

Abstract
G protein‐coupled receptor kinase 5 (GRK5) is present in endothelial cells (ECs) and has the potential to regulate EC function through seven transmembrane‐spanning receptor (7TMR) signaling. Recently, it has been appreciated that GRKs can affect receptor tyrosine kinases (RTKs). VEGF, an RTK, is one of the most potent mediators for EC function and angiogenesis; therefore, we determined the role GRK5 plays in VEGF signaling in human coronary artery ECs (HCAECs). GRK5 levels were increased by VEGF treatment in HCAECs. Adenoviral overexpression of GRK5 inhibited migration and proliferation of HCAECs in response to VEGF. GRK5 overexpression in HCAECs significantly suppressed both acute and late activation of Akt and extracellular signal‐related kinase (ERKs) as well as the phosphorylationof GSK‐3β, an endogenous substrate of Akt. Coimmunoprecipitations revealed that GRK5 is physically associated with Akt. This study shows for the first time that GRK5 negatively regulates VEGF signaling in HCAECs and suggests that targeted intervention of GRK5 in ECs might be a novel therapeutic strategy to prevent and treat disorders involving altered EC function.
Keywords: G protein‐coupled receptor, G protein‐coupled receptor kinase 5, endothelial cell, vascular endothelial cell growth factor, signal transduction
Introduction
Seven transmembrane‐spanning receptors (7TMRs) play an important role in the regulation of cardiovascular function. After being stimulated by an agonist, 7TMRs are desensitized and downregulated primarily by a group of serine/threonine kinases, G protein‐coupled receptor kinases (GRKs) that induce receptor phosphorylation and desensitization. 1 Of the GRKs present in endothelial cells (ECs), GRK5 is the most abundant. 2 Importantly, GRK5 plays a key role in endothelial barrier function, at least in part, through its phosphorylation and desensitization of the thrombin PAR‐1 receptor. 2
Vascular endothelial cell growth factor (VEGF) is also a major mediator in maintaining vasculature integrity and blood vessel formation, including angiogenesis and vasculogenesis. 3 VEGF is a multifunctional cytokine that is expressed in a variety of cells including ECs. 4 In ischemic organ/tissues, VEGF is abundantly expressed and is involved in the activation of ECs and angiogenesis. VEGF exerts its biological eff ects through two high affinity receptors with tyrosine kinase activity: VEGF receptor 1 (VEGFR‐1, also known as flt‐1) and VEGF receptor 2 (VEGFR‐2, also known as Flk‐1 or KDR). 3 In ECs, these receptors are found at the surface of ECs, and VEGFR‐2 is considered the principal receptor of VEGF‐dependent angiogenic signals. 3
Recently, it has been shown that GRKs can have actions toward receptors other than 7TMRs. GRK2 is involved in a negative feedback regulation of non‐7TMRs including transforming growth factor‐β (TGF‐β) 5 and platelet‐derived growth factor receptor‐β (PDGF‐β). 6 GRK2 phosphorylates and desensitizesthese receptors, thus having an important impact on the cell in a non‐7TRM‐mediated manner. Whether this is a property shared by other GRKs is, of yet, unknown. However, if GRK5 were also able to affect non‐7TMR signaling, especially VEGF, it would provide evidence for another mechanism whereby GRK5 could have a profound effect on EC function.
In this study, we reveal for the first time that GRK5 is regulated by VEGF in human coronary artery ECs (HCAECs). Importantly, we also show that GRK5 attenuates VEGF‐evoked signaling and functionally inhibits VEGF‐induced EC migration and proliferation. These results suggest an intriguing role for GRK5 in the regulation of the non‐7TMR receptor for VEGF in ECs and broaden our understanding of GRK5 involvement in cardiovascular regulation.
Methods
Cell culture, reagents, and materials
Primary HCAECs were purchased from Cambrex (San Diego, CA, USA). The cells were cultured in EGM2‐MV medium containing EC growth supplements (Cambrex). Cultures within 10 passages were used in all experiments. Antibodies were from various providers: anti‐GRK5 (C‐20) and anti‐pERKs (C‐4) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); anti‐extracellular signal‐related kinases (ERKs), ‐Akt, ‐phospho‐Akt (Ser473), and anti‐phospho‐GSK‐3β (Ser9) from Cell Signaling (Danvers, MA, USA); rat anti‐CD31 (PECAM‐1) from BD Pharmingen (San Diego, CA, USA); anti‐GAPDH from RDI Research Diagnostics (Concord, MA, USA); secondary antibodies for immunoblotting were goat IRDye 800 CW conjugated anti‐mouse IgG from Rockland Immunochemicals, Inc. (Gilbertsville, PA, USA) and Alexa Fluor 680 goat anti‐mouse IgG from Molecular Probes (Eugene, OR, USA); Alexa Fluor 488 goat anti‐rabbit antibody and Alexa Fluor 594 goat anti‐rat antibody for immunostaining were obtained from Molecular Probes (Eugene, OR, USA). Horse radish peroxidase (HRP)‐conjugated secondary antibody for immunohistochemistry and Vectashield mounting medium with DAPI and 3,3′‐diaminobenzidine (DAB) substrate kit for peroxidase were obtained from Vector Lab (Burlingame, CA, USA). VEGF165 was obtained from PeproTech (Rocky Hill, NJ, USA).
Construction of recombinant virus
Adenovirus expressing bovine GRK5 (adeno‐GRK5) was constructed, prepared, and expanded as described previously. 7 Th e constitutively active Akt construct, which is HA‐tagged myristylated Akt (HA‐myr‐Akt) with the c‐Src myristylation sequence fused in‐frame to the N‐terminus of the HA‐Akt coding sequence, was described previously (kindly provided by Dr Tung Chan, Th omas Jeff erson University). 8 Adenovirus expressing GFP and LacZ adenovirus, as described elsewhere, were used as control viruses. 7 , 9
Immunoblot analysis
Cultured HCAECs were harvested in lysis buff er containing 10 mM Tris‐HCl, pH 6.8, 2% (v/v) SDS, 2 mM EDTA, 50 mM DTT, 6% glycerol, 1 mM PSMF, 1 μg/mL leupeptin, and 1μg/mL aprotinin. Whole‐cell lysates were subjected to SDS‐PAGE and immunoblotting according to standard protocols and visualized using Infrared Odyssey Imaging System (Li‐Cor, Inc., Lincoln, NE, USA).
Coimmunoprecipitaion for Akt and GRK5 interaction
HCAECs were infected with both adeno‐GRK5 and HA‐myr‐Akt for 20 hours prior to further treatment. Cells were lysed in IP buffer containing 50 mM HEPES, 125 mM NaCl, 1 mM EDTA, 0.5% (v/v) NP‐40, 1 mM NaF, 5% glycerol, 1 mM PSMF, 1 μg/ mL leupeptin, and 1 ìg/mL aprotinin. The supernatants, with equal amounts of protein, were incubated with monoclonal HA antibody‐conjugated sepharose beads or with GRK5 antibody followed by incubation with protein A‐sepharose beads. Precipitated immune complexes were subjected to three washes with IP washing buff er containing 0.25% NP‐40, resuspended in sample loading buffer, and subjected to immunoblot analysis, as described above.
Real‐time quantitative PCR
Total RNA was extracted from HCAECs using Qiagen RNeasy Kit, and cDNA was synthesized by reverse transcription of the RNA with Superscript II (Invitrogen, Carlsbad, CA, USA). Real‐time PCR was performed in triplicate with the cDNA on a MyIQ realtime PCR detection system (Bio‐Rad, Hercules, CA, USA) with SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Gene‐specific mRNA was normalized to 18S rRNA as an internal control. Sequences of the primer sets used were as follows: GRK5, 5′‐ACCTGAGGGGAGAACCATTC‐3 and 5′‐TGGACTCCCCTTTCCTCT TT‐3′; 18S rRNA, 5′‐TCAA GAACG AAAGTCGGAGG‐3′, and 5′‐GGACA TCTAAGGGCATCAC‐3′. PCR conditions were 95°C for 3 minutes and 40 cycles of 95°C for 10 seconds and 63.5°C for 45 seconds. Specificity of PCR products was confirmed by gelelectrophoresis, sequencing, and melt curve analysis. For visualization, additional PCR reactions were stopped mid‐log phase to verify fluorescence output.
Cell migration and proliferation assays
HCAEC migration was quantitatively measured using both modified Boyden chamber and outgrowth models, as previously described. 10 For the outward growth migration model, a cell cloning cylinder, 6 mm in diameter, was set on a cell culture dish, and 1.0 × 105 HCAECs infected with GRK5 or GFP virus, as above, were seeded inside the cylinder in 100 μL EGM2 basal growth medium containing 0.5% FBS. Two hours after seeding, the cylinder was removed, the edges of cell nest marked, and migration was measured as the distance away from the edge of the cell nest.
Cell cycle assay
DNA synthesis was assessed by flow cytometry. A total of 2 × 104 cells were analyzed with a FACScan flow cytometer. Cell cycle modeling and proportion of cells in the G0/1, S, and G2/M phases were determined accordingly, as previously described. 10
Statistical analyses
The results are expressed as mean ± standard deviation (SD) from independent experiments. The data were analyzed by a 2‐tailed Student's t‐test. Values of p < 0.05 are considered to be statistically significant.
Results
Regulation of GRK5 by VEGF in HCAECs
In this study, we investigated whether VEGF, an important effector for the biological actions of ECs, can regulate GRK5 in HCAECs. We used VEGF165, the most potent isoform of VEGF in terms of inducing angiogenesis. Our results show that GRK5 protein expression was increased aft er VEGF treatment in HCAECs in a time‐dependent manner such that there was a doubling in GRK5 levels at 1 hour of VEGF treatment ( Figure 1A ). In further experiments, we infected HCAECs with an adenovirus overexpressing GRK5 driven by a CMV promoter and challenged the cells with VEGF for different periods of time ( Figure 1B ). We found that exogenous GRK5 protein levels were also increased by VEGF. GRK5 mRNA expression was unchanged by VEGF as detected by RT quantitative PCR ( Figure 1C ).
Figure 1.
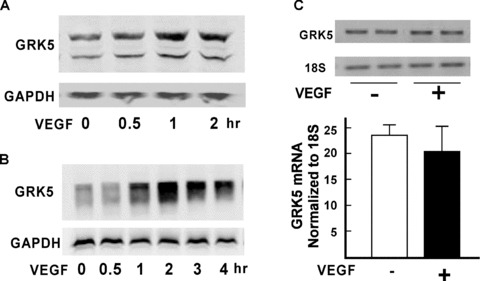
VEGF regulation of GRK5 in HCAECs. (A) HCAECs starved for 20 hours were treated with VEGF (20 ng/ mL) and harvested at different time points as indicated. Whole‐cell lysates were subjected to SDS‐PAGE and analyzed by Western blotting with anti‐GRK5 antibody. The blot was stripped and reprobed with GAPDH as a loading control. (B) HCAECs were infected with GRK5 or GFP adenovirus for 20 hours as described in Methods and then treated with VEGF (20 ng/mL) for indicated times. Whole‐cell lysates were subjected to Western blotting for GRK5 and GAPDH, as described above. (C) HCAECs were starved for 20 hours and challenged with VEGF (20 ng/mL). Total RNA was extracted. Expression of GRK5 mRNA was quantified by real‐time PCR. Data are mean ± SD for mRNA encoding GRK5 relative to that of 18S rRNA in triplicate.
Eff ect of GRK5 on HCAEC migration and proliferation in response to VEGF stimulation
Cell migration and proliferation are two key events for ECs to maintain vascular integrity and neovascular formation. Results already presented suggest that GRK5 expression is increased by VEGF. To examine the functional implication of increased GRK5 expression in HCAECs in response to VEGF, we overexpressed GRK5 in these cells using recombinant adenovirus and challenged them with VEGF. The results demonstrate that GRK5 overexpression significantly suppressed VEGF‐induced HCAEC migration as compared with control viral GFP counterparts in both Boyden chamber ( Figure 2A and 2B ) and outgrowth migration assays ( Figure 2C and 2D ).
Figure 2.
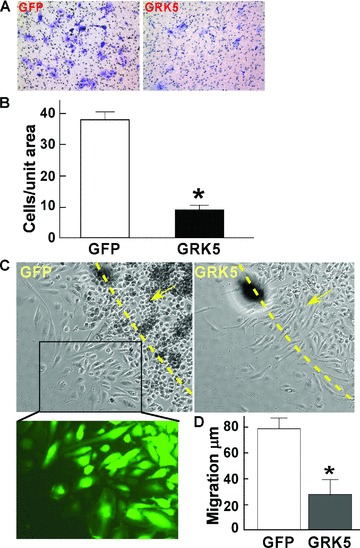
GRK5 inhibits HCAEC migration. (A and B) HCAECs were infected with GRK5 or GFP adenovirus for 18 hours. Cells were then trypsinized and reseeded (1 · 104) in endothelial basal medium containing 0.5% FBS to the upper chamber of the Transwell system for 2 hours and then transferred to wells preloaded with 0.6 mL basal medium containing 0.5% FBS plus 20 ng/mL VEGF. Twenty‐four hours later, the inserts were fi xed and stained with crystal violet. The cells in the upper side of the insert were removed. Representative images of stained cells on the undersurface of the porous membrane as shown (A) were counted (n= 6) (B). (C and D) HCAECs were infected with GRK5 or GFP adenovirus as above and then trypsinized and reseeded in a cell clone cylinder for outward migration assay in the presence of VEGF (20 ng/mL). Representative images of cell migration and GFP expression are shown (C). Migration distances (μm) were measured 24 hours later (D) (n= 10). Data represent mean ± SD. *p < 0.001 versus control.
To examine EC proliferation in response to VEGF in the presence of increased expression of GRK5, we used FACS flow cytometry. GFP adenovirus was used as a control. Following stimulation of HCAECs with VEGF for 24 hours, there was an increase in ECs found in the Sphase with a corresponding change in G0/G1 phase ( Figure 3A ). Following stimulation with VEGF in the presence of GRK5, there was an approximate 56% decrease in the number of cells in S phase as compared to the GFP/VEGF group ( Figure 3A ). Therefore, these data suggest that overexpression of GRK5 expression is sufficient to decrease cell cycle progression in response to an EC proliferation agonist, VEGF. We also detected the number of ECs per unit area following overexpression of GRK5 as another indicator of EC proliferation. We found that GRK5 overexpression resulted in a 40% decrease in cell number, again indicating a GRK5‐mediated decrease in proliferation ( Figure 3B ).
Figure 3.
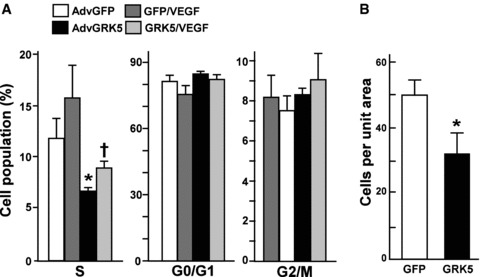
GRK5 inhibits HCAEC proliferation. HCAECs were infected with GRK5 or GFP adenovirus for 20 hours and treated with VEGF (20 ng/mL). EC cell cycle progression was analyzed by flow cytometry (A). Percentage of cells in each of the cell cycle phases is shown (n= 4). (B) HCAECs were infected with GRK5 or GFP adenovirus, as described above, then stimulated with VEGF for 24 hours. Cell number in each dish was counted and cells per unit area was determined (n= 10). Data represent mean ± SD. *p < 0.01.
Regulation of VEGF signaling pathways by GRK5 in HCAECs
Our results reveal that GRK5 is inhibitory for both migration and proliferation of HCAECs treated with VEGF. To explore the underlying molecular mechanisms, we investigated the effects of GRK5 on VEGF‐elicited signal transduction in HCAECs by focusing on Akt and mitogen‐activated protein kinase (MAPK) (ERK1/2) pathways, which are well elucidated for their critical role in mediating EC migration, proliferation, and angiogenesis. 12 We infected HCAECs with AdvGRK5 or one of two diff erent control viruses, GFP and LacZ, to rule out the possible eff ects of the adenovirus itself on the signaling pathway ( Figure 4 ). Our results illustrate that GRK5 inhibits activation of Akt (∼40%) and ERK (∼30%) in response to VEGF treatment for either short ( Figure 4A ) or long ( Figure 4B ) time courses, as demonstrated by decreased phosphorylation of both molecules following VEGF stimulation in the presence of increased GRK5 expression as compared with controls ( Figure 4 ). Both control viruses showed similar patterns in terms of the VEGF‐stimulated Akt and ‐ERK1/2 activation ( Figure 4A ). Consistent with the inhibition of Akt activation, the phosphorylation of the endogenous Akt substrate, GSK‐3β, was also inhibited in both short and long time course experiments ( Figure 4 ). Th is suggests that GRK5 overexpression is sufficient to decrease intracellular signaling pathways downstream of VEGF stimulation and can have a direct impact on migration and proliferation of ECs. It remains to be determined whether GRK5 has a direct impact on VEGF receptor desensitization similar to GRK2‐mediated phosphorylation and desensitization of PDGF‐β receptors, 6 and/or whether there are direct interactions between GRK5 and the signaling molecules themselves.
Figure 4.
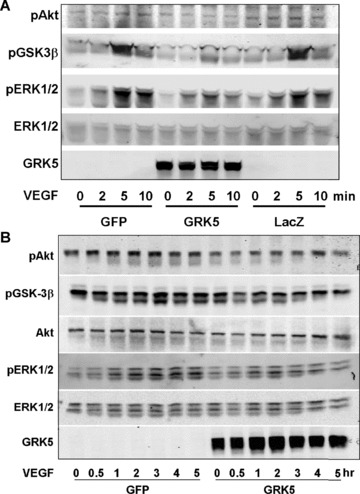
Effect of GRK5 on VEGF‐induced signaling in HCAECs. (A and B) HCAECs were infected with GRK5 adenovirus or one of the control viruses, GFP or LacZ. After 20 hours of starvation, the cells were treated with VEGF (20 ng/mL) for the indicated times. Whole‐cell lysates were subjected to SDS‐PAGE and Western blot analysis using antiphospho‐Akt and antiphospho‐ERK1/2 antibody. Total Akt and total ERK were detected as internal controls. Phosphorylation of GSK‐3β was also detected using the antiphospho‐GSK‐3β antibody. GRK5 overexpression is confirmed.
Association of GRK5 with Akt in HCAECs
Our data illustrate that GRK5 significantly inhibited Akt phosphorylation as well as Akt activity in response to VEGF, which is indicated by the decreased phosphorylation of GSK‐3β in HCAECs. We therefore investigated whether GRK5 could aff ect Akt by directly interacting with it. We coinfected HCAECs with a virus expressing an HA‐tagged, constitutively active form of Akt, HA‐myr‐Akt,8 together with AdvGRK5. Immunoprecipitation with either anti‐GRK5 or anti‐HA pulled down HA‐Myr‐Akt or GRK5, respectively ( Figure 5A and 5B ). Importantly, there was a decrease in this association with VEGF treatment ( Figure 5 ). Further, we infected HCAECs with a single virus expressing either GRK5 or HA‐Myr‐Akt and performed coimmunoprecipitation of overexpressed GRK5 or Akt with endogenous Akt or GRK5. Th e results consistently show that GRK5 and Akt directly interact with each other in HCAECs ( Figure 5C and 5D ).
Figure 5.
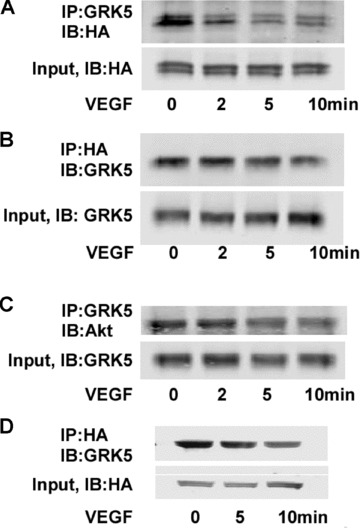
Coimmunoprecipitation of GRK5 and Akt in HCAECs. (A and B) HCAECs were coinfected with GRK5 and HA‐myr‐Akt adenoviruses. Cells were lysed and GRK5 or HA‐myr‐Akt was immunoprecipitated from the lysate using (A), rabbit anti‐GRK5 followed by protein (A) sepharose beads, or (B), monoclonal mouse anti‐HA sepharose beads. Proteins were determined by Western/immunoblotting (IB) using antibodies as indicated. The amounts of input GRK5 or HA‐myr‐Akt are also shown. (C and D) HCAECs were infected with either GRK5 (C) or HA‐myr‐Akt (D) adenovirus. Cells were lysed and GRK5 or HA‐myr‐Akt was immunoprecipitated from the lysate by rabbit anti‐GRK5 followed by C, protein A sepharose beads or D, monoclonal mouse anti‐HA sepharose beads. Endogenous Akt or GRK5 was determined by Western/IB using antibodies as indicated. The amounts of input GRK5 or HA‐Akt are also shown.
Discussion
In this study, we provide the first piece of evidence that GRK5 can regulate biological functions and signal transduction of HCAECs in response to a non‐7TMR agonist, VEGF. Th is is supported by the following findings: (1) GRK5 is expressed in HCAECs and its protein level can be regulated by VEGF; (2) functionally, GRK5 inhibits migration and proliferation of HCAECs in response to VEGF; (3) overexpression of GRK5 inhibits VEGF‐induced activation of Akt and ERK1/2; (4) inhibition of Akt could, at least in part, result from an interaction between Akt and GRK5.
Our results show that VEGF treatment led to an increase of GRK5 protein level in HCAECs. However, GRK5 mRNA expression was not changed by VEGF, suggesting that VEGF regulates GRK5 protein through posttranscriptional modification and stabilization. To further support this notion, we also find that following VEGF stimulation, exogenously expressed GRK5 levels using a viral‐mediated delivery were also increased. Although posttranslational modification and degradation for other GRKs have been investigated, evidence for the modifi cation of GRK5 and the involvement of protein stability is still lacking. 12 Therefore, further studies are needed in order to explore the mechanisms underlying VEGF‐induced increases in GRK5 protein expression.
VEGF is a multifunctional cytokine and one of the most potent mediators in blood vessel formation. 3 VEGF elicits signal transduction and exerts its biological effects through the cognate receptors with tyrosine kinase activity, VEGFR‐1 and VEGFR‐2. 3 Among the signaling molecules activated by VEGF, Akt and ERKs are well established for their critical role in EC migration and proliferation. 11 Therefore, to understand the mechanisms responsible for the inhibitory eff ects of GRK5 on VEGF‐mediated HCAEC function, we explored the effects of GRK5 on VEGF activation of Akt and ERKs. We found that VEGF signal transduction could be negatively regulated by GRK5. Importantly, this is a novel role for GRK5, which to date has been described as a 7TMR kinase. Th e results also show that GRK5 could inhibit both acute and chronic VEGF‐mediated activation of ERKs and Akt. Th erefore, we have found that VEGF increases GRK5 in HCAECs, which in turn suppresses VEGFmediated cellular function and signal transduction. Hence, it appears that a feedback regulation of VEGF biological functions by GRK5 exists in HCAECs.
Feedback or feedforward circuits are important mechanisms in cellular networking. Recently, it has been shown that GRK2 is involved in the negative feedback regulation of signal transduction. TGF‐β can induce GRK2 expression, which acts in a negative feedback loop to control TGF‐β biological responses.5 Activated PDGFR‐β upregulates GRK2, which then through feedback inhibits the signaling of the PDGFR‐β. 6 Our study uncovered a feedback loop for the regulation of VEGF biological functions by GRK5. Lately, more details of GRK‐regulating cell signaling have been unveiled. 13 GRK2 can negatively regulate chemokine CCL2‐induced ERK activation at the level of the MEK‐ERK interface. 14 It is also now appreciated that Akt physically interacts with GRK2 in liver sinusoidal ECs, and this interaction inhibits Akt phosphorylation and the activity of its downstream eff ector eNOS in the pathogenesis of portal hypertension. 15 Similarly, our study showed that GRK5 can form a complex with Akt, and that this interaction appears to be weakened by VEGF treatment. The GRK5‐Akt interaction could, at least in part, contribute to the negative regulation of Akt phosphorylation and its activity and GRK5 inhibition of VEGF‐induced migration and proliferation. Importantly, we also found that GRK5 inhibited ERK activation; hence, the role of GRK5 and its impact on molecules other than Akt in the VEGF signaling cascade, including VEGF receptors and downstream effectors such as MEK, will be interesting targets for further studies.
Conclusion
Our study uncovers a novel mechanism whereby GRK5 plays a key role in the regulation of signaling and biological functions of the non‐7TMR agonist VEGF in HCAECs. Hence, targeted intervention of GRK5 in ECs could provide us with a new therapeutic target for the prevention and treatment of diseases involving altered EC functions.
Acknowledgments
Th ese studies were funded by the NIH NHLBI (Andrea D. Eckhart) and the WW Smith Charitable Trust (Andrea D. Eckhart). This project was funded, in part, by a grant from the Pennsylvania Department of Health. The department specifically disclaims responsibility for any analyses, interpretations, or conclusions. Th ere are no disclosures to report.
References
- 1. Hata JA, Koch WJ. Phosphorylation of G protein‐coupled receptors: GPCR kinases in heart disease. Mol Interv. 2003; 3: 264–272. [DOI] [PubMed] [Google Scholar]
- 2. Tiruppathi C, Yan W, Sandoval R, Naqvi T, Pronin AN, Benovic JL, Malik AB. G protein‐coupled receptor kinase‐5 regulates thrombin‐activated signaling in endothelial cells. Proc Natl Acad Sci USA. 2000; 97: 7440–7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luttun A, Autiero M, Tjwa M, Carmeliet P. Genetic dissection of tumor angiogenesis: are PlGF and VEGFR‐1 novel anti‐cancer targets? Biochim Biophys Acta. 2004; 1654: 79–94. [DOI] [PubMed] [Google Scholar]
- 4. Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004; 56: 549–580. [DOI] [PubMed] [Google Scholar]
- 5. Ho J, Cocolakis E, Dumas VM, Posner BI, Laporte SA, Lebrun JJ. The G protein‐coupled receptor kinase‐2 is a TGF‐β‐inducible antagonist of TGF‐β signal transduction. EMBO J. 2005; 24: 3247–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu JH, Goswami R, Kim LK, Miller WE, Peppel K, Freedman NJ. The platelet‐derived growth factor receptor‐beta phosphorylates and activates G protein‐coupled receptor kinase‐2. A mechanism for feedback inhibition. J Biol Chem. 2005; 280: 31027–31035. [DOI] [PubMed] [Google Scholar]
- 7. Drazner MH, Peppel KC, Dyer S, Grant AO, Koch WJ, Lefkowitz RJ. Potentiation of beta a drenergic signaling by adenoviral‐mediated gene transfer in adult rabbit ventricular myocytes. J Clin Invest. 1997; 99: 288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fujio Y, Guo K, Mano T, Mitsuuchi Y, Testa JR, Walsh K. Cell cycle withdrawal promotes myogenic induction of Akt, a positive modulator of myocyte survival. Mol Cell Biol. 1999; 19: 5073–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pleger ST, Remppis A, Heidt B, Volkers M, Chuprun JK, Kuhn M, Zhou RH, Gao E, Szabo G, Weichenhan D, Muller OJ, Eckhart AD, Katus HA, Koch WJ, Most P. S100A1 gene therapy preserves in vivo cardiac function after myocardial infarction. Mol Ther. 2005; 12: 1120–1129. [DOI] [PubMed] [Google Scholar]
- 10. Zhou RH, Yao M, Lee TS, Zhu Y, Martins‐Green M, Shyy JY. Vascular endothelial growth factor activation of sterol regulatory element binding protein: a potential role in angiogenesis. Circ Res. 2004; 95: 471–478. [DOI] [PubMed] [Google Scholar]
- 11. Dimmeler S, Dernbach E, Zeiher AM. Phosphorylation of the endothelial nitric oxide synthase at ser‐1177 is required for VEGF‐induced endothelial cell migration. FEBS Lett. 2000; 477: 258–262. [DOI] [PubMed] [Google Scholar]
- 12. Willets JM, Challiss RA, Nahorski SR. Non‐visual GRKs: are we seeing the whole picture? Trends Pharmacol Sci. 2003; 24: 626–633. [DOI] [PubMed] [Google Scholar]
- 13. Penela P, Ribas C, Mayor F Jr. Mechanisms of regulation of the expression and function of G protein‐coupled receptor kinases. Cell Signal. 2003; 15: 973–981. [DOI] [PubMed] [Google Scholar]
- 14. Jimenez‐Sainz MC, Murga C, Kavelaars A, Jurado‐Pueyo M, Krakstad BF, Heijnen CJ, Mayor F Jr, Aragay AM. G protein‐coupled receptor kinase 2 negatively regulates chemokine signaling at a level downstream from G protein subunits. Mol Biol Cell. 2006; 17: 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu S, Premont RT, Kontos CD, Zhu S, Rockey DC. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat Med. 2005; 11: 952–958. [DOI] [PubMed] [Google Scholar]