( Figure 2A ), we observed no difference in tumor rejection between the 100 nmol (660 μg) and 33.3 nmol (220 μg) doses of CpG DNA coadministered twice in equal doses 7 days apart with DCs. However, some mice developed toxicities, including death, at the 33.3‐nmol dose level and progressive tumors at the 10‐nmol dose level. Therefore, in two subsequent series, we empirically fixed the priming dose of CpG DNA at intermediate doses of either 20 nmol ( Figure 2B ) or 15 nmol ( Figure 2C ) and again varied the day‐7 booster doses. Our strategy was to evaluate a series of potential dosages and to select, on the basis of ranking the responses, a dose tier for a confirmatory study. Because of the large number of experimental treatments, each with three mice per group, hypotheses tests were not conducted. Instead a ranking and selection experimental design was used in which a single treatment would be selected from a large panel of candidate therapies that are ranked by apparent efficacy. The final step would then be to conduct a confirmatory study of the winning treatment with a formal hypothesis test. From these experiments, we noted that mice treated with DC and CpG DNA at the lower 15‐nmol (injections 1 and 2) dose level (i.e., DC + CpG 15/15) and the DC + CpG 15/10‐treated mice ( Figure 2C ) had complete tumor rejection, whereas other dose groups did not. We then undertook a validation experiment using DC + CpG 15/10 and five mice per group ( Figure 3) with survival as the endpoint. From this study, we observed that all five mice treated with DC + CpG 15/10 had complete tumor rejection and survived tumor‐free for 240 days, whereas all five mice treated with DCs alone had progressive tumors and died within 40 days (p= 0.002, log‐rank test). This systemic dose of CpG DNA (15 + 10 = 25 nmol) was 42‐fold higher than that required to activate DCs in vitro (3.9 μg or 0.6 nmol). Mice treated with CpG DNA alone appeared to have intermediate survival (p= 0.13), attesting to the potency of CpG DNA in immune‐mediated tumor rejection. No statistically significant difference existed in survival between mice treated with DCs alone compared with PBS. Surviving tumor‐free mice from both the DC + CpG 15/10 and the CpG 15/10‐alone treatment groups were subsequently challenged with fresh TS/A tumor (5 × 104 cells) approximately 65 days after initial tumor implantation. After 240 days, there was no evidence of tumor growth in either group.
Figure 2.
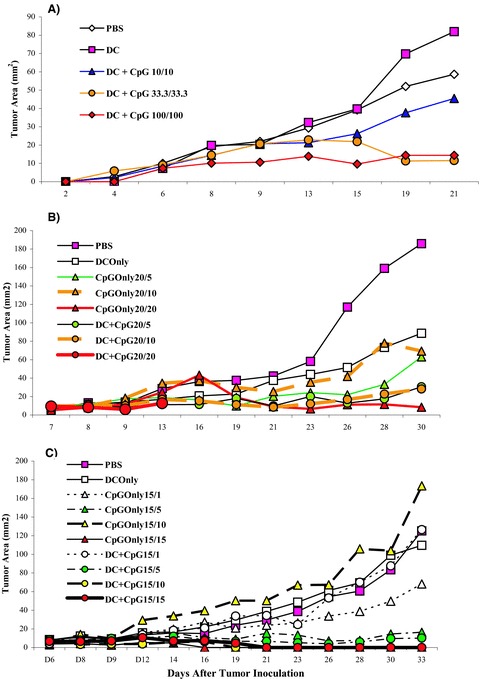
Dose‐finding studies of CpG DNA in combination with tumor lysate‐loaded DC in BALB/c mice bearing an established TS/A tumor. Individual groups of three BALB/c mice were inoculated subcutaneously in their right flanks with 5 × 104 TS/A tumor cells. Tumors were allowed to grow for 7 days until they were palpable (approximately 1–3 mm2). (A) On day 7, post tumor implantation, TS/A tumor‐bearing mice were then treated twice, 7 days apart, by subcutaneous injections in the left flank with 100 μL of PBS, TS/A antigen‐loaded DCs alone (5 × 105 DCs), or TS/A antigen‐loaded DC + CpG DNA at the test dose (nanomoles), where the DCs and CpG DNA were mixed in the same syringe just prior to injection. (B and C) CpG DNA alone was added as a control. Tumors were then measured with calipers twice a week in a blinded fashion, and tumor area (mm2) was determined by calculating the product of the two longest perpendicular diameters. Dosing of CpG DNA is represented as “x”/“y”, where “x” and “y” are the doses (nanomoles) of CpG DNA administered with the first and, 7 days later, the second injection, respectively. Note: the curves for 15/15 and 15/10 overlap.
Figure 3.
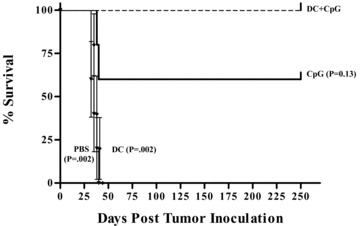
Survival of TS/A tumor‐bearing BALB/c mice. Tumor therapy was performed as detailed in Materials and Methods. Four groups of five mice bearing established subcutaneous TS/A tumors were treated 7 days after tumor implantation with DC + CpG 15/10, CpG 15/10 alone, DCs alone, or PBS given twice, 7 days apart, by subcutaneous injection in the flank opposite the tumor nodule. Data are represented as % survival at 240 days after initial tumor implantation, p values presented with respect to DC + CpG 15/10 using the log‐rank test.
DC + CpG DNA‐mediated tumor rejection is associated with both CD4+ and CD8+ T cells
In a separate experiment, we induced systemic depletion of TS/A tumor‐bearing mice of both CD4+ and CD8+ T cells and measured tumor‐free survival. In total, 5 of 6 treated control mice (DC + CpG DNA without T‐cell depletion) survived compared with 2 of 10 and 0 of 9 following administration of sequential anti‐CD4 and anti‐CD8 antibodies, respectively ( Figure 4 ). These proportions were significantly different from the control group (p= 0.035 and 0.002, respectively, exact chi‐squared test).
Figure 4.
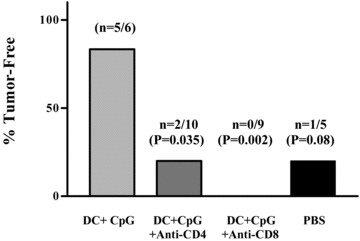
Both CD4+ and CD8+ T cells are required for DC + CpG DNA‐mediated tumor rejection. Tumor therapy was performed as detailed in Materials and Methods. Three groups of tumor‐bearing mice were treated with either DC + CpG 15/10 alone or DC + CpG 15/10 in mice depleted in vivo of either CD4+T cells or CD8+ T cells. Data are represented as the percentage of mice free from tumor at 33 days after tumor implantation, p values presented with respect to DC + CpG 15/10 using an exact two‐tailed chi‐squared test for the equality of proportions.
Discussion
We have verified our hypothesis that an optimized systemic dose of CpG DNA establishes Thl immunity to enhance the DC‐mediated rejection of an established and poorly immunogenic TS/A mammary tumor in Th2‐biased and Tregrich BALB/c mice with no apparent toxicity. The dose of CpG DNA required to enhance DC‐mediated tumor rejection in vivo was 42‐fold higher than that required to activate DCs in vitro. To our knowledge, this is the first dose‐finding study of CpG DNA for DC‐based therapy in tumor‐bearing mice. Furthermore, once an optimal dose of CpG DNA was identified, only one priming and one booster immunization with DCs and CpG DNA were required to effect complete tumor regression in vivo, thereby obviating the need for repeat booster injections. Interestingly, we noted that pre‐activation of DCs with CpG DNA in vitro did not improve in vivo tumor kill and, in fact, showed a nonstatistically significant decline in tumor regression compared with unprimed DCs coinjected with CpG DNA (data not shown). Although the reason for this decrease in tumor kill efficacy is uncertain, it may result from DC “exhaustion” secondary to protracted stimulation by CpG DNA. 29
It was necessary to undertake this CpG DNA dose‐finding study for two reasons. First, as mentioned, the dose of CpG DNA required to maximally activate BALB/c DCs in vitro (3 μM, or 3.9 μg/200 μl) was insufficient to mediate tumor rej ection in vivo. Second, repetitive higher doses of CpG DNA (>15 nmol or >99 μg) were toxic to mice. In our studies, tumor‐bearing mice received two treatments, 7 days apart. We discovered that we could administer a higher dose of CpG DNA (i.e., >15 nmol, up to 100 nmol, or 660 μg) with the first treatment with no apparent toxicity. However, after such initial high doses were followed 7 days later by the administration of even minute doses of CpG DNA (1 nmol or 6.6 μg) (data not shown), the mice became acutely ill, with some deaths. Therefore, to avoid this toxicity, it was important to achieve a fine dosing balance between the efficacy and toxicity for CpG DNA.
Acute inflammatory responses to the administration of CpG DNA have been described, 30 , 31 , 32 , 33 , 34 but one study has shown that such systemic toxicity might be averted by the direct intranodal inj ection of low‐dose CpG DNA. 35 In a tumor protection model, this study showed that four intranodal injections of low‐dose CpG DNA with tumor peptide were successful in protecting mice against a challenge with peptide‐pulsed tumor. In our tumor therapy model, once we established an optimal dose for subcutaneously administered CpG DNA, the mice tolerated the therapy with no toxicity and required only two treatments with DC + CpG DNA to achieve complete tumor rejection. Also, all tumor‐free mice resisted a subsequent challenge with fresh tumor with no evidence of late tumor recurrences (240 days), suggesting the development of robust systemic antitumor immune memory. Interestingly, given that DCs are potent APCs, it was surprising to observe that tumor lysate‐pulsed DCs by themselves were no better than PBS in mediating tumor rejection in our model. Many studies have addressed the optimal source of antigen for tumor immunization, including peptides, apoptotic bodies, DC–tumor fusions, and others. We elected to use tumor lysate because of the ease in obtaining antigen by thermal disruption of tumor cells and because lysate provides a broad spectrum of tumor antigen for immune priming. 36 , 37 , 38 , 39 , 40 , 41 Nevertheless, our lysate‐loaded DCs were completely ineffective in mediating tumor rejection in vivo, except when combined with CpG DNA. This observation suggests that the actual source of tumor antigen might not be as important as the state of DC activation after having acquired antigen. This issue of DC activation is of primary importance in our model because BALB/c mice are rich in immunosuppressive Tregs, 26 and immature DCs have been shown to preferentially activate Tregs, 42 , 43 whereas highly activated DCs circumvent the activity of such cells. 44 Nevertheless, our data clearly demonstrate that the mere activation of DCs, as stimulated in vitro by low‐dose CpG DNA, was insufficient to mediate tumor rejection in vivo. Rather, there was the added requirement for the generation of systemic Thl immunity by a higher optimized dose of CpG DNA.
Conclusion
Our results have important implications for the design of DC‐based therapy of immunosuppressed patients with poorly immunogenic tumors. That is, we have observed the complete rejection of an established poorly immunogenic TS/A tumor in a Th2‐biased, Tregrich BALB/c mouse using the “low‐tech” approach of subcutaneously administered DCs with CpG DNA. Once such an optimized dose of CpG DNA was identified, no more than one priming and one booster dose of CpG DNA with DCs were required to effect complete tumor rejection in vivo, thereby obviating the need for repeat booster injections. Also, there was no a priori requirement to deplete mice of Tregs 45 or modify TS/A tumor cells or DCs 45 , 46 to enhance tumor rejection, thereby considerably simplifying any comparable application to humans. However, although our study demonstrates a dose‐response relationship for CpG DNA in tumor‐bearing mice, such a relationship has not yet been demonstrated in human cancer therapy 47 , 48 This may relate to a different biology of CpG DNA in humans, a higher level of immunosuppression in patients with advanced cancer, or underdosing of CpG DNA. Therefore, given that a subset of human DCs is activated by CpG DNA, 15 , 16 , 17 any clinical trial combining such pulsed DCs with CpG DNA will require a formal dose‐finding schema for CpG DNA that evolves beyond mere local activation of DCs to induce systemic Thl immunity, as demonstrated in our study.
Acknowledgments
The authors thank Dr. Olja Finn, University of Pittsburgh, for careful review of this manuscript. This work was supported by grants from the Department of Defense Breast Cancer Clinical Translational Research Award (DAMD17‐01‐1‐0372),Susan G. Komen Foundation (BCTR0201426) Award, and Herb Jacob Memorial Fund.
References
- 1. Paczesny S, Ueno H, Fay J, Banchereau J, Palucka AK. Dendritic cells as vectors for immunotherapy of cancer. Semin Cancer Biol. 2003; 13: 439–447. [DOI] [PubMed] [Google Scholar]
- 2. Zhou U, Tedder TF. A distinct pattern of cytokine gene expression by human CD83+ blood dendritic cells. Blood. 1995; 86: 3295–3301. [PubMed] [Google Scholar]
- 3. Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, Wysocka M, Trinchieri G, Murphy KM, O'Garra A. Dendritic cells produce IL‐12 and direct the development of Thl cells from naive CD4+ T cells. J Immunol. 1995; 154: 5071–5079. [PubMed] [Google Scholar]
- 4. Celluzzi CM, Mayordomo JI, Storkus WJ, Lotze MT, Falo LD Jr. Peptide‐pulsed dendritic cells induce antigen‐specific, CTL‐mediated protective tumor immunity. J Exp Med. 1994; 183: 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mayordomo JI, Zorina T, Storkus WJ, Zitvogel L, Celluzzi C, Falo LD, Melief CI, Ildstad ST, Kast WM, Deleo AB, Lotze MT. Bone marrow derived dendritic cells pulsed with synthetic tumor peptides elicit protective and therapeutic antitumor immunity. Nature Med. 1995; 1: 1297–1302. [DOI] [PubMed] [Google Scholar]
- 6. Zitvogel L, Mayordomo JI, Tjandrawan T, DeLeo AB, Clarke MR, Lotze MT, Storkus WJ. Therapy of murine tumors with tumor peptide‐pulsed dendritic cells: dependence on T cells, B7 costimulation, and T helper cell‐associated cytokines. J Exp Med. 1996; 183: 87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heckelsmiller K, Beck S, Rail K, Sipos B, Schlamp A, Tuma E, Rothenfusser S, Endres S, Hartmann G. Combined dendritic cell‐ and CpG oligonucleotide‐based immune therapy cures large murine tumors that resist chemotherapy. Eur J Immunol. 2002; 32: 3235–3245. [DOI] [PubMed] [Google Scholar]
- 8. Chagnon F, Tanguay S, Ozdal OL, Guan M, Ozen ZZ, Ripeau JS, Chevrette M, Elhilali MM, Thompson‐Snipes LA. Potentiation of a dendritic cell vaccine for murine renal cell carcinoma by CpG oligonucleotides. Clin Cancer Res. 2005; 11: 1302–1311. [PubMed] [Google Scholar]
- 9. Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R. Vaccination of patients with B‐cell lymphoma using autologous antigen‐pulsed dendritic cells. Nature Med. 1996;2:52–55. [DOI] [PubMed] [Google Scholar]
- 10. Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide‐ or tumor lysate‐pulsed dendritic cells. Nature Med. 1998;4:328–332. [DOI] [PubMed] [Google Scholar]
- 11. Chakraborty NG, Sporn JR, Tortora AF, Kurtzman SH, Yamase H, Ergin MT, Mukherji B. Immunization with a tumor‐cell‐lysate‐loaded autologous‐antigen‐presenting‐cell‐based vaccine in melanoma. Cancer Immunol Immunother. 1998; 47: 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salgaller ML, Tjoa BA, Lodge PA, Ragde H, Kenny G, Boynton A, Murphy GP. Dendritic cell‐based immunotherapy of prostate cancer. Crit Rev Immunol. 1998; 18: 109–119. [DOI] [PubMed] [Google Scholar]
- 13. Avigan D, Vasir B, Gong J, Borges V, Wu Z, Uhl L, Atkins M, Mier J, McDermott D, Smith T, Giallambardo N, Stone C, Schadt K, Dolgoff J, Tetreault JC, Villarroel M, Kufe D. Fusion cell vaccination of patients with metastatic breast and renal cancer induces immunological and clinical responses. Clin Cancer Res. 2004; 10: 4699–4708. [DOI] [PubMed] [Google Scholar]
- 14. Klinman DM, Currie D, Gursel I, Verthelyi D. Use of CpG oligodeoxynucleotides as immune adjuvants. Immunol Rev. 2004; 199:201–216. [DOI] [PubMed] [Google Scholar]
- 15. Hartmann G, Weiner G, Krieg AM. CpG DNA: a potent signal for growth, activation, and maturation of human dendritic cells. Proc Natl Acad Sci USA. 1999; 96: 9305–9310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krug A, Towarowski A, Britsch S, Rothenfusser S, Hornung V, Bals R, Giese T, Engelmann H, Endres S, Krieg AM, Hartmann G. Toll‐like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL‐12. Eur J Immunol. 2001; 31: 3026–3037. [DOI] [PubMed] [Google Scholar]
- 17. Gursel M, Verthelyi D, Klinman DM. CpG oligodeoxynucleotides induce human monocytesto mature into functional dendritic cells. Eur J Immunol. 2002; 32: 2617–2622. [DOI] [PubMed] [Google Scholar]
- 18. Weiner GJ, Liu HM, Wooldridge JE, Dahle CE, Krieg AM. Immunostimulatory oligodeoxynucleotides containing the CpG motif are effective as immune adjuvants in tumor antigen immunization. Proc Natl Acad Sci USA. 1997; 94: 10833–10837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jakob T, Walker PS, Krieg AM, Udey MC, Vogel JC. Activation of cutaneous dendritic cells by CpG‐containing oligodeoxynucleotides: a role for dendritic cells in the augmentation of Thl responses of immunostimulatory DNA. J Immunol. 1998; 161: 3042–3049. [PubMed] [Google Scholar]
- 20. Sparwasser T, Koch ES, Vabulas RM, Heeg K, Lipford GB, Ellwart JW, Wagner H. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur J Immunol. 1998; 28: 2045–2054. [DOI] [PubMed] [Google Scholar]
- 21. Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci USA. 1996; 93: 2879–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Halpern MD, Kurlander RJ, Pisetsky DS. Bacterial DNA induces murine interferon‐gamma production by stimulation of interleukin‐12 and tumor necrosis factor‐alpha. Cell Immunol. 1996; 167:72–78. [DOI] [PubMed] [Google Scholar]
- 23. Cowdery JS, Chace JH, Yi AK, Krieg AM. Bacterial DNA induces NK cells to produce IFN‐gamma in vivo and increases the toxicity of lipopolysaccharides. J Immunol. 1996; 156: 4570–4575. [PubMed] [Google Scholar]
- 24. Ballas ZK, Rasmussen WL, Krieg AM. Induction of NK activity in murine and human cells by CpG motifs in oligodeoxynucleotides and bacterial DNA. J Immunol. 1996; 157: 1840–1845. [PubMed] [Google Scholar]
- 25. Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B‐cell activation. Nature. 1995; 374: 546–549. [DOI] [PubMed] [Google Scholar]
- 26. Yi AK, Chace JH, Cowdery JS, Krieg AM. IFN‐gamma promotes IL‐6 and IgM secretion in response to CpG motifs in bacterial DNA and oligodeoxynucleotides. J Immunol. 1996; 156: 558–564. [PubMed] [Google Scholar]
- 27. Chen X, Oppenheim JJ, Howard OM. BALB/c mice have more CD4+CD25+ T regulatory cells and show greater susceptibility to suppression of their CD4+CD25‐responder T cells than C57BL76 mice. J Leukoc Biol. 2005;78: 114–121. [DOI] [PubMed] [Google Scholar]
- 28. Nanni P, De Giovanni C, Lollini PL, Nicoletti G, Prodi G. TS/A: a new metastasizing cell line from a BALB/c spontaneous mammary adenocarcinoma. Clin Exp Metastasis. 1983; 1: 373–380. [DOI] [PubMed] [Google Scholar]
- 29. Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol. 2000; 1:311–316. [DOI] [PubMed] [Google Scholar]
- 30. Heikenwalder M, Polymenidou M, Junt T, Sigurdson C, Wagner H, Akira S, Zinkernagel R, Aguzzi A. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat Med. 2004; 10: 187–192. [DOI] [PubMed] [Google Scholar]
- 31. Wagner H, Lipford GB, Hacker H. The role of immunostimulatory CpG DNA in septic shock. Springer Semin Immunopathol. 2000; 22: 167–171. [DOI] [PubMed] [Google Scholar]
- 32. Lipford GB, Sparwasser T, Zimmermann S, Heeg K, Wagner H. CpG‐DNA‐mediated transient lymphadenopathy is associated with a state of Thl predisposition to antigen‐driven responses. J Immunol. 2000; 165: 1228–1235. [DOI] [PubMed] [Google Scholar]
- 33. Schmidt U, Wagner H, Miethke T. CpG‐DNA upregulates the major acute‐phase proteins SAA and SAP. Cell Microbiol. 1999; 1: 61–67 [DOI] [PubMed] [Google Scholar]
- 34. Sparwasser T, Miethke T, Lipford G, Borschert K, Hacker H, Heeg K, Wagner H. Bacterial DNA causes septic shock. Nature. 1997; 386: 336–337 [DOI] [PubMed] [Google Scholar]
- 35. Von Beust BR, Johansen P, Smith KA, Bot A, Storni T, Kundig TM. Improving the therapeutic index of CpG oligodeoxynucleotides by intralymphatic administration. Eur J Immunol. 2005; 35: 1869–1876. [DOI] [PubMed] [Google Scholar]
- 36. Asavaroengchai W, Kotera Y, Koike N, Pilon‐Thomas S, Mule JJ. Augmentation of antitumor immune responses after adoptive transfer of bone marrow derived from donors immunized with tumor lysate‐pulsed dendritic cells. Biol Blood Marrow Transplant. 2004; 10: 524–533. [DOI] [PubMed] [Google Scholar]
- 37. Gatza E, Okada CY. Tumor cell lysate‐pulsed dendritic cells are more effective than TCR Id protein vaccines for active immunotherapy of T cell lymphoma. J Immunol. 2002; 169: 5227–5235. [DOI] [PubMed] [Google Scholar]
- 38. Griffioen M, Borghi M, Schrier PI, Osanto S, Schadendorf D. Analysis of T‐cell responses in metastatic melanoma patients vaccinated with dendritic cells pulsed with tumor lysates. Cancer Immunol Immunother. 2004; 53: 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kandil H, Bachy V, Williams DJ, Helmi R, Gotch FM, Ibrahim MA. Regulation of dendritic cell interleukin‐12 secretion by tumourcell necrosis. Clin Exp Immunol. 2005; 140: 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chang GC, Lan HC, Juang SH, Wu YC, Lee HC, Hung YM, Yang HY, Whang‐Peng J, Liu KJ. A pilot clinical trial of vaccination with dendritic cells pulsed with autologous tumor cells derived from malignant pleural effusion in patients with late‐stage lung carcinoma. Cancer. 2005; 103: 763–771. [DOI] [PubMed] [Google Scholar]
- 41. Fields RC, Shimizu K, Mule JJ. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc Natl Acad Sci USA. 1998; 95: 9482–9487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10‐producing, nonproliferating CD4+ T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med. 2000; 192: 1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen‐specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001; 193:233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pasare C, Medzhitov R. Toll pathway‐dependent blockade of CD4+CD25+ T cell‐mediated suppression by dendritic cells. Science. 2003; 299: 1033–1036. [DOI] [PubMed] [Google Scholar]
- 45. Mortara L, Castellani P, Meazza R, Tosi G, De Lerma Barbara A, Procopio FA, Comes A, Zardi L, Ferrini S, Accolla RS. CIITA‐induced MHC class II expression in mammary adenocarcinoma leads to a Thl polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin Cancer Res. 2006; 12: 3435–3443. [DOI] [PubMed] [Google Scholar]
- 46. Comes A, Rosso O, Orengo AM, Di Carlo E, Sorrentino C, Meazza R, Piazza T, Valzasina B, Nanni P, Colombo MP, Ferrini S. CD25+ regulatory T cell depletion augments immunotherapy of micrometastases by an IL‐21‐secreting cellular vaccine. J Immunol. 2006; 176: 1750–1758. [DOI] [PubMed] [Google Scholar]
- 47. Link BK, Ballas ZK, Weisdorf D, Wooldridge JE, Bossier AD, Shannon M, Rasmussen WL, Krieg AM, Weiner GJ. Oligodeoxynucleotide CpG 7909 delivered as intravenous infusion demonstrates immunologic modulation in patients with previously treated non‐Hodgkin lymphoma. J Immunother. 2006; 29(5): 558–568. [DOI] [PubMed] [Google Scholar]
- 48. Thompson JA, Kuzel T, Bukowski R, Masciari F, Schmalbach T. Phase Ib trial of a targeted TLR9 CpG immunomodulator (CPG 7909) in advanced renal cell carcinoma (RCQ). J Clin Oncol. 2004; 22(Suppl 14): 4644. [Google Scholar]