

Abstract
The growth and morphological differentiation of dendrites are critical events in the establishment of proper neuronal connectivity and neural function. One extrinsic factor, BMP7, has been shown to specifically affect dendritic morphogenesis; however, the underlying mechanism by which this occurs is unknown. Here we show that LIM kinase 1 (LIMK1), a key downstream effector of Rho GTPases, colocalizes with the BMP receptor, BMPRII, in the tips of neurites and binds to BMPRII. This interaction is required for BMP-dependent induction of the dendritic arbor in cortical neurons. Furthermore, we demonstrate that the physical interaction of LIMK1 with BMPRII synergizes with the Rho GTPase, Cdc42, to activate LIMK1 catalytic activity. These studies thus define a Smad-independent pathway that directly links the BMP receptor to regulation of actin dynamics and provides insights into how extracellular signals modulate LIMK1 activity to permit fine spatial control over cytoskeletal remodelling during dendritogenesis.
Keywords: BMP, dendritogenesis, LIM kinase, neuronal cells
Introduction
Bone morphogenetic proteins (BMPs) regulate many aspects of the differentiation and morphogenesis of neurons in metazoans (Munoz-Sanjuan and Brivanlou, 2002). BMP7 in particular is noted for its ability to stimulate formation of the dendritic arbor in cultured sympathetic, cerebral cortical and hippocampal neurons (Lein et al, 1995; Mehler et al, 1997; Ebendal et al, 1998; Le Roux et al, 1999; Withers et al, 2000; Jan and Jan, 2001; Luo, 2002; Whitford et al, 2002; Miller and Kaplan, 2003). BMPs are members of the TGFβ superfamily of ligands, all of which signal through receptor complexes comprised of type I and II Ser/Thr kinases (Attisano and Wrana, 2002). In general, ligand binding induces formation of a heteromeric receptor complex in which the type II receptor phosphorylates and thereby activates the type I receptor. The activated type I receptor then propagates the signal through the Smad family of intracellular mediators. The BMP type II receptor, BMPRII, is thought to be an important mediator of BMP signals in neuronal lineages as it is predominantly expressed in brain, and in flies the BMPRII counterpart, wishful thinking (Wit), regulates synaptic growth and function (Mehler et al, 1997; Ebendal et al, 1998; Aberle et al, 2002; Marqués et al, 2002). Unlike the other vertebrate type II receptors, but similar to Wit, BMPRII contains a long carboxy-terminal extension of unknown function. Deletion of this region does not impair Smad-dependent signals (Nishihara et al, 2002), leading to the idea that this carboxy-terminal domain may have an important role in activating previously undefined signalling pathways. Although the molecular function of the tail is not known, mutations in this domain as well as throughout the BMPRII receptor are associated with familial and sporadic primary pulmonary hypertension in humans (Lane et al, 2000; De Caestecker and Meyrick, 2001).
Dendrites are critical for receiving and interpreting synaptic inputs, yet little is known of the molecular basis of dendrite development (Jan and Jan, 2001; Scott and Luo, 2001; Luo, 2002; Whitford et al, 2002; Miller and Kaplan, 2003). A key structure in the elaboration of dendrites, as well as axons, is the growth cone, which is localized to the tips of neurites. The highly motile nature of the growth cone is controlled by extracellular signals that mediate rapid reorganization of the actin cytoskeleton through poorly understood pathways that regulate the Rho GTPases, RhoA, Cdc42 and Rac1 (da Silva and Dotti, 2002; Whitford et al, 2002; Miller and Kaplan, 2003). Accumulating evidence indicates that RhoA promotes neurite retraction, whereas Cdc42 and Rac1 are positive regulators of neurite outgrowth. One key target of Rho GTPases in the growth cone is the ADF/cofilin family of F-actin binding proteins that can both depolymerize actin and increase the number of filaments through their severing activity (Meyer and Feldman, 2002; Gungabissoon and Bamburg, 2003). ADF/cofilin activity is regulated by reversible phosphorylation on a conserved serine residue, Ser3. LIM kinase (LIMK) is responsible for phosphorylating this site, and thereby inactivates ADF/cofilin, whereas phosphatases such as slingshot (Niwa et al, 2002) can activate ADF/cofilin by dephosphorylating Ser3. Abundant evidence now indicates that it is the rate of turnover of this reversible phosphorylation that is critical for ADF/cofilin to regulate rapid changes in actin cytoskeleton associated with neurite dynamics (Meyer and Feldman, 2002; Gungabissoon and Bamburg, 2003). This pathway is particularly important in dendrites, as LIMK1 null mice display defects in the formation of actin-based dendritic spines in hippocampal neurons and decreased levels of cofilin phosphorylation (Meng et al, 2002). Furthermore, loss of LIMK1 is thought to contribute to Williams syndrome, a disorder characterized by defects in visuospatial cognition (Morris and Mervis, 2000). How LIMK-dependent regulation of ADF/cofilin activity is connected to extracellular signals that control dendrite formation has remained elusive. Here, we demonstrate that the BMP receptor, BMPRII, directly binds and regulates LIMK1 activity and that this pathway is critical for BMP-dependent dendritogenesis.
Results
The BMPRII tail is required for BMP-induced dendritogenesis
Abundant evidence indicates that BMPs regulate many aspects of neural development, but it is becoming increasingly clear that BMPs can also influence the development of postmitotic neurons (Mehler et al, 1997; Ebendal et al, 1998; Jan and Jan, 2001; Luo, 2002; Munoz-Sanjuan and Brivanlou, 2002; Whitford et al, 2002; Miller and Kaplan, 2003). In particular, the closely related BMPs, BMP5, 6 and 7, can stimulate dendritic growth in cultured sympathetic, cortical and hippocampal neurons. However, the molecular mechanisms whereby BMPs elicit these biological effects are not known. BMPs signal through Ser/Thr kinase receptors and the BMP type II receptor is abundantly expressed in the brain and neuron derived tissues (Mehler et al, 1997; Ebendal et al, 1998; Attisano and Wrana, 2002). Thus, to investigate how BMPs regulate dendrite growth, we examined the contribution of the BMP receptor, BMPRII, in BMP-induced dendrite formation, using low-density short-term (5–7 days) cultures of E16.5 primary mouse cortical neurons. For this, cells were infected with recombinant adenoviruses encoding BMPRII constructs and a GFP marker. Dendrite formation was determined in GFP-positive cells counterstained with a Map2 (a+b) antibody that recognizes the Map2 isoforms specifically expressed in dendrites. In cultures infected with the GFP-encoding adenovirus alone, BMP7 induced a 50% increase in dendrite formation as compared to control cells (Figure 1A) and the dendrites were longer and displayed more branching as reported in previous studies in cortical, sympathetic and hippocampal neurons (Lein et al, 1995; Le Roux et al, 1999; Withers et al, 2000). Similar BMP-dependent effects on dendrite formation and growth were also observed in cells overexpressing full-length BMPRII (Figure 1A). Unlike other TGFβ superfamily receptors, BMPRII contains a 512-amino-acid carboxy-terminal extension that is also found in the Caenorhabditis elegans and Drosophila counterparts (Mehler et al, 1997; Ebendal et al, 1998; Aberle et al, 2002; Marqués et al, 2002). Thus, we examined the effect of BMPRII lacking this tail (BMPRII ΔT) on dendrite formation. We note that PCR analysis confirmed that the reported splice variant of BMPRII that lacks this tail (Liu et al, 1995) is not expressed in the cortical cell cultures (Supplementary Figure 1). In sharp contrast to control or full-length BMPRII-infected cells, expression of BMPRII ΔT blocked the BMP-dependent increase in dendrite formation (Figure 1A). This suggests that the truncated receptor functions in a dominant-negative manner to block BMP-induced dendrite outgrowth and that the tail domain is important for this activity. As the tail domain is not required for Smad-dependent transcriptional events, our finding also indicates that the tail may mediate BMP signalling through a Smad-independent pathway.
Figure 1.
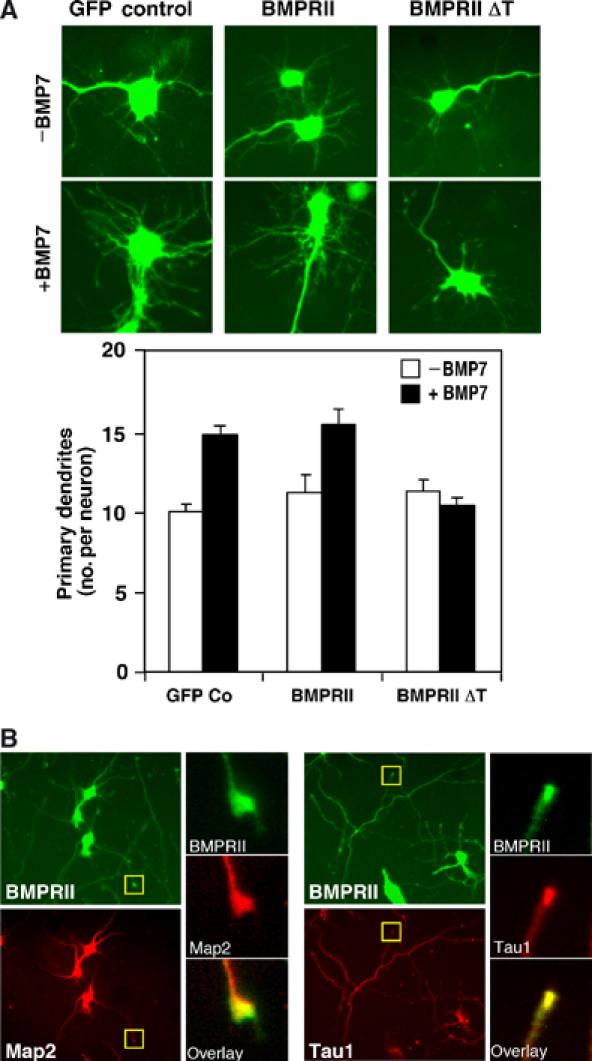
Role of BMPRII in mouse cortical neurons. (A) Deletion of the BMPRII tail blocks BMP-dependent dendritogenesis. Primary cortical neurons infected with adenoviral vectors encoding full-length (FL) or truncated (ΔT) BMPRII were incubated with or without BMP7 after 4 DIV. The number of dendrites/neuron in infected (GFP-positive) cells (left) that costained with the dendrite-specific Map2 (a+b) antibody (not shown) was determined in three independent experiments. Quantitation (mean±s.e.) of a representative experiment is shown (right). (B) Localization of endogenous BMPRII to the tips of neurites in primary cortical neurons was visualized by immunofluorescence microscopy using anti-BMPRII tail primary and Alexa Fluor 488-conjugated secondary antibodies. Dendrites were identified using Map2 (a+b) primary antibodies and axons using Tau1 primary antibodies followed by Alexa Fluor 546-conjugated secondary antibodies. Expanded views of neurite tips (yellow box) demonstrate localization of BMPRII to growth cones.
BMPRII is localized to dendrite and axon tips
To determine whether BMPRII mediates BMP7 signals, we first confirmed its expression in cortical neurons by RT–PCR (data not shown) and next examined its subcellular localization by immunofluorescence microscopy using affinity-purified antibodies directed toward the tail domain. Dendrites were identified with Map2 (a+b) antibodies and axons with primary antibodies directed toward the axon-enriched isoform of Tau1 (Mandell and Banker, 1996). BMPRII was localized in the cell body, and in dendrites and axons was particularly prominent at the tips, where the neuronal growth cone is localized (Figure 1B). We note that incubation of cultures with BMP7 did not enhance BMPRII localization to the tips of neurites (data not shown). The presence of BMPRII in the growth cone of dendrites is consistent with a role for this receptor in BMP-dependent enhancement of dendrite formation and growth.
BMP7 activates the Rho GTPase, Cdc42, and induces phosphorylation of cofilin
Although Smads have emerged as critical effectors of TGFβ superfamily signals (Attisano and Wrana, 2002), BMPRII ΔT binds ligand and activates the Smad pathway in a manner indistinguishable from that of its full-length counterpart (Nishihara et al, 2002). This suggests that Smad-independent pathways are important for BMP-dependent dendritogenesis. The Rho family of GTPases are major regulators of cytoskeletal dynamics, a process that underlies neurite formation, extension and branching. In neuronal cells, the Rho GTPases, Cdc42 and Rac1, are positive regulators of neurite formation (Threadgill et al, 1997; Dickson, 2001; da Silva and Dotti, 2002; Etienne-Manneville and Hall, 2002; Meyer and Feldman, 2002; Whitford et al, 2002; Gungabissoon and Bamburg, 2003; Miller and Kaplan, 2003). Thus, we determined whether BMP regulates these Rho GTPases in N1E115 cells, a BMP-responsive mouse neuroblastoma cell line that forms neurites upon serum withdrawal. N1E115 cells were transiently transfected with Flag-tagged Cdc42 and GTP-bound Cdc42 isolated using GST-CRIB pull-down assays (Bernard et al, 1999). Treatment of cells with BMP increased the levels of active GTP-bound Cdc42 between 5 and 15 min of ligand addition (Figure 2A). Similar results were obtained when endogenous Cdc42 was analyzed (Figure 2B). In contrast, active GTP-bound Rac1 levels were not altered (Figure 2B). Thus, BMPs can induce Cdc42, but not Rac1, activation.
Figure 2.
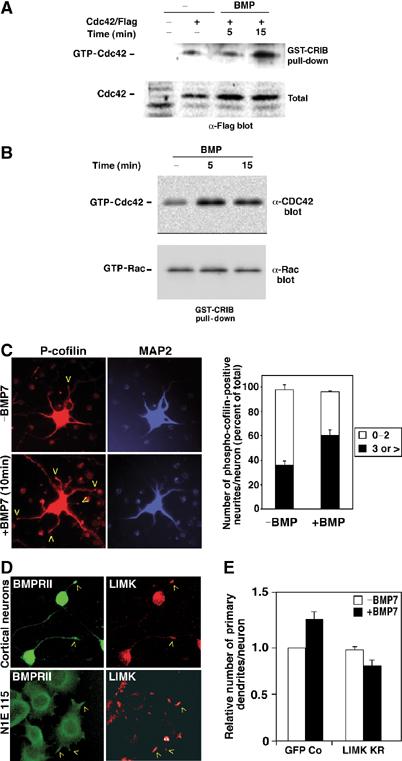
LIMK1 is required for BMP-induced dendritogenesis. (A, B) BMP activates Cdc42. Levels of GTP-bound Cdc42 and Rac1 were determined in lysates of serum-deprived control or BMP-treated N1E115 cells (B), or N1E115 cells transiently transfected with Flag-tagged Cdc42 (A), using GST-CRIB pull-down assays followed by anti-Flag, anti-Cdc42 or anti-Rac1 immunoblotting. (C) BMP enhances cofilin phosphorylation. Primary cortical neurons were incubated with or without BMP7 and localization of phosphorylated cofilin at the tips of neurites in primary cortical neurons was determined by immunofluorescence microscopy using rabbit anti-phospho-cofilin primary and goat anti-rabbit Alexa Fluor 546-conjugated secondary antibodies. Map2 was detected using anti-Map2 primary antibody and anti-mouse Cy5-conjugated secondary antibody. The number of phospho-cofilin-positive neurites in each Map2-positive neuron was counted and is plotted as a percent of total. Shown is the mean+s.e.m. for three independent experiments with a minimum of 50 neurons per condition determined in each experiment (right). (D) BMPRII and LIMK1 subcellular localization was determined in primary cortical neurons (top) and in N1E115 cells (bottom) by confocal microscopy. BMPRII was visualized with anti-BMPRII tail antibodies followed by Alexa Fluor 488-conjugated secondary antibodies and LIMK1 detected using anti-LIMK primary followed by Alexa Fluor 546-conjugated secondary antibodies. (E) Dominant-negative LIMK1 blocks BMP-induced dendritogenesis. Primary cortical neurons infected with adenoviral vectors encoding kinase-deficient LIMK(KR) or GFP empty vector control were incubated with or without BMP7 as in Figure 1A. The number of dendrites/neuron in infected (GFP-positive) cells that costained with the dendrite-specific Map2 (a+b) antibody was determined in a minimum of 20 neurons/condition. Fold changes in dendrite numbers relative to the unstimulated GFP empty vector control (Co) from three independent experiments (mean±s.e.) are shown.
Rho GTPases regulate neurite dynamics through effector molecules that directly modulate the actin cytoskeleton (Etienne-Manneville and Hall, 2002; Luo, 2002; Whitford et al, 2002; Miller and Kaplan, 2003). One such Cdc42 downstream effector, the ADF/cofilin family of proteins, is the key regulator of actin dynamics in neuronal cells (Dickson, 2001; da Silva and Dotti, 2002; Etienne-Manneville and Hall, 2002; Meyer and Feldman, 2002; Whitford et al, 2002; Gungabissoon and Bamburg, 2003; Miller and Kaplan, 2003). ADF/cofilin is a depolymerizing factor that is essential for the rapid turnover of actin filaments and its activity is regulated by reversible phosphorylation on Ser3. Thus, we next determined whether BMP7 treatment alters cofilin phosphorylation and/or its subcellular distribution in primary cortical neurons. In control cells, phospho-cofilin staining was occasionally observed in the tips of dendrites (Figure 2C). However, in cells treated with BMP7 for 10 min, there was a marked increase in phospho-cofilin staining at the tips of neurites (Figure 2C). To quantitate this effect, the number of phospho-cofilin-positive dendrites in each Map2-positive neuron was determined. In cortical neurons incubated with BMP7, we observed a 50% increase in cells with three or greater phospho-cofilin-positive dendrites as compared to controls. Unlike our immunofluorescence analysis, we did not observe significant changes in BMP-dependent phosphorylation of the total cofilin pool when assayed by immunoblotting of total cell lysates (data not shown), suggesting that BMP-induced increases in phospho-cofilin are most prominent in neurite tips where BMPRII is also found to reside. Thus, our findings indicate that BMPs can stimulate Cdc42 activation and can induce phosphorylation of cofilin. Since LIMKs are downstream effectors of Cdc42 and as they are the only kinases known to induce phosphorylation of cofilin (Arber et al, 1998; Yang et al, 1998; Edwards et al, 1999), these observations also suggest that BMPs may regulate LIMK activity.
LIMK1 activity is required for BMP-induced dendritogenesis
LIMK1 is a neuronal specific LIMK family member that has been implicated in dendrite formation. Genetic deletion of LIMK1 results in behavioral abnormalities that are associated with the abnormal formation of dendritic spines and reduced levels of ADF/cofilin phosphorylation (Meng et al, 2002). Since BMP7 treatment increases the levels of phosphorylated cofilin, we investigated how BMPs regulate LIMK by first examining LIMK subcellular localization. LIMK was found in the growth cones of primary cortical neurons where BMPRII was also found to reside (Figure 2D). A similar, colocalization of LIMK and BMPRII was also observed at the tips of neurites in N1E115 cells (Figure 2D). Incubation of either cell type with BMP7 did not visibly alter the subcellular distribution of LIMK (data not shown). To determine whether LIMK activity was required for BMP-induced dendritogenesis, primary cortical neurons were infected with recombinant adenoviruses encoding a kinase-deficient version of LIMK (LIMK KR) and a GFP marker. As described above (Figure 1A), BMP7 induced an increase in dendrite formation in cultures infected with the GFP-encoding adenovirus alone (Figure 2E). This increase was abrogated in cells infected with the dominant-negative version of LIMK. These results thereby indicate that LIMK activity is required for BMP-induced dendritogenesis.
LIMK1 interacts with the tail domain of BMPRII
As BMPRII, phospho-cofilin and LIMK are similarly localized, we next investigated whether LIMK1 and BMPRII physically interact. Immunoprecipitation of BMPRII from cell lysates of transiently transfected COS cells revealed an association between LIMK1 and BMPRII (Figure 3A). This interaction occurred via the tail domain of BMPRII, as deletion of this region resulted in a loss of LIMK1 association. A similar interaction between LIMK1 and the BMPRII tail has recently been reported (Foletta et al, 2003). Unlike LIMK1, which is primarily expressed in neuronal lineages, we did not detect interaction of the ubiquitously expressed LIMK2 with the BMPRII receptor (Figure 3B). This interaction was direct, as purified LIMK1 directly interacted with a bacterially produced tail fragment (construct 10, see Figure 4A) of BMPRII (Figure 3C). To determine whether the tail domain was sufficient for association with LIMK1, we expressed a portion of the BMPRII tail as a GST bacterial fusion protein (construct 6, see Figure 4A) and examined its ability to interact with endogenous LIMK1. Cell lysates from N1E115 cells were incubated with a bacterially expressed GST-BMPRII tail fusion protein and bound proteins were visualized by anti-LIMK1 immunoblotting. This analysis revealed that endogenous LIMK1 interacted with the BMPRII tail domain. In contrast, no interaction was detected in control lysates from NMuMG cells, which do not express LIMK1 (Figure 3D).
Figure 3.
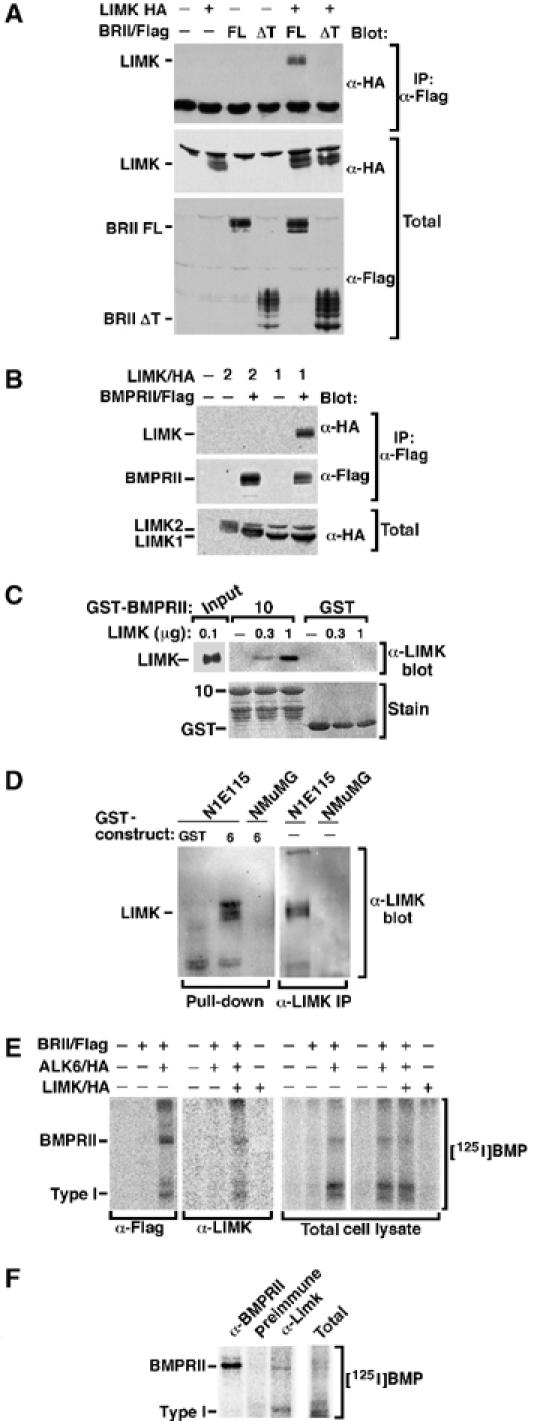
LIMK associates with BMPRII. (A, B) BMPRII was immunoprecipitated from transiently transfected COS-1 cells and associated LIMK1 or LIMK2 visualized by anti-HA immunoblotting. Expression of transfected proteins was confirmed by immunoblotting. (C) Purified LIMK1 was incubated with GST-BMPRII tail construct 10 and direct association of LIMK1 with BMPRII confirmed by α-LIMK1 immunoblotting. (D) Cell lysates from N1E115 or NMuMG cells (10 × 100 mm dishes) were incubated with GST-BMPRII tail construct 6 and association of endogenous LIMK1 was determined by anti-LIMK1 immunoblotting (left). Expression of LIMK in N1E115 but not NMuMG cells was confirmed (right). (E) Transiently transfected COS-1 cells were affinity-labelled with [125I]BMP2 and receptor complexes collected by anti-Flag or anti-LIMK1 immunoprecipitation and visualized by phosphorimaging. (F) N1E115 cells (3 × 100 mm dishes) were affinity-labelled with [125I]BMP2 and endogenous receptor complexes collected by anti-BMPRII or anti-LIMK1 immunoprecipitation and visualized by phosphorimaging.
Figure 4.
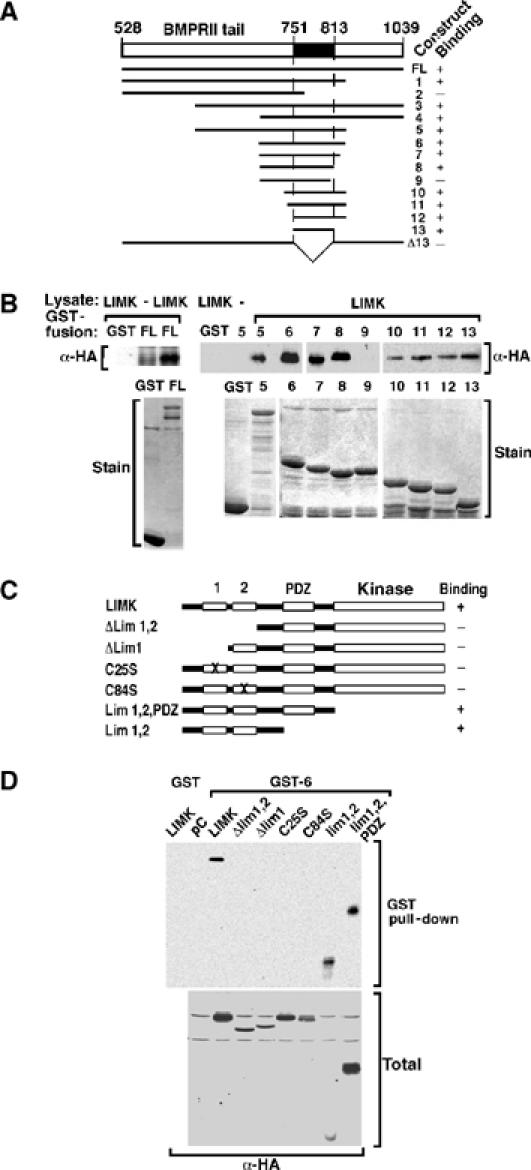
Mapping of the interaction domains in LIMK1 and BMPRII. (A) A schematic representation of GST fusion constructs of the BMPRII tail and a summary of their interaction with LIMK1 is shown. (B) COS-1 cells were transiently transfected with LIMK1/HA and cell lysates incubated with bacterially expressed GST fusion proteins. The interaction was visualized by anti-HA immunoblotting. Levels of GST fusion proteins were confirmed by Coomassie blue staining. (C) A schematic representation of mutant versions of LIMK1 and a summary of their interactions with BMPRII is shown. (D) COS-1 cells were transfected with LIMK1/HA constructs, and cell lysates were incubated with bacterially expressed BMPRII tail construct 6. The interaction and total LIMK1 expression levels were visualized by anti-HA immunoblotting.
BMP signalling requires formation of a heteromeric receptor complex in which the type II receptors phosphorylate and thereby activate the type I receptors (Attisano and Wrana, 2002). We observed that LIMK1 associates with BMPRII in the absence of ligand (Figure 3A) and this interaction was unaltered when a kinase-deficient version of BMPRII was analyzed (data not shown, but see Figure 5B). Thus, we next determined whether LIMK1 association was maintained upon formation of an active BMP receptor signalling complex. For this, COS cells expressing BMPRII, the type I receptor, ALK6 and LIMK1 were affinity-labelled with [125I]BMP2 and cell lysates were subjected to anti-Flag or anti-LIMK1 immunoprecipitation. For these experiments, we used BMP2, another isoform of BMP that also binds to BMPRII, since it affinity-labels these receptors more efficiently; however, similar results were obtained with [125I]BMP7 (data not shown). Coexpression of BMPRII with the type I receptor, ALK6, results in formation of a receptor complex that can be detected in BMPRII (anti-Flag) immunoprecipitates (Figure 3E). A similar complex was observed in anti-LIMK1 immunoprecipitates, but not in controls lacking LIMK1 expression. Furthermore, use of this approach revealed the association of endogenous LIMK with endogenous BMPRII-containing receptor complexes in N1E115 cells (Figure 3F). These results demonstrate that LIMK1 can interact with BMPRII receptor alone as well as with the activated BMP heteromeric receptor complex comprised of BMPRII and a type I receptor.
Figure 5.
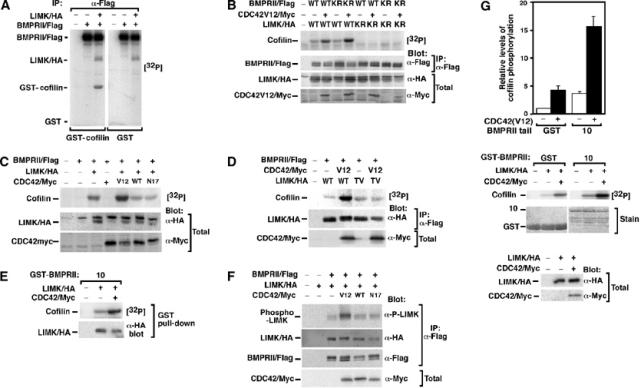
BMPRII-bound LIMK1 synergizes with Cdc42 to phosphorylate cofilin. (A–C) Lysates from transiently transfected COS-1 cells were immunoprecipitated with anti-Flag antibodies and the activity of associated LIMK1 was determined using an in vitro kinase assay with GST-cofilin as substrate. (A) BMPRII-bound LIMK1 phosphorylates cofilin. (B) The kinase activity of LIMK1 but not BMPRII is required for phosphorylation of cofilin. (C) Coexpression of constitutively active (V12) but not wild-type (WT) or dominant-negative (N17) Cdc42 enhances BMPRII-bound LIMK1 activity. (D) Cdc42-dependent enhancement of LIMK1 is lost in an LIMK1 mutated in the activation site residue, Thr508 (LIMK TV). (E) LIMK1 coexpressed with or without Cdc42(V12) in transiently transfected COS-1 cells was isolated using the GST-BMPRII tail construct 10 and the ability of bound LIMK1 to phosphorylate cofilin was determined using an in vitro kinase assay. (F) Lysates from transiently transfected COS-1 cells were immunoprecipitated with anti-Flag antibodies and activation of associated LIMK1 was determined by anti-phospho-LIMK immunoblotting. (G) LIMK1, immunoprecipitated from transiently transfected COS-1 cells, was separated into two equal aliquots and then was incubated for 60 min with bacterially expressed BMPRII tail fragment or GST alone prior to an in vitro kinase assay. Phosphorylation of cofilin was quantitated (top panel) in duplicate from a representative experiment. Protein levels were confirmed by Coomassie staining and immunoblotting (middle and bottom panels).
Mapping of the regions that mediate BMPRII and LIMK1 interaction
To identify the domains in BMPRII that mediate association with LIMKl, we expressed LIMK1 in COS cells and examined its ability to interact with various deletion constructs of the BMPRII tail domain expressed as bacterial fusion proteins (Figure 4A). In particular, a construct encoding 63 amino acids from residue 751 to 813 (construct 13) interacted with LIMK1 (Figure 4B). This finding is consistent with a recent study showing that LIMK binds to a 190-amino-acid region spanning amino acids 742–932 of BMPRII (Foletta et al, 2003). Thus, we have defined the LIMK1 binding region (LBR) on BMPRII as amino acids 751–813 and have shown that this region when expressed alone is sufficient for association with LIMK1.
To determine the domains in LIMK1 that mediate interaction with BMPRII, we expressed various HA-tagged versions of LIMK1 in COS cells (Figure 4C) and examined their interaction with the BMPRII tail construct 6, expressed as a bacterial fusion protein. A protein comprised only of LIM1 and LIM2 domains efficiently associated with the BMPRII tail protein as previously reported (Foletta et al, 2003). Furthermore, deletion of LIM1 alone, the LIM1/LIM2 domains or the introduction of point mutations Cys25Ser or Cys84Ser that disrupt LIM domain structure in either LIM1 or LIM2, respectively (Edwards and Gill, 1999), abrogated interaction of LIMK1 with the BMPRII tail (Figure 4D). Thus, we show that both LIM1 and LIM2 domains are necessary and sufficient for interaction with the BMPRII receptor.
LIMK1 bound to BMPRII is activated by Cdc42
We next wanted to explore how the kinase activity of LIMK1 was regulated by binding to BMPRII. So we first determined whether BMPRII-bound LIMK1 was capable of phosphorylating its substrate cofilin. COS cells transiently transfected with BMPRII/Flag and LIMK1/HA were subjected to anti-Flag immunoprecipitation followed by an in vitro kinase assay using bacterially expressed cofilin as a substrate. We observed that LIMK1 that was bound to BMPRII was capable of phosphorylating GST-cofilin but not GST expressed alone (Figure 5A). Cofilin phosphorylation was unaffected when we used a kinase-deficient version of BMPRII but was abolished with kinase-deficient LIMK1 (Figure 5B), confirming that cofilin phosphorylation is dependent on the kinase activity of LIMK1 but not BMPRII. Since LIMK1 activity is regulated by Cdc42, we next determined whether BMPRII-bound LIMK could be activated by Cdc42. Coexpression of a constitutively activated Cdc42, Cdc42(V12), enhanced LIMK1-dependent phosphorylation of cofilin, whereas the wild-type (WT) or the dominant-negative N17 version of Cdc42 did not (Figure 5C). Cdc42-dependent activation of LIMK occurs through a PAK1-dependent pathway that results in phosphorylation of Thr508 (Edwards and Gill, 1999; Edwards et al, 1999). Thus, we mutated this residue to Val in LIMK1 and examined the effect of coexpression of Cdc42(V12). As expected, this mutation abrogated Cdc42-dependent activation of BMPRII-bound LIMK1 but did not affect basal LIMK1 activity (Figure 5D). In these experiments, we isolated BMPRII from intact cells and then examined the activity of the associated LIMK1. In addition, we also examined whether Cdc42-activated LIMK1 expressed in mammalian cells was able to bind bacterially expressed BMPRII tail using a GST pull-down assay. We observed that the Cdc42-activated LIMK1 bound the bacterially produced BMPRII tail and that this bound LIMK1 was capable of phosphorylating cofilin (Figure 5E). Finally, we examined whether LIMK1 associated with BMPRII was directly activated prior to the in vitro kinase assay using an anti-phospho-LIMK antibody that recognizes the phosphorylated T508 and thus the activated form LIMK. We observed that LIMK1 associated with BMPRII was phosphorylated in the presence of Cdc42 V12 but not with WT or dominant-negative (N17) Cdc42 (Figure 5F).
Binding of LIMK1 to BMPRII increases LIMK1 catalytic activity
Previous work has demonstrated that the LIM1/LIM2 domains of LIMK1 can associate with its kinase domain in an autoinhibitory manner, thereby limiting the activity of LIMK1 toward its substrate cofilin (Nagata et al, 1999). As LIMK1 associates with BMPRII via the LIM1/2 domains, we next considered whether this association might affect LIMK activity. For this, LIMK1 was immunoprecipitated and then incubated with or without bacterially expressed BMPRII tail fragments prior to an in vitro kinase assay. Incubation of LIMK1 with the BMPRII tail construct 10 resulted in a four-fold increase in LIMK1 activity that was not apparent in samples incubated with GST protein alone (Figure 5G). A similar increase in activity was observed when LIMK1 T508V was analyzed (data not shown). Next, we coexpressed activated Cdc42(V12) with LIMK1, and observed a four-fold increase in LIMK1 basal activity, consistent with previous reports (Edwards et al, 1999). Strikingly, when Cdc42-activated LIMK1 was incubated with the LIMK1 binding region of BMPRII (construct 10), we observed more than a 16-fold enhancement in LIMK1 activity. Thus, our results indicate that association of LIMK1 with BMPRII synergizes with Cdc42 to drive high levels of LIMK1 activity on the BMPRII tail.
BMPRII lacking the LIMK1 binding region blocks BMP-induced cofilin phosphorylation and dendritogenesis
BMPRII lacking the entire tail domain functions as a dominant negative to block BMP-induced dendritogenesis (Figure 1A). Thus, we next determined whether this effect could be attributed to the loss of LIMK1 binding to the BMPRII receptor. The minimal region identified as sufficient for LIMK1 association was comprised of amino acids 751–813 (Figure 4). Thus, we deleted this LBR in the context of the full-length BMPRII receptor (BMPRII ΔLBR) and tested its association with LIMK1 in cells. Consistent with our expectations, we observed a loss of the association of LIMK1 with BMPRII in transfected COS cells (Figure 6A).
Figure 6.
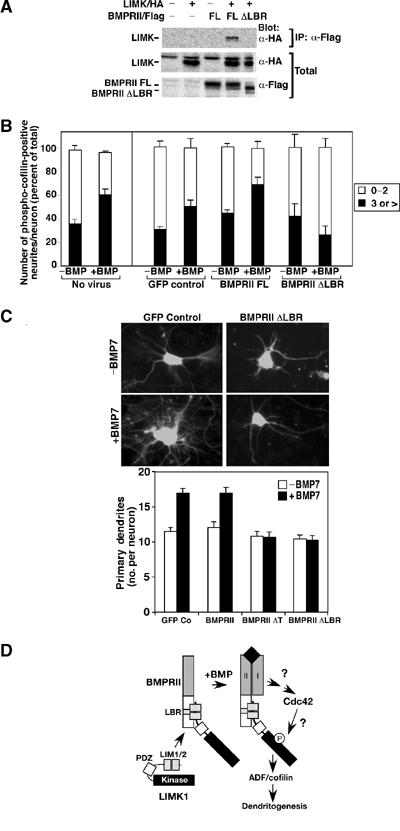
Disruption of LIMK1 interaction with BMPRII blocks cofilin phosphorylation and dendritogenesis. (A) Cell lysates from COS-1 cells transiently transfected with full-length BMPRII or a version lacking the LIMK1 binding region (BMPRII ΔLBR) were subjected to anti-Flag immunoprecipitation and associated LIMK1 was detected by anti-HA immunoblotting. (B) Primary cortical neurons were infected with the indicated adenoviral constructs and the number of phospho-cofilin-positive dendrites per neuron was determined as in Figure 2C. Shown is the mean±s.e.m. for three independent experiments with a minimum of 20 neurons/condition determined in each experiment (right). For direct comparison, the results obtained using noninfected neurons (from Figure 2C) are shown on the left. (C) The number of dendrites/neuron in primary cortical neurons was determined as in Figure 1A. Immunofluorescent images of GFP-containing neurons (top) and quantitation of a representative experiment are shown (bottom). (D) A model for BMP-dependent regulation of dendrite formation. BMP-induced activation of Cdc42 cooperates with binding of LIMK1 to the BMPRII tail to provide for high levels of LIMK1 activity. The mechanism of BMP receptor-induced activation of Cdc42 and the pathway leading from Cdc42 to LIMK1 are not known.
We next examined the effect of expressing this receptor on BMP-induced cofilin phosphorylation in primary mouse cortical neurons. In cultures infected with a GFP-control adenovirus, BMP7 induced a 65% increase in neurons containing three or more phospho-cofilin-positive neurites (Figure 6B) similar to that observed in uninfected controls (Figure 2C; reproduced in Figure 6B (left side) for comparison). A similar increase in the number of phospho-cofilin-positive neurites was also observed in cells infected with an adenoviral construct expressing the full-length BMPRII receptor. In marked contrast, this BMP-dependent increase in phospho-cofilin-positive neurites was lost in cultures infected with BMPRII ΔLBR. We next determined whether expression of BMPRII ΔLBR altered BMP-induced dendritogenesis in the mouse cortical cultures. Infection of neurons with adenovirus encoding BMPRII ΔLBR blocked the BMP-dependent increases in dendritogenesis in a manner similar to that observed for the tailless BMPRII receptor (Figure 6C). Together, these results indicate that BMP-dependent increases in cofilin phosphorylation and induction of dendritogenesis requires LIMK1 association with BMPRII.
Discussion
Remodelling of the actin cytoskeleton is a key aspect of neuronal morphogenesis, yet, how extracellular signals regulate this process to control elaboration of the dendritic arbor is poorly understood. The Rho GTPases transmit extracellular cues through various effector molecules and Cdc42 and Rac1 have been shown to promote neurite extension in part through a pathway involving the activation of LIMKs and its downstream substrate, the actin depolymerizing factor, ADF/cofilin (da Silva and Dotti, 2002; Meyer and Feldman, 2002; Gungabissoon and Bamburg, 2003). BMP7 regulates dendritogenesis and here, we have investigated the molecular mechanism for this effect. We show that BMPRII is localized in growth cones and that LIMK1 interacts with BMPRII. Furthermore, we demonstrate that while binding to the BMPRII tail or activation of Cdc42 individually can stimulate LIMK1, together they act synergistically to yield high levels of LIMK1 catalytic activity. This yields a BMPRII/LIMK1 complex that is poised in the growth cone to receive and then transmit extracellular BMP signals directly to the actin cytoskeleton via regulation of ADF/cofilin (Figure 6D). Consistent with this, we observed a BMP-dependent induction of phospho-cofilin in the growth cones of cortical cells. Moreover, since BMP-dependent dendritogenesis was blocked upon disruption of the LBR, our findings suggest that physical interaction of LIMK1 with BMPRII is key for BMP-induced formation of the dendritic arbor.
BMPs are important regulators of neuronal cell differentiation and during early development in vertebrates they block default pathways for neural differentiation (Munoz-Sanjuan and Brivanlou, 2002). BMPs regulate these pathways through heteromeric Ser/Thr kinase receptor complexes and require signalling through the Smad pathway. Furthermore, in Drosophila, BMP-like molecules regulate neuromuscular synapse morphology and neuropeptide cell identity via the BMPRII-like receptor, wishful thinking (Wit) and Smad pathways (Aberle et al, 2002; Marqués et al, 2002; McCabe et al, 2003). However, Smad-independent signalling pathways emanating from Ser/Thr kinase receptors and their role in regulating neuronal function have not been previously defined. Our studies now indicate that in addition to these Smad-dependent functions, there are also key roles for BMPs in regulating dendritogenesis in neurons via a Smad-independent pathway that involves LIMK1 activation. Furthermore, although BMPRII binds LIMK1 in the absence and presence of ligand, we observed BMP-dependent activation of phospho-cofilin in cortical cells. Thus, the ligand dependence of this pathway is likely conferred by BMP-dependent regulation of Cdc42 that functions synergistically with LIMK1 bound to the BMPRII tail (Figure 6D). In contrast to our study, a recent report appeared, which suggests that LIMK1 binding to BMPRII inhibits LIMK1 activity and that BMPs act by promoting dissociation of LIMK from the receptor complex (Foletta et al, 2003). However, this model is not consistent with our demonstration that LIMK1 is present in the BMP-bound receptor complex, that binding of LIMK1 to the BMPRII tail enhances its activity and that this bound LIMK1 can be further activated by Cdc42 to phosphorylate its substrate cofilin. Despite these mechanistic differences however, both studies concur in the conclusion that BMPs promote activation of LIMK1 and that this leads to cytoskeletal rearrangements.
Although LIM domains in general mediate intermolecular protein–protein interactions, the LIM1/2 domains in LIMK have been shown to regulate negatively the kinase activity of LIMK1 via an intramolecular interaction of the LIM domains with the carboxy-terminal kinase domain (Edwards and Gill, 1999; Nagata et al, 1999). In our study, we observed that LIMK1 bound to the BMPRII tail has an enhanced ability to phosphorylate cofilin. This suggests that interaction with the LBR of BMPRII, which occurs via the LIM domains, relieves the autoinhibitory effect of LIM1/2 on the catalytic domain (Figure 6D). This functions synergistically with Cdc42, which can in parallel activate LIMK1 via PAK-dependent phosphorylation of T508. Consistent with this model, we found that the catalytic activity of LIMK1, harboring a Thr508 to Val mutation that prevents its activation by Rho GTPases, was stimulated by the LBR to a similar degree as WT LIMK1, but failed to show synergistic activation in response to Cdc42 (data not shown). Thus, our data suggest that maximal LIMK1 activity can be achieved through a two-step mechanism (Figure 6C). First, binding of LIMK1 to BMPRII relieves an autoinhibitory effect on the catalytic domain and, second, BMP-dependent activation of Cdc42 results in phosphorylation of the activation loop Thr residue, thereby increasing LIMK1 catalytic activity. Currently, we do not know the pathway that leads to BMP-dependent Cdc42 activation, but TGFβ also activates Cdc42 (Bhowmick et al, 2001; Edlund et al, 2002); so it is possible that both BMP and TGFβ receptor complexes might utilize a common pathway to target this Rho GTPase. However, the absence of a C-terminal tail on TGFβ receptors would prevent strong synergistic activation of LIMK1 by TGFβ. Consistent with this, TGFβ-like factors do not induce dendritogenesis in cortical neurons (Le Roux et al, 1999). Thus, a two-step mechanism to regulate LIMK1 likely also ensures specific, rapid and localized activation of LIMK1 in response to BMP. It is interesting to note that although we observed colocalization of BMPRII and LIMK1 in the growth cones of both dendrites and axons, BMP7 does not appear to alter axonal growth (Higgins et al, 1997). So, the role of the BMPRII–LIMK1 pathway in axons is not clear; however, it may modulate other regulators of axon dynamics.
Phosphorylation of ADF/cofilin blocks its actin depolymerizing activity, whereas dephosphorylation by phosphatases, such as slingshot, allows actin binding and promotion of actin disassembly (Meyer and Feldman, 2002; Gungabissoon and Bamburg, 2003). This turnover of ADF/cofilin phosphorylation is thought to be a key determinant in actin remodelling at the leading edge of cells. Furthermore, it has become increasingly clear that dynamic reorganization of the actin cytoskeleton requires that the activation and inactivation of ADF/cofilin be restricted both in a spatial and temporal manner. For instance, overexpression of either positive or negative regulators of ADF/cofilin such as LIMK and slingshot results in disorganized growth cones and interferes with growth cone motility and morphology (Endo et al, 2003). Recent work has demonstrated that Semaphorin 3A, a chemorepulsive axonal guidance molecule, sequentially induces phosphorylation and then dephosphorylation of cofilin in dorsal root ganglion cells thereby providing evidence for the temporal control of ADF/cofilin phosphorylation (Aizawa et al, 2001). However, an understanding of how spatial control of ADF/cofilin might be achieved has remained elusive. We postulate that anchoring of LIMK1 to the BMPRII receptor may provide a means to restrict ADF/cofilin phosphorylation to specific regions. Spatial confinement of LIMK1 activity may thereby result in localized regions of actin polymerization, which might then drive neurite outgrowth. We speculate that this spatial restriction combined with localized delivery of BMPs may be involved in controlling the timing of dendritogenesis during developmental processes and in adults may contribute to neural plasticity by altering dendritic growth. A similar, but not yet described mechanism whereby extracellular signals also control localization of ADF/cofilin dephosphorylating enzymes such as slingshot, might thereby provide for the required spatial and perhaps temporal control essential for the tightly regulated actin remodelling required in the motile neuronal growth cone.
Materials and methods
Isolation of primary cortical neurons and dendritogenesis
Cortices were dissected from embryonic day (E) 16.5 mice, digested with 0.25% trypsin at 37°C for 30 min and dissociated with a fire-polished Pasteur pipette in Neurobasal medium containing 10% FBS. Neurons were plated on poly-L-lysine- and laminin-coated dishes or slides in Neurobasal medium supplemented with B27, N2, L-glutamine and penicillin/streptomycin. Neurons were incubated at 37°C in 5% CO2. For adenoviral studies, cortical neurons were plated on 48-well dishes. After 3 days in vitro (DIV), neurons were infected with various dilutions of recombinant adenoviruses ranging from approximately 1 × 106 to 1 × 107 viral particles per well. At 24 h postinfection, neurons were treated with 100 ng/ml BMP7 and at 72 h postinfection (7 DIV), neurons were fixed with 4% paraformaldehyde in phosphate-buffered saline for 10 min and processed for neurite quantitation. GFP-expressing adenoviral infected neurons were analyzed with a Leica DMIRB inverted microscope equipped with fluorescence optics. Images of neurons were taken with a CCD camera (Photometrics SenSys) and analyzed with Metamorph Meta Imaging software. Dendrite numbers were quantitated by blind counting of the number of dendrites extending from Map2-positive cell soma in at least 16 neurons per treatment condition. For each adenovirus, at least three independent experiments were performed.
Constructs and vectors
Flag-tagged BMPRII, provided by M Kawabata, and its mutant derivatives were subcloned into pCMV5 for mammalian cell work, pGEX4T1 for bacterial expression and pAdTrack-CMV shuttle vector containing GFP (Q-Biogene) for adenoviral infections. BMPRII short (BMPRII ΔT) construct in a mammalian expression vector was provided by J Massagué (Liu et al, 1995). Expression constructs for LIMK1 (a gift of E Robertson), LIMK2 (IMAGE: 3496836) and cofilin (NIA clone) were epitope-tagged and subcloned into pCMV5 or pGEX4T1. Cdc42 constructs in pCMV5 were provided by H-R Wang. To test for expression of the BMPRII short splice variant, RNA from cortical cultures was extracted and subjected to RT–PCR analysis using primers located within exons 11 and 13.
Biochemical assays
Polyclonal rabbit anti-BMPRII antibodies were raised to bacterially expressed BMPRII tail construct 3 (Primm, Milan, Italy) and were affinity-purified using an AminoLink Kit (Pierce). Antibody specificity was confirmed in transiently transfected cells by immunoblotting and immunofluorescence microscopy. Affinity-labelling, immunoprecipitation, immunoblotting and GST pull-down assays were conducted as described previously (Labbé et al, 2000; Di Guglielmo et al, 2003). Phospho-LIMK antibody was purchased from Cell Signalling and purified LIMK1 was obtained from Upstate Biotechnology. For GST-CRIB pull-down assays, cells were lysed in Mg2+/Lysis/Wash Buffer (MLB, Upstate Biotech.) and cleared supernatants were incubated with 30 μg GST-PAK CRIB fusion protein (provided by B Ozdamar) for 30 min at 4°C. For in vitro kinase assays, immunoprecipitates from cell lysates were incubated with 5 μg of bacterially expressed GST-cofilin at 37°C for 20 min in 20 μl of assay buffer containing 0.1 mM ATP and 5 μCi [32P]ATP.
Immunofluorescent microscopy
For subcellular localization studies, primary cortical neurons or N1E115 cells were plated on coated Permanox chamber slides and fixed as described above and then permeabilized with 0.2% Triton X-100. Cells were stained with monoclonal Map2 (a+b) (Sigma), monoclonal Tau1 (Chemicon), monoclonal LIMK1 (BD Transduction Labs), polyclonal phospho-cofilin (Santa Cruz) or polyclonal BMPRII tail primary antibodies and the indicated fluorescently tagged secondary antibodies (Molecular Probes) as described previously (Di Guglielmo et al, 2003). Images of cortical neurons were obtained with a Leica TCS Sp2 confocal microscope equipped with fluorescence optics and multiphoton lasers (Spectral Physics, California) and were analyzed with Leica TCS confocal software. N1E115 cell images were obtained using a Bio-Rad MRC-600 confocal fluorescence microscope equipped with a krypton/argon laser and were analyzed with OpenLab3 Software (Improvision).
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank Michal Opas and the members of his lab for assistance with confocal microscopy, Jim Fawcett for advice on cortical neuron cultures, Perry Howard for advice on adenoviral generation, Landis Black for technical assistance and Cris Silvestri for advice. This work was supported by grants from the Canadian Institute for Health Research (CIHR, LA and JLW), a Heart and Stroke/CIHR studentship (STL-H) and a National Cancer Institute of Canada postdoctoral fellowship (CGC). LA and JLW are CIHR Investigators and JLW is an International Scholar of the Howard Hughes Medical Institute.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhaes TR, Goodman CS (2002) wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron 33: 545–558 [DOI] [PubMed] [Google Scholar]
- Aizawa H, Wakatsuki S, Ishii A, Moriyama K, Sasaki Y, Ohashi K, Sekine-Aizawa Y, Sehara-Fujisawa A, Mizuno K, Goshima Y, Yahara I (2001) Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nat Neurosci 4: 367–373 [DOI] [PubMed] [Google Scholar]
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P (1998) Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393: 805–809 [DOI] [PubMed] [Google Scholar]
- Attisano L, Wrana JL (2002) Signal transduction by the TGF-β superfamily. Science 296: 1646–1647 [DOI] [PubMed] [Google Scholar]
- Bernard V, Bohl BP, Bokock GM (1999) Characterization of Rac and Cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J Biol Chem 274: 13198–13204 [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassim M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL (2001) Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell 12: 27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva JS, Dotti CG (2002) Breaking the neuronal sphere: regulation of the actin cytoskeleton in neuritogenesis. Nat Rev Neurosci 3: 694–704 [DOI] [PubMed] [Google Scholar]
- De Caestecker M, Meyrick B (2001) Bone morphogenetic proteins, genetics and the pathophysiology of primary pulmonary hypertension. Respir Res 2: 193–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL (2003) Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol 5: 410–421 [DOI] [PubMed] [Google Scholar]
- Dickson BJ (2001) Rho GTPases in growth cone guidance. Curr Opin Neurobiol 11: 103–110 [DOI] [PubMed] [Google Scholar]
- Ebendal T, Bengtsson H, Soderstrom S (1998) Bone morphogenetic proteins and their receptors: potential functions in the brain. J Neurosci Res 51: 139–146 [DOI] [PubMed] [Google Scholar]
- Edlund S, Landstrom M, Heldin CH, Aspenstrom P (2002) Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell 13: 902–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DC, Gill GN (1999) Structural features of LIM kinase that control effects on the actin cytoskeleton. J Biol Chem 274: 11352–11361 [DOI] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN (1999) Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol 1: 253–259 [DOI] [PubMed] [Google Scholar]
- Endo M, Ohashi K, Sasaki Y, Goshima Y, Niwa R, Uemura T, Mizuno K (2003) Control of growth cone motility and morphology by LIM kinase and Slingshot via phosphorylation and dephosphorylation of cofilin. J Neurosci 23: 2527–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420: 629–635 [DOI] [PubMed] [Google Scholar]
- Foletta VC, Lim MA, Soosairaiah J, Kelly AP, Stanley EG, Shannon M, He W, Das S, Massague J, Bernard O (2003) Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol 162: 1089–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gungabissoon RA, Bamburg JR (2003) Regulation of growth cone actin dynamics by ADF/cofilin. J Histochem Cytochem 51: 411–420 [DOI] [PubMed] [Google Scholar]
- Higgins D, Burack M, Lein P, Banker G (1997) Mechanisms of neuronal polarity. Curr Opin Neurobiol 7: 599–604 [DOI] [PubMed] [Google Scholar]
- Jan Y-N, Jan LY (2001) Dendrites. Genes Dev 15: 2627–2641 [DOI] [PubMed] [Google Scholar]
- Labbé E, Letamendia A, Attisano L (2000) Association of Smads with LEF1/TCF mediates cooperative signalling by the TGFβ and Wnt pathways. Proc Natl Acad Sci USA 97: 8358–8363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA III, Loyd JE, Nichols WC, Trembath RC (2000) Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet 26: 81–84 [DOI] [PubMed] [Google Scholar]
- Lein P, Johnson M, Guo X, Rueger D, Higgins D (1995) Osteogenic protein-1 induces dendritic growth in rat sympathetic neurons. Neuron 15: 597–605 [DOI] [PubMed] [Google Scholar]
- Le Roux P, Behar S, Higgins D, Charette M (1999) OP-1 enhances dendritic growth from cerebral cortical neurons in vitro. Exp Neurol 160: 151–163 [DOI] [PubMed] [Google Scholar]
- Liu F, Ventura F, Doody J, Massagué J (1995) Human type II receptor for bone morphogenetic proteins (BMPs): extension of the two-kinase receptor model to the BMPs. Mol Cell Biol 15: 3479–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L (2002) Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol 18: 601–635 [DOI] [PubMed] [Google Scholar]
- Mandell JW, Banker GA (1996) A spatial gradient of Tau protein phosphorylation in nascent axons. J Neurosci 16: 5727–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marqués G, Bao H, Haerry TE, Shimell MJ, Ducheck P, Zhang B, O'Connor MB (2002) The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron 33: 529–543 [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB (2003) The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron 39: 241–254 [DOI] [PubMed] [Google Scholar]
- Mehler MF, Mabie PC, Zhang D, Kessler JA (1997) Bone morphogenetic proteins in the nervous system. Trends Neurosci 20: 309–317 [DOI] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z (2002) Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35: 121–133 [DOI] [PubMed] [Google Scholar]
- Meyer G, Feldman EL (2002) Signaling mechanisms that regulate actin-based motility processes in the nervous system. J Neurochem 83: 490–503 [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR (2003) Signaling mechanisms underlying dendrite formation. Curr Opin Neurobiol 13: 391–398 [DOI] [PubMed] [Google Scholar]
- Morris CA, Mervis CB (2000) Williams syndrome and related disorders. Annu Rev Genomics Hum Genet 1: 461–484 [DOI] [PubMed] [Google Scholar]
- Munoz-Sanjuan I, Brivanlou AH (2002) Neural induction, the default model and embryonic stem cells. Nat Rev Neurosci 3: 271–280 [DOI] [PubMed] [Google Scholar]
- Nagata K, Ohashi K, Yang N, Mizuno K (1999) The N-terminal LIM domain negatively regulates the kinase activity of LIM-kinase 1. Biochem J 343 (Part 1): 99–105 [PMC free article] [PubMed] [Google Scholar]
- Nishihara A, Watabe T, Imamura T, Miyazono K (2002) Functional heterogeneity of bone morphogenetic protein receptor-II mutants found in patients with primary pulmonary hypertension. Mol Biol Cell 13: 3055–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T (2002) Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108: 233–246 [DOI] [PubMed] [Google Scholar]
- Scott EK, Luo L (2001) How do dendrites take their shape? Nat Neurosci 4: 359–365 [DOI] [PubMed] [Google Scholar]
- Threadgill R, Bobb K, Ghosh A (1997) Regulation of dendritic growth and remodeling by Rho, Rac, and Cdc42. Neuron 19: 625–634 [DOI] [PubMed] [Google Scholar]
- Whitford KL, Dijkhuizen P, Polleux F, Ghosh A (2002) Molecular control of cortical dendrite development. Annu Rev Neurosci 25: 127–149 [DOI] [PubMed] [Google Scholar]
- Withers GS, Higgins D, Charette M, Banker G (2000) Bone morphogenetic protein-7 enhances dendritic growth and receptivity to innervation in cultured hippocampal neurons. Eur J Neurosci 12: 106–116 [DOI] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K (1998) Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393: 809–812 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1