

Abstract
Recent evidence indicates that the transactivation of estrogen receptor α (ERα) requires estrogen-dependent receptor ubiquitination and degradation. Here we show that estrogen-unbound (unliganded) ERα is also ubiquitinated and degraded through a ubiquitin–proteasome pathway. To investigate this ubiquitin–proteasome pathway, we purified the ubiquitin ligase complex for unliganded ERα and identified a protein complex containing the carboxyl terminus of Hsc70-interacting protein (CHIP). CHIP preferentially bound to misfolded ERα and ubiquitinated it to induce degradation. Ligand binding to the receptor induced the dissociation of CHIP from ERα. In CHIP−/− cells, the degradation of unliganded ERα was abrogated; however, estrogen-induced degradation was observed to the same extent as in CHIP+/+ cells. Our findings suggest that ERα is regulated by two independent ubiquitin–proteasome pathways, which are switched by ligand binding to ERα. One pathway is necessary for the transactivation of the receptor and the other is involved in the quality control of the receptor.
Keywords: estrogen receptor, nuclear receptors, transcription, ubiquitination
Introduction
The effects of estrogen are mediated through the estrogen receptors ERα and ERβ, which function as ligand-induced transcriptional factors and belong to the nuclear receptor superfamily (Beato et al, 1995; Mangelsdorf et al, 1995; Chambon, 1996; McKenna and O'Malley, 2002). Estrogen binding to its receptor induces the ligand-binding domain (LBD) to undergo a characteristic conformational change, whereupon the receptor dimerizes, binds to DNA and subsequently stimulates the gene expression. ERα is stimulated by two distinct activation regions, activation function-1 (AF-1) and AF-2, which are located in the C-terminal LBD and exert ligand-dependent transcriptional activity. Cellular response to estrogen is tightly controlled, and a large number of ERα-interacting proteins have been described as coactivators or corepressors that modify ERα transcriptional activity (Shang et al, 2000; Yanagisawa et al, 2002; Metivier et al, 2003).
Crystal-structural analysis of ERα and other nuclear receptors has revealed the presence of 12 conserved helices in their LBD (Shiau et al, 1998). The LBD forms a structure described as a sandwich of 12 α-helices (Helices 1–12) with a central hydrophobic ligand-binding pocket. Helix 12, the most C-terminal of these helices, has been identified as the critical core (AD core) of the AF-2 function of the receptor and plays an important role in coactivator binding to the ligand-bound receptor. In the presence of the ligand, the hinge region between Helices 11 and 12 moves closer to Helices 3 and 5, and Helix 12 is positioned over the ligand-binding pocket formed by Helices 3–5. The repositioned Helix 12 forms a hydrophobic groove with Helices 3 and 5. This hydrophobic groove is known to be important for the interaction with LXXLL motifs found in coactivator molecules (Heery et al, 1997).
The activation of nuclear receptors appears to be coupled with the degradation of these proteins by the ubiquitin–proteasome pathway (Boudjelal et al, 2000; Dace et al, 2000; Blanquart et al, 2002). Several recent studies have focused on the involvement of the ubiquitin–proteasome pathway in the estrogen-dependent degradation of ERα, which can be blocked with specific inhibitors of proteasome function, such as MG132 and lactacystin. It has also been reported that the 26S proteasome is essential for estrogen-dependent ERα transcription activity (Nawaz et al, 1999a; Lonard et al, 2000; Reid et al, 2003). Furthermore, several components of the ubiquitin–proteasome pathway have been identified as nuclear receptor-interacting proteins, including SUG1/TRIP1 (Lee et al, 1995), RSP5/RPF1 (Imhof and McDonnell, 1996), E6-AP (Nawaz et al, 1999b) and UBC9 (Poukka et al, 1999). These observations suggest that the ubiquitin–proteasome pathway may play an important role in regulating nuclear receptor levels and restricting the duration and magnitude of receptor activity in response to ligands. Nonetheless, mechanisms governing ERα protein levels remain poorly understood.
Here we show that, in the absence of estrogen, ERα is also ubiquitinated and degraded via a ubiquitin–proteasome pathway. The observation that estrogen-dependent ubiquitination of the receptor required the AD core region within the ERαLBD, whereas the ubiquitination of the unliganded receptor did not, raised the possibility that the ubiquitin ligase for unliganded ERα might differ from the ligase involved in estrogen-dependent ubiquitination. Therefore, we purified the ubiquitin-ligase complex for unliganded ERα and identified a chaperone complex containing the carboxyl terminus of Hsc70-interacting protein (CHIP) (Ballinger et al, 1999; Dai et al, 2003). CHIP selectively bound to and ubiquitinated misfolded ERα and stimulated the degradation of these receptors. This model was further supported by an experiment using CHIP-deficient mouse (CHIP−/−) embryonic fibroblast cells. The unliganded ERα was degraded in CHIP+/+ cells but not in CHIP−/− cells under thermally stressed conditions. In contrast, estrogen-dependent degradation was observed in both CHIP+/+ and CHIP−/− cells, supporting the idea that the inactive and active forms of the receptor are regulated by two independent ubiquitin–proteasome pathways. Our findings shed light on the ubiquitin–proteasome network regulating nuclear receptors.
Results
Unliganded ERα is degraded through a ubiquitin–proteasome pathway
As shown in Figure 1A, addition of estrogen to MCF-7 cells reduced the level of ERα protein. The reduction of ERα was inhibited by the proteasome inhibitors MG132 or lactacystin. In the absence of estrogen, MG132 or lactacystin treatment also resulted in ERα accumulation (Figure 1A, lanes 3 and 5), suggesting that not only estrogen-bound ERα but also unliganded ERα is degraded through proteasomes. In ubiquitination assay, ERα was ubiquitinated in both the presence and absence of estrogen (Figure 1B, lanes 3 and 4), indicating that this process is mediated through ubiquitin–proteasome pathways.
Figure 1.

Unliganded ERα was degraded through a ubiquitin–proteasome pathway. (A) ERα was degraded in the absence of estrogen. The MCF-7 cells were cultured in the presence or absence of estrogen (10−8 M), or the proteasome inhibitor MG132 or lactacystin (10−6 M). ERα level was analyzed by Western blotting using anti-ERα monoclonal antibody. (B) ERα was ubiquitinated in the absence of estrogen. MCF-7 cells were cultured in the presence or absence of estrogen (10−8 M) or MG132 (10−6 M). ERα was immunoprecipitated using anti-ERα antibody. The ubiquitination status of ERα was analyzed by Western blotting using anti-ubiquitin antibody. (C) ERαΔAD was selectively degraded in the absence of estrogen. 293 cells were transfected with either ERα or ERαΔAD (500 ng). At 24 h post-transfection, the cells were cultured in the presence or absence of estrogen (10−8 M) or MG132 (10−6 M). ERα or ERαΔAD protein levels were analyzed by Western blotting using anti-ERα antibody. (D) ERαΔAD was ubiquitinated in the absence of estrogen. Flag-tagged ERαΔAD (500 ng) was transfected into 293 cells in the presence or absence of estrogen (10−8 M) or MG132 (10−6 M). Flag-tagged ERαΔAD was immunoprecipitated using anti-Flag M2 antibody. The ubiquitination status of ERαΔAD was analyzed by Western blotting using anti-ubiquitin antibody.
We next determined whether the degradation of unliganded and liganded ERα is regulated by the same ubiquitin–proteasome pathway. It has been reported that truncated ERα, ERαΔAD, which does not have an AD core domain, does not exhibit estrogen-dependent degradation (Lonard et al, 2000). Thus, we examined the ubiquitination and degradation of ERαΔAD. ERα and ERαΔAD were transfected into 293 cells and the ERα protein level was examined by Western blot analysis. While the ERα degradation was observed regardless of estrogen treatment, ERαΔAD was stabilized by ligand binding, as it accumulates in response to estrogen. MG132 treatment increases the levels of ERαΔAD in the absence of the ligand but does not affect its estrogen-induced accumulation (Figure 1C). We next tested whether ERαΔAD turnover is mediated through ubiquitination. In the absence of MG132, we detected almost no or little ubiquitination of ERαΔAD in the presence and absence of estrogen (Figure 1D, lanes 2 and 4). However, in the presence of MG132, we observed smeary bands of ubiquitin-conjugated ERαΔAD products in the absence of estrogen (Figure 1D, lane 3). These results indicate that while ERαΔAD shows no ligand-dependent ubiquitination, unliganded ERαΔAD is still degraded through ubiquitin–proteasome pathways. According to these results, there are possibly two independent ubiquitination pathways for ERα.
Unliganded ERα associates with a protein complex containing CHIP
We then investigated the region responsible for the degradation of unliganded ERα. The protein level of truncated ERα was examined by Western blotting in the presence or absence of estrogen. As shown in Figure 2A, all of the deletion mutants containing the E domain accumulated with estrogen treatment. MG132 treatment increased the levels of these mutants, indicating that they were degraded through proteasome (Figure 2A, lower panel). These results suggest that the region responsible for the degradation of unliganded ERα is located within ERαLBD. From these results, we speculated that an E3 ubiquitin ligase specifically binds and conjugates ubiquitin to the unliganded ERαLBD. We therefore attempted to identify the putative ubiquitin ligase for unliganded ERα. A HeLa cell extract-derived fraction was incubated with glutathione-S-transferase (GST)-fused ERαLBD in the presence or absence of estrogen. Proteins that interacted with ERαLBD were separated by SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and silver stained (Figure 2B). To identify the proteins that selectively bound to unliganded ERαLBD, we performed peptide mass fingerprinting, and revealed that the 35 kDa protein eluted from the unliganded ERαLBD column consisted of CHIP (Figure 2B). The result obtained from peptide mass fingerprinting was confirmed by Western blotting using a specific antibody against CHIP (Figure 2B, lower panel).
Figure 2.

The unliganded ERα associated with a protein complex containing CHIP and Hsc/Hsp70. (A) The E region of ERα was sufficient for the degradation of unliganded ERα. Indicated Flag-tagged ERα deletion mutants (500 ng) were transfected into 293 cells. These cells were cultured in the presence or absence of estrogen (10−8 M) (upper panel) or MG132 (10−6 M) (lower panel). To evaluate the protein level of ERα mutants, Western blot analysis was performed using anti-Flag M2 antibody. (B) Purification and identification of ERαLBD-interacting proteins. Extracts prepared from HeLa S3 cells were incubated with immobilized GST-ERαLBD in the presence or absence of estrogen (10−6 M). ERα-interacting proteins were eluted from the GST-ERαLBD column by N-lauroyl sarkosin and subjected to SDS–PAGE followed by silver staining. The fractions eluted from unliganded GST-ERαLBD column (lane 1) and liganded GST-ERαLBD column (lane 2) are shown. Proteins eluted from both columns were examined by mass spectrometry. *Hsc70. (C) Interaction between unliganded ERα and CHIP in vivo. MCF-7 cells were lysed and subjected to immunoprecipitation using either anti-CHIP or anti-ERα antibody in the presence or absence of indicated ligands (estrogen (10−8 M); OHT: 4-hydroxytamoxifen (10−6 M); ICI: ICI182,780 (10−7 M)). The precipitates were Western blotted with antibodies for CHIP, ERα and Hsc70. MCF-7 whole-cell extract is shown in lane 1 (WCE).
CHIP is known to possess E3 ubiquitin-ligase activity mediated by its carboxy-terminal U-box domain and has the ability to bind to chaperones Hsp/Hsc70 by means of its tetratricopeptide repeat (TPR) domain (Scheufler et al, 2000; Connell et al, 2001; Imai et al, 2002). Mass spectrometric analysis also identified chaperone proteins Hsp/Hsc70 (Figure 2B), indicating that CHIP binds unliganded ERαLBD as a protein complex containing Hsp/Hsc70. Thus, we examined the interaction between ERα and CHIP/Hsp/Hsc70 complex using a co-immunoprecipitation method. As shown in Figure 2C, CHIP is selectively co-immunoprecipitated with unliganded ERα and Hsc70. Cell treatment with either 4-hydroxytamoxifen (OHT), a partial antagonist of ERα, or ICI182,780 (ICI), a pure antagonist of ERα, abrogated the binding between ERα and CHIP. CHIP was also detected in the immunoprecipitation performed with an anti-ERα antibody in the absence of ligands, confirming the interaction between ERα and CHIP in vivo. The same results were obtained in the human endometrial adenocarcinoma cell line Ishikawa (data not shown).
To better characterize and identify other components of the CHIP–Hsc70 complex, we generated HeLa cell lines stably expressing Flag-HA double-tagged CHIP. The protein complex containing CHIP was precipitated and separated by SDS–PAGE. Protein identification of the purified proteins by mass spectrometric analysis identified KIAA0678, Hsp90, Hsc70, Hsp70, Hsp40 and CHIP (Figure 3A). The protein components of the CHIP complex were confirmed by Western blotting using specific antibodies. Hsp90, Hsc70, Hsp70, Hsp40 and BAG-1 in the CHIP complex are shared with the chaperone components, whereas other chaperone components, Hip, Hop and p23, were undetectable by Western blot analysis (Figure 3B). To investigate whether this protein complex binds to unliganded ERα, Flag-tagged ERα expressed in 293 cells was immunoprecipitated using anti-Flag monoclonal antibody. As shown in Figure 3C, all of the components detected in the CHIP complex by Western blotting existed in the precipitant (Figure 3C, left panel). Next, to investigate whether this protein complex has the same composition in physiological conditions, ERα was immunoprecipitated from MCF-7 cells using a specific antibody for ERα. In the absence of estrogen, the protein complex purified from MCF-7 contained the same components as the complex in 293 cells (Figure 3C, right panel), suggesting that this protein complex exists in the physiological conditions.
Figure 3.

Purification and identification of a protein complex containing CHIP. (A, B) HeLa S3 cells (Mock) or HeLa S3 cells constitutively expressing Flag/HA double-tagged CHIP (Flag/HA-CHIP) were subjected to sequential immunoprecipitation using anti-Flag M2 and anti-HA antibody as described in Materials and methods. The purified fractions were subjected to SDS–PAGE followed by silver staining (A). Proteins eluted from these columns were examined by mass spectrometry (A) and Western blotting (B). Total HeLa cell extract is shown in lane 1 (WCE) (B). (C) Unliganded ERα interacted with a protein complex containing chaperones and CHIP. Flag-ERα-transfected 293 cells (ERα), untransfected cells (Mock) or MCF-7 cells were subjected to immunoprecipitation using either anti-Flag M2 (left panel) or anti-ERα (right panel) antibody and then Western blotted using indicated antibodies. The whole-cell extract is shown in lane 1 (WCE).
CHIP ubiquitinates and degrades unliganded ERα
To test whether CHIP is involved in the ubiquitination and degradation of unliganded ERα, either ERα or ERαΔAD was transfected into 293 cells with or without CHIP. Western blot analysis revealed that, in the absence of estrogen, the steady-state levels of ERα and ERαΔAD were decreased when CHIP was expressed (Figure 4A; 293, lanes 3 and 5). In contrast, in the presence of estrogen, the expression of CHIP exhibited little or no effect on the protein level of ERα and ERαΔAD (Figure 4A; 293, lanes 4 and 6). Endogenous ERα in MCF-7 cells was also decreased by CHIP expression (Figure 4A; MCF-7). Cell treatment with MG132 or lactacystin blocked CHIP-dependent ERα degradation, indicating that the degradation is mediated through proteasome pathways (Figure 4A, lower panel). We further determined the CHIP function by developing MCF-7 cells in which endogenous CHIP expression was suppressed by the introduction of a small interfering RNA (siRNA) complementary to sequences present in the CHIP mRNA. The introduction of the siRNA vector into MCF-7 cells resulted in the suppression of CHIP mRNA (data not shown) and protein expression, and the accumulation of ERα protein (Figure 4B). In contrast, a control vector failed to alter the CHIP or ERα protein level. In addition, either OHT or ICI treatment abrogated CHIP-induced ERα degradation (Figure 4C). Considering the observation that OHT- or ICI-bound ERα showed no interaction with CHIP, it is suggested that the degradation requires binding between ERα and CHIP.
Figure 4.
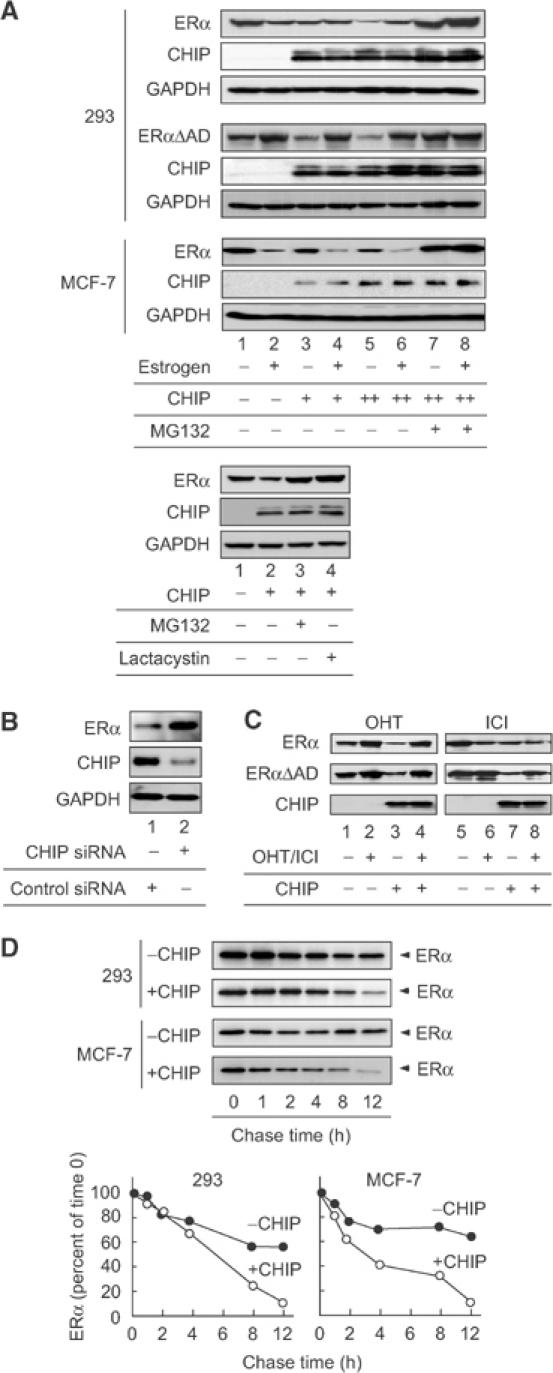
CHIP ubiquitinated and degraded unliganded ERα. (A) CHIP facilitated the degradation of unliganded ERα. HA-tagged CHIP (250 ng) was cotransfected into 293 or MCF-7 cells with or without ERα or ERαΔAD (500 ng) and in the absence or presence of estrogen (10−8 M), MG132 or lactacystin (10−6 M). The protein level of ERα was examined by Western blotting using anti-ERα antibody. (B) siRNA-mediated suppression of endogenous CHIP. The plasmid containing siRNA specific for CHIP or control vector was introduced into MCF-7 cells. Transfected cells were selected by puromycin. Protein levels of CHIP and ERα were assessed by immunoblotting of whole-cell lysate with the specific antibodies as indicated. (C) CHIP did not alter the steady-state level of ERα in the presence of OHT or ICI. Either ERα or ERαΔAD (500 ng) was cotransfected into 293 cells with or without HA-CHIP (250 ng) in the absence or presence of the indicated ligands. The protein level of ERα was examined by Western blotting using specific antibodies for ERα. (D) Pulse-chase assay. 293 cells transfected with CHIP (250 ng) and ERα (500 ng) or MCF-7 cells transfected with CHIP (2 μg) were pulse-labeled with [35S]methionine and then chased for the indicated times in media containing unlabeled methionine. 35S-labeled ERα in anti-ERα immunoprecipitate was quantified by phosphoimaging, and the levels in control cells (closed circle) and CHIP-expressing cells (open circle) were plotted relative to the amount present at time 0.
To confirm that CHIP enhances unliganded ERα degradation, pulse-chase experiments were performed. In the absence of CHIP, the half-life of unliganded ERα exceeded 12 h (Figure 4D; 293), whereas, in the presence of CHIP, the turnover of unliganded ERα increased and exhibited a half-life of approximately 6 h (Figure 4D; 293). The half-life of estrogen-bound ERα was not changed by the expression of CHIP (data not shown). In MCF-7 cells, CHIP also enhanced the turnover of endogenous ERα in the absence of estrogen (Figure 4D; MCF-7). To test the specificity of this effect, we created constructs in which the TPR and U-box domains of CHIP were deleted (ΔTPR and ΔUbox). CHIP binds to Hsp/Hsc70 by means of its TPR motif, while also displaying E3 ubiquitin-ligase activity mediated by its U-box domain. Although the expression of these proteins was similar to that of wild-type CHIP (data not shown), the deletion of either of these domains abolished the effects of CHIP on ERα or ERαΔAD protein level (Figure 5A). The requirement of a TPR motif indicates that CHIP may need to interact with Hsc70 to promote ERα degradation. Functional requirement of the U-box implies that CHIP regulates ERα ubiquitination. In order to validate this model, we evaluated the presence of Hsp/Hsc70 and ERα in complexes containing CHIPΔTPR or CHIPΔUbox. As shown in Figure 5B, CHIPΔTPR did not have the ability to form a complex with Hsc70 and ERα, indicating that Hsc70 mediates the interaction between ERα and CHIP. Finally, we tested whether CHIP enhances ERα turnover through ubiquitination. When ERα was coexpressed with CHIP, we observed the appearance of smeary bands of ubiquitin-conjugated ERα products (Figure 5C, lanes 3 and 5). In the presence of estrogen, CHIP did not enhance the conjugation of ubiquitin to ERα (Figure 5C, lanes 2 and 4). Overall, these observations indicate that the ubiquitination and degradation of unliganded ERα is mediated by a protein complex containing CHIP ubiquitin ligase.
Figure 5.

CHIP-dependent ubiquitination and degradation of ERα required its TPR and U-box domain. (A) Both the TPR and U-box domain in CHIP were necessary for ERα degradation. CHIP, CHIPΔTPR or CHIPΔUbox (250 ng) was transfected into 293 cells with or without ERα or ERαΔAD (500 ng). Protein levels of ERα and ERαΔAD were examined by Western blotting using anti-ERα antibody. (B) The TPR domain of CHIP is necessary for binding to Hsc70 and ERα. HA-tagged CHIP or CHIP mutants were expressed in 293 cells and immunoprecipitated with anti-HA antibody in the absence of estrogen. Precipitates were Western blotted with antibodies for CHIP, ERα and Hsc70. (C) CHIP induced the ubiquitination of unliganded ERα. Flag-tagged ERα (500 ng) was transfected into 293 cells with or without CHIP (250 ng) or CHIPΔUbox (250 ng) in the presence or absence of estrogen (10−8 M). Flag-tagged ERα was immunoprecipitated using anti-Flag M2 antibody. The ubiquitination status of ERα was analyzed by Western blotting using anti-ubiquitin antibody.
CHIP preferentially recognizes and degrades misfolded ERα
To investigate the effect of CHIP on the transcriptional activity of ERα, a luciferase assay was performed as shown in Figure 6A. While the protein level of ERα was reduced by the expression of CHIP (Figure 6B, upper panel), the transcriptional activity of ERα was slightly enhanced by CHIP expression (Figure 6B, lower panel, compare lane 2 with lanes 5 and 8). Therefore, we next estimated the level of transcriptional activity per ERα protein amount. When ERα was coexpressed with CHIP, the level of transcriptional activity per ERα protein was about two-fold higher than ERα alone (Figure 6B, lower panel, compare lane 3 with lanes 6 and 9).
Figure 6.
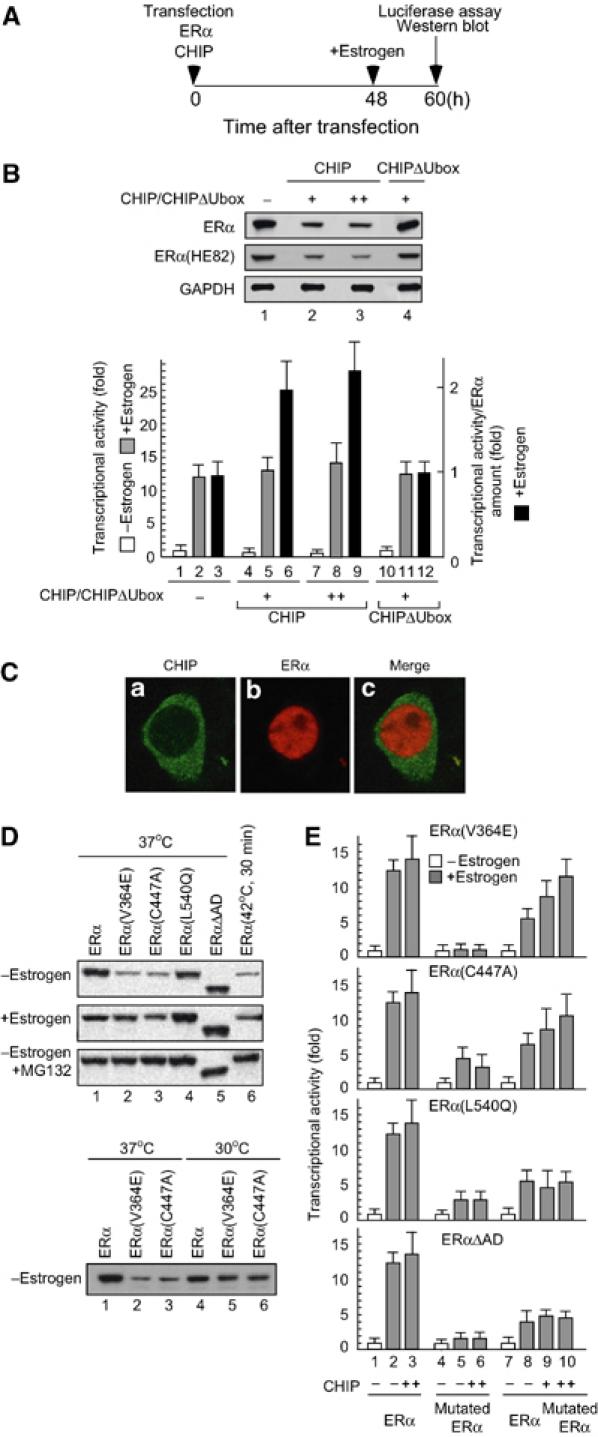
CHIP preferentially recognized and degraded misfolded ERα. (A) The time schedule for luciferase assay and Western blot analysis. 293 cells were transfected with indicated plasmids. At 48 h after transfection, cells were treated with estrogen (10−8 M) for an additional 12 h and harvested for luciferase assay and Western blotting. (B) The level of transcriptional activity per ERα protein amount was enhanced by CHIP. Upper panel: The steady-state level of ERα or ERα(HE82) was reduced by the expression of CHIP but not by CHIPΔUbox. Lower panel: Transcriptional activity of ERα was slightly enhanced by CHIP. ERα (100 ng) and either CHIP or CHIPΔUbox (100 ng) were cotransfected into 293 cells with ERE-TATA-Luc (100 ng) and pRSVβGAL (100 ng), and cell extracts were used in a luciferase assay. The protein amount of ERα was quantified by phosphoimaging. The levels of transcriptional activity per ERα protein amount were plotted relative to the level in control cells. (C) Immunocytochemistry of CHIP and ERα. 293 cells were transiently transfected with HA-tagged CHIP and ERα. The mounted cells were examined by immunofluorescence microscopy as described in Materials and methods. Green represents immunofluorescence for HA-CHIP and red ERα. The distribution of CHIP in a cell body is shown in panel a, and panel b shows the distribution of ERα. Panel c shows the merge images of panels a and b. (D) Temperature-sensitive mutants of ERα degraded faster than wild-type ERα in the absence of ligands. ERα(V364E), ERα(C447A), both of which are temperature sensitive, and ERα(L540Q) were generated by amino-acid substitutions of wild-type ERα. Indicated ERα or ERα mutants (500 ng) were transfected into 293 cells in the presence or absence of estrogen (10−8 M) and MG132 (10−6 M) at 30°C (permissive temperature; lower panel), 37°C (normal/nonpermissive temperature; upper panel) or under thermally stressed conditions (42°C for 30 min; upper panel). Protein levels of ERα or mutants were analyzed by Western blotting using anti-ERα antibody. (E) CHIP recovered the transcriptional activity of ERα suppressed by coexpression of ERα mutants. ERα (100 ng), ERE-TATA-Luc (100 ng) and pRSVβGAL (100 ng) were cotransfected into 293 cells with or without either ERα(V364E), ERα(C447A), ERα(L540Q), ERαΔAD (100 ng) or CHIP (100 ng), and cell extracts were used in a luciferase assay.
Our results show that CHIP binds to unliganded but not to liganded ERα. In addition, CHIP was localized mainly in the cytoplasm (Figure 6C). From these observations, it is difficult to believe that CHIP acts as a coactivator for ERα in the nucleus. Furthermore, ERα(HE82), which has three amino-acid substitutions in the DNA-binding region (C domain) in ERα and has almost no ability to bind DNA (Mader et al, 1989), was also degraded by CHIP, suggesting that the CHIP-dependent degradation of ERα does not require DNA binding. From these results and previous reports (Hohfeld et al, 2001; Meacham et al, 2001; Murata et al, 2001; Goldberg, 2003), we hypothesized that CHIP preferentially ubiquitinates misfolded ERα proteins to eliminate them. CHIP expression may selectively reduce the protein level of unfolded or misfolded ERα, which has less activity than the normal form. Consequently, CHIP could enhance the level of transcriptional activity per ERα protein.
To test this hypothesis, amino-acid substitutions were introduced into ERα to induce protein misfolding. In the absence of ligands, ERα(V364E) (McInerney et al, 1996) and ERα(C447A) (Reese and Katzenellenbogen, 1992), both of which have an amino-acid substitution in the LBD and exhibit temperature sensitivity, were unstable and degraded faster than wild-type protein at a nonpermissive temperature (37°C). Wild-type ERα also degraded to the same extent as temperature-sensitive mutants when cells were cultured under thermally stressed conditions (cells were cultured at 42°C for 30 min) (Figure 6D, upper panel, compare lane 1 with lanes 2, 3 and 6). In contrast, ERα(L540Q) (Ince et al, 1995) and ERαΔAD, which have either an amino-acid substitution or truncation in the flexible Helix 12 region, exhibited the same stability as wild type at 37°C (Figure 6D, upper panel, compare lane 1 with lanes 4 and 5). Under a permissive temperature (30°C), the protein stability of ERα(V364E) and ERα(C447A) was comparable with that of the wild type (Figure 6D, lower panel).
In a luciferase assay, these four mutated ERα proteins showed a loss or reduction of transcriptional activity compared to the wild type (Figure 6E, lane 5), and they were able to suppress wild-type activity when coexpressed with wild-type ERα (Figure 6E, lane 8). CHIP did not enhance the ERα activity suppressed by ERα(L540Q) or ERαΔAD; however, transcriptional activity suppressed by ERα(V364E) or ERα(C447A) was recovered by CHIP expression (Figure 6E, lanes 9 and 10). These results suggest that CHIP may preferentially ubiquitinate ERα(V364E) and ERα(C447A) to degrade these mutants.
If CHIP is directly involved in the hydrolysis of abnormal or mutant forms of ERα, then it should be able to form specific complexes with mutated or misfolded ERα. ERα or mutated forms of ERα were immunoprecipitated from transfected cells and the presence of CHIP and chaperone proteins was detected using specific antibodies. At a permissive temperature (30°C), the amount of CHIP in the precipitate pellets with ERα(V364E) or ERα(C447A) was almost the same in precipitates with the wild type (Figure 7A, right panel). However, at a nonpermissive temperature (37°C), CHIP and BAG-1, a co-chaperone that binds to both Hsc70 and the proteasome, preferentially co-immunoprecipitated with ERα(V364E) and ERα(C447A), while the amount of other chaperone components in precipitants was unchanged (Figure 7A, left panel). In addition, thermally stressed conditions (42°C for 30 min) also increased the CHIP and BAG-1 levels in the precipitated pellet (Figure 7A, left panel, lane 6). Consistent with the results obtained from the degradation and interaction experiments, the polyubiquitination of the temperature-sensitive mutants or thermally denatured ERα was enhanced at nonpermissive temperature (Figure 7B, compare left panel with right panel).
Figure 7.

The misfolding of ERα induced the recruitment of CHIP and BAG-1 to the complex. (A) CHIP and BAG-1 preferentially recognized and bound misfolded ERα. Flag-tagged ERα, ERα(V364E) or ERα(C447A) (100 ng) was transfected into 293 cells. These cells were cultured with MG132 (10−6 M) at 30°C (permissive temperature; right panel), 37°C (normal/nonpermissive temperature; left panel) or under thermally stressed conditions (42°C for 30 min; left panel). Extracts prepared from these cells (lanes 3–6) or untransfected cells (Mock) were subjected to immunoprecipitation using anti-Flag M2 antibody and then Western blotted using antibodies as indicated. The whole-cell extract is shown in lane 1 (WCE). (B) The ubiquitination status of the temperature-sensitive mutants or heat-shocked ERα was enhanced. Flag-tagged ERα, ERα(V364E) or ERα(C447A) (500 ng) was transfected into 293 cells. These cells were cultured with MG132 (10−6 M) at 30°C (right panel), 37°C (left panel) or under thermally stressed conditions (42°C for 30 min; left panel). Extracts prepared from these cells (lanes 2–5) or untransfected cells (Mock) were subjected to immunoprecipitation using anti-Flag M2 antibody. The ubiquitination status of ERα and mutants was analyzed by Western blotting using anti-ubiquitin antibody.
Liganded but not unliganded ERα degradation is observed in CHIP−/− cells
To firmly establish the importance of the observation of CHIP-dependent ERα degradation, we isolated mouse embryonic fibroblast (MEF) cells from either CHIP−/−, CHIP+/− mice or wild-type littermates, CHIP+/+, and determined the protein level of ERα. To induce misfolding of ERα protein, these cells were cultured under thermally stressed conditions. In the absence of estrogen, the thermally stress conditions reduced ERα levels in both CHIP+/+ and CHIP+/− cells but not in CHIP−/− cells (Figure 8A, lanes 4–6). MG132 induced the accumulation of ERα in CHIP+/+ and CHIP+/− cells, indicating that ERα was degraded through proteasome pathways in these cells. These observations provide further support for a model in which CHIP preferentially binds misfolded ERα proteins and degrades them to maintain the quality of ERα protein in cells. Co-immunoprecipitation experiments showed the existence of ERα/Hsc70/CHIP complex in CHIP+/+ cells but not in CHIP−/− cells (Figure 8B). Furthermore, estrogen treatment induced ERα degradation in CHIP−/− cells to the same extent as in CHIP+/+ cells (Figure 8C), suggesting that CHIP is not involved in estrogen-dependent degradation, and supporting the idea that there are two independent ubiquitin–proteasome pathways for ERα (Figure 8D).
Figure 8.

Liganded but not unliganded ERα degradation was observed in CHIP−/− MEF cells. (A) Thermally induced degradation of ERα was not observed in CHIP−/− cells. MEF cells were isolated from CHIP−/−, CHIP+/− mice and wild-type littermates (CHIP+/+). MEF cells were cultured under normal conditions (37°C) or thermally stressed conditions (42°C for 30 min) without estrogen. Extracts prepared from the MEF cells were subjected to Western blotting using the indicated antibody. (B) CHIP+/+ or CHIP−/− cells were lysed and subjected to immunoprecipitation using anti-ERα antibody in the absence of estrogen. Precipitates were Western blotted with antibodies for ERα, Hsc70 and CHIP. (C) Estrogen induced degradation of ERα in CHIP−/− cells. MEF cells were cultured in the presence or absence of estrogen (10−8 M), and cell extracts prepared from these cells were subjected to Western blotting using anti-ERα antibody. (D) ERα degradation may be regulated by two independent ubiquitin–proteasome pathways.
Discussion
Estrogen receptor is regulated by two independent ubiquitin–proteasome pathways
Several studies have mentioned that the AD core region of ERα is essential not only for transactivation but also for estrogen-dependent ERα degradation (Lonard et al, 2000). These reports are in good agreement with our result that ERαΔAD, which has no AD core region, does not show estrogen-dependent degradation. Interestingly, however, MG132 had no effect on ligand-bound ERαΔAD; the steady-state level of ERαΔAD in the absence of estrogen is accumulated in the presence of MG132. These results indicate that unliganded ERαΔAD is still degraded through proteasome pathways. According to these observations, it is possible that the degradation pathway for the unliganded receptor differs from that for liganded. ERαΔAD might be able to recruit a degradation machinery for the unliganded receptor but not for the liganded. Otherwise, there may be a change in the conformation of the receptor, which would protect the receptor from degradation. Reid et al (2003) also demonstrated that unliganded ERα is subject to proteasome-mediated turnover, which is mechanistically different from the turnover of liganded ERα.
Several lines of evidence indicate that estrogen, progesterone and glucocorticoid receptors (GRs) are degraded in the presence of their cognate ligands (Nawaz et al, 1999a; Wallace and Cidlowski, 2001). However, this is contrasted with observations of androgen and vitamin D receptors, which are accumulated in the presence of their agonist ligands (Li et al, 1999). From our results, these inconsistent observations might be explained by the balance between the two degradation pathways in the cells. When the degradation pathway for unliganded receptors is more active than that for liganded receptors, these receptors would stabilize in the presence of ligands. In contrast, when the liganded receptor degradation pathway is stronger than the unliganded receptor degradation pathway, the protein level of receptors is downregulated by ligand treatment.
CHIP containing a protein complex specifically binds and ubiquitinates unliganded estrogen receptor
To address the mechanism of the ubiquitination and degradation of unliganded ERα, we purified proteins using GST-fused ERαLBD, and identified CHIP, which specifically bound to unliganded ERαLBD. Our findings indicate that CHIP binds unliganded ERα as a protein complex containing Hsp90, Hsc70, Hsp70, Hsp40 and BAG-1, all of which are known to possess or assist chaperoning functions, and a Dna J-like protein, KIAA0678. Dna J is a member of the Hsp40 family of molecular chaperones, which regulate the activity of Hsp70s. Dna J-like proteins that contain regions closely resembling a Dna J domain are suggested to regulate the activity of Dna J proteins during protein translocation, assembly and disassembly (Cheetham and Caplan, 1998).
CHIP expression with ERα enhanced the conjugation of ubiquitin to the receptors and stimulated degradation. Receptor ubiquitination and degradation was abrogated when cells were treated with estrogen. These results are in good agreement with the results obtained from binding experiments. Furthermore, OHT and ICI, both of which inhibited the interaction between CHIP and ERα, reduced the CHIP-mediated degradation of ERα. These findings confirmed the idea that unliganded ERα ubiquitination is mediated by CHIP. In immunostaining, CHIP was largely detected in the cytoplasm (Figure 6C). The localization of CHIP was not changed when cells were cultured under heat-stressed conditions (data not shown). According to these results, CHIP-dependent ERα ubiquitination may occur mainly in the cytoplasm. However, we cannot exclude the possibility that a small amount of CHIP is involved in the ubiquitination of ERα in the nucleus.
Recently, CHIP was reported to induce ubiquitination of the GR bound to Hsp90 for proteasomal degradation (Connell et al, 2001). While our findings indicate that CHIP selectively binds to unliganded ERα and ubiquitinates it, CHIP-mediated GR degradation is observed in the presence of ligands. Recent reports indicate that in the presence of ligands, nuclear receptors do not remain permanently bound at a promoter, but rather undergo cycles of binding and unbinding (Shang et al, 2000; Stenoien et al, 2001; Galigniana et al, 2004). The cycling of ligand-bound ERα requires proteasomal activity (Reid et al, 2003). Together with these reports and our observations, it is possible that the binding of estrogen to ERα induces the dissociation of CHIP and the association of other ubiquitin ligases, which are involved in receptor cycling at a promoter. The ligand-dependent cycling of GR is known to be much faster than that of ERα and both chaperones and proteasomes are thought to be important for GR cycling since the disruption of either leads to alterations in the exchange rate (Galigniana et al, 2004). According to these results, it is possible that, while the chaperone complex containing CHIP mainly resides in the cytoplasm, it may translocate into the nucleus and regulate the cycling of liganded GR.
CHIP is involved in the quality control of estrogen receptor
Since CHIP selectively bound to and ubiquitinated unliganded ERα, CHIP seemed not to be directly involved in transcriptional regulation. Recently, it was shown that CHIP is involved in the ubiquitination of the immature cystic fibrosis transmembrane conductance regulator (CFTR) in the endoplasmic reticulum-associated degradation (ERAD) pathway (Wickner et al, 1999; Meacham et al, 2001). Based on these findings, it is speculated that CHIP may be a new category of E3 enzyme responsible for the quality control of cellular proteins linked to the function of molecular chaperones. However, there is no experimental evidence to show that CHIP indeed acts as E3 ubiquitin ligase capable of distinguishing the non-native states from native states of target proteins in vivo.
In this study, we have shown that temperature-sensitive mutants of ERα preferentially recruited CHIP to ubiquitinate and degrade these receptors under nonpermissive temperatures. In addition, the ubiquitination and degradation of unliganded ERα was enhanced when cells were cultured under thermally stressed conditions. These observations suggest that CHIP preferentially induces the hydrolysis of abnormal or mutant forms. Using MEF cells derived from CHIP−/− or wild-type littermates, we confirmed the importance of the observation of CHIP-mediated unfolded ERα degradation. These observations provide direct in vivo evidence that CHIP selectively ubiquitinated thermally denatured ERα. Our observations provide the first in vivo evidence that CHIP functions as ‘quality-control E3' involved in the selective ubiquitination of target proteins by recognizing the non-native state in a molecular chaperone-assisted manner. Furthermore, estrogen treatment induced the degradation of ERα in CHIP−/− cells to the same extent as in CHIP+/+ cells, suggesting that CHIP is not involved in estrogen-dependent degradation, and supporting the idea that there are two independent ubiquitin–proteasome pathways for ERα. Considering that nuclear receptors have conserved LBDs and that some are known to associate with a chaperone complex, our findings raise the possibility that other members of the nuclear receptor family may also be regulated by two independent ubiquitin–proteasome pathways.
Materials and methods
Expression vectors, antibodies, cell culture and transfection
These are available as Supplementary data at The EMBO Journal Online.
Co-immunoprecipitation and Western blotting
293 cells were transfected with the indicated plasmids, lysed in TNE (10 mM Tris–HCl (pH 7.8), 1% NP-40, 0.15 M NaCl, 1 mM EDTA, 1 μM phenylmethylsulfonyl fluoride (PMSF), 1 μg/ml aprotinin) buffer. Extracted proteins were immunoprecipitated with the antibody-coated protein A/G Sepharose (Amersham) or anti-Flag M2 agarose (Sigma). The bound proteins were separated by SDS–PAGE, transferred onto polyvinylidine difluoride membranes (Millipore) and detected with indicated antibodies, and secondary antibodies conjugated with horseradish peroxidase. Specific proteins were detected using enhanced chemiluminescence (ECL) Western blot detection system (Amersham).
Ubiquitination assay
MCF7 and 293 cells, which were transfected with or without Flag-tagged ERα and HA-tagged CHIP, were lysed with radioimmunoprecipitation (RIPA) buffer (50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with COMPLETE protease inhibitor mixture (Roche) and kept for 20 min on ice. The extracts clarified by centrifugation were immunoprecipitated with anti-Flag agarose for 1 h at 4°C. After washing the resin with RIPA buffer, the bound proteins were eluted by incubation for 1 h at 4°C with Flag peptide in RIPA buffer (0.4 mg/ml). Immunoprecipitates were immunoblotted with the indicated antibody.
Protein purification
Immobilized GST-ERαLBD fusion proteins were preincubated for 1 h at 4°C in GST-binding buffer (20 mM Tris–HCl (pH 7.9), 180 mM KCl, 0.2 mM EDTA, 0.5 mM PMSF, 1 mM DTT) containing BSA (1 mg/ml) with or without estrogen (10−6 M). Bead-immobilized proteins were then incubated at 4°C for 6–10 h with HeLa cell extracts in the presence or absence of 10−6 M estrogen. After washing with GST buffer (GST-binding buffer with 0.1% NP-40) three times, the beads were further washed with GST buffer containing 0.2% N-lauroyl sarkosine. Proteins bound to ERα were eluted with 15 mM reduced glutathione in elution buffer (50 mM Tris–HCl (pH 8.3), 150 mM KCl, 0.5 mM EDTA, 0.5 mM PMSF, 5 mM NaF, 0.08% NP-40, 0.5 mg/ml BSA, 10% glycerol). For purification of the Flag/HA-CHIP complex, HeLa cells stably expressing Flag/HA-CHIP were extracted with TNE buffer and extracted proteins were incubated with anti-Flag M2 agarose for 2 h at 4°C. After washing the resin with TNE buffer, the bound proteins were eluted by incubation for 1 h at 4°C with Flag peptide in TNE buffer (0.4 mg/ml). For further purification, eluted fractions were incubated with anti-HA agarose for 2 h at 4°C. After washing with TNE buffer, the bound proteins were eluted with a small aliquot of HA peptide in TNE buffer (0.05 mg/ml).
Pulse chase
MCF7 and 293 cells were transfected with or without ERα and CHIP, and 48 h post-transfection, the cells were labeled for 30 min at 37°C with 50 μCi [35S]methionine per ml in methionine-free Dulbecco's modified Eagle's medium (DMEM). The cells were then washed twice and incubated in DMEM containing 10% FBS for the indicated time periods (chase). At each time point of the chase, cell lysates were immunoprecipitated with anti-ERα antibody. The immunoprecipitates were resolved by SDS–PAGE and visualized by autoradiography. Phosphoimager was used to quantify the metabolically labeled ERα present at each time point.
Immunofluorescence
The 293 cells were grown on poly-L-lysine-coated eight-well chamber culture slides, and transfected with plasmids. At 24 h post-transfection, the cells were fixed with 4% paraformaldehyde in PBS for 10 min and permeabilized with Triton buffer (50 mM Tris–HCl (pH 7.5), 0.5% Triton X-100, 150 mM NaCl, 2 mM EDTA) for 15 min. The cells in each well were blocked with PBS containing 1% BSA and 0.5% goat serum for 3 h at 37°C. The cells were incubated with anti-HA and ERα antibody in PBS containing 1% BSA for 2 h at 37°C. After washing with PBS, the cells were incubated with Alexa fluor 488 goat anti-rat IgG and Alexa fluor 594 goat anti-mouse IgG (Molecular Probes) for 1 h at 37°C and washed with PBS. The sample was mounted in VECTASHIELD mounting medium (Vecter Labs) and analyzed with Leica TCS SP2 spectral confocal scanning system.
RNAi
MCF7 cells maintained in the DMEM medium containing charcoal-stripped FBS were cotransfected with CHIP siRNA vector or luciferase siRNA vector (control) and pUC19 vector carrying puromycin-resistant gene. At 24 h post-transfection, the transfected cells were changed to the medium containing 1 μg/ml of puromycin. At 48 h after puromycin selection, the puromycin-resistant cells were harvested and lysed with TNE buffer. The equal amounts of extracted protein were subjected to Western blotting.
Supplementary Material
Supplementary Data
Acknowledgments
We thank Dr Akiyoshi Fukamizu and his laboratory staff for providing materials and instruments. This work was supported by the 21st Century COE Program from the Ministry of Education, Culture, Sports, Sciences, and Technology (MEXT).
References
- Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol 19: 4535–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beato M, Herrlich P, Schutz G (1995) Steroid hormone receptors: many actors in search of a plot. Cell 83: 851–857 [DOI] [PubMed] [Google Scholar]
- Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C (2002) Peroxisome proliferator-activated receptor alpha (PPARalpha) turnover by the ubiquitin–proteasome system controls the ligand-induced expression level of its target genes. J Biol Chem 277: 37254–37259 [DOI] [PubMed] [Google Scholar]
- Boudjelal M, Wang Z, Voorhees JJ, Fisher GJ (2000) Ubiquitin/proteasome pathway regulates levels of retinoic acid receptor gamma and retinoid X receptor alpha in human keratinocytes. Cancer Res 60: 2247–2252 [PubMed] [Google Scholar]
- Chambon P (1996) A decade of molecular biology of retinoic acid receptors. FASEB J 10: 940–954 [PubMed] [Google Scholar]
- Cheetham ME, Caplan AJ (1998) Structure, function and evolution of DnaJ: conservation and adaptation of chaperone function. Cell Stress Chaperones 3: 28–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C (2001) The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol 3: 93–96 [DOI] [PubMed] [Google Scholar]
- Dace A, Zhao L, Park KS, Furuno T, Takamura N, Nakanishi M, West BL, Hanover JA, Cheng S (2000) Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proc Natl Acad Sci USA 97: 8985–8990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V, Li HH, Madamanchi N, Xu W, Neckers L, Cyr D, Patterson C (2003) CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J 22: 5446–5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galigniana MD, Harrell JM, Housley PR, Patterson C, Fisher SK, Pratt WB (2004) Retrograde transport of the glucocorticoid receptor in neurites requires dynamic assembly of complexes with the protein chaperone hsp90 and is linked to the CHIP component of the machinery for proteasomal degradation. Brain Res Mol Brain Res 123: 27–36 [DOI] [PubMed] [Google Scholar]
- Goldberg AL (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426: 895–899 [DOI] [PubMed] [Google Scholar]
- Heery DM, Kalkhoven E, Hoare S, Parker MG (1997) A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387: 733–736 [DOI] [PubMed] [Google Scholar]
- Hohfeld J, Cyr DM, Patterson C (2001) From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep 2: 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI, Takahashi R (2002) CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity. Mol Cell 10: 55–67 [DOI] [PubMed] [Google Scholar]
- Imhof MO, McDonnell DP (1996) Yeast RSP5 and its human homolog hRPF1 potentiate hormone-dependent activation of transcription by human progesterone and glucocorticoid receptors. Mol Cell Biol 16: 2594–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince BA, Schodin DJ, Shapiro DJ, Katzenellenbogen BS (1995) Repression of endogenous estrogen receptor activity in MCF-7 human breast cancer cells by dominant negative estrogen receptors. Endocrinology 136: 3194–3199 [DOI] [PubMed] [Google Scholar]
- Lee JW, Ryan F, Swaffield JC, Johnston SA, Moore DD (1995) Interaction of thyroid-hormone receptor with a conserved transcriptional mediator. Nature 374: 91–94 [DOI] [PubMed] [Google Scholar]
- Li XY, Boudjelal M, Xiao JH, Peng ZH, Asuru A, Kang S, Fisher GJ, Voorhees JJ (1999) 1,25-Dihydroxyvitamin D3 increases nuclear vitamin D3 receptors by blocking ubiquitin/proteasome-mediated degradation in human skin. Mol Endocrinol 13: 1686–1694 [DOI] [PubMed] [Google Scholar]
- Lonard DM, Nawaz Z, Smith CL, O'Malley BW (2000) The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell 5: 939–948 [DOI] [PubMed] [Google Scholar]
- Mader S, Kumar V, de Verneuil H, Chambon P (1989) Three amino acids of the oestrogen receptor are essential to its ability to distinguish an oestrogen from a glucocorticoid-responsive element. Nature 338: 271–274 [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM (1995) The nuclear receptor superfamily: the second decade. Cell 83: 835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney EM, Ince BA, Shapiro DJ, Katzenellenbogen BS (1996) A transcriptionally active estrogen receptor mutant is a novel type of dominant negative inhibitor of estrogen action. Mol Endocrinol 10: 1519–1526 [DOI] [PubMed] [Google Scholar]
- McKenna NJ, O'Malley BW (2002) Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108: 465–474 [DOI] [PubMed] [Google Scholar]
- Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM (2001) The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol 3: 100–105 [DOI] [PubMed] [Google Scholar]
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F (2003) Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115: 751–763 [DOI] [PubMed] [Google Scholar]
- Murata S, Minami Y, Minami M, Chiba T, Tanaka K (2001) CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep 2: 1133–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz Z, Lonard DM, Dennis AP, Smith CL, O'Malley BW (1999a) Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci USA 96: 1858–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz Z, Lonard DM, Smith CL, Lev-Lehman E, Tsai SY, Tsai MJ, O'Malley BW (1999b) The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol Cell Biol 19: 1182–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poukka H, Aarnisalo P, Karvonen U, Palvimo JJ, Janne OA (1999) Ubc9 interacts with the androgen receptor and activates receptor-dependent transcription. J Biol Chem 274: 19441–19446 [DOI] [PubMed] [Google Scholar]
- Reese JC, Katzenellenbogen BS (1992) Characterization of a temperature-sensitive mutation in the hormone binding domain of the human estrogen receptor. Studies in cell extracts and intact cells and their implications for hormone-dependent transcriptional activation. J Biol Chem 267: 9868–9873 [PubMed] [Google Scholar]
- Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F (2003) Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol Cell 11: 695–707 [DOI] [PubMed] [Google Scholar]
- Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I (2000) Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell 101: 199–210 [DOI] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103: 843–852 [DOI] [PubMed] [Google Scholar]
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL (1998) The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95: 927–937 [DOI] [PubMed] [Google Scholar]
- Stenoien DL, Patel K, Mancini MG, Dutertre M, Smith CL, O'Malley BW, Mancini MA (2001) FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat Cell Biol 3: 15–23 [DOI] [PubMed] [Google Scholar]
- Wallace AD, Cidlowski JA (2001) Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem 276: 42714–42721 [DOI] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, Gottesman S (1999) Posttranslational quality control: folding, refolding, and degrading proteins. Science 286: 1888–1893 [DOI] [PubMed] [Google Scholar]
- Yanagisawa J, Kitagawa H, Yanagida M, Wada O, Ogawa S, Nakagomi M, Oishi H, Yamamoto Y, Nagasawa H, McMahon SB, Cole MD, Tora L, Takahashi N, Kato S (2002) Nuclear receptor function requires a TFTC-type histone acetyl transferase complex. Mol Cell 9: 553–562 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data