

Abstract
Forkhead transcription factors of the FOXO class are negatively regulated by PKB/c-Akt in response to insulin/IGF signalling, and are involved in regulating cell cycle progression and cell death. Here we show that, in contrast to insulin signalling, low levels of oxidative stress generated by treatment with H2O2 induce the activation of FOXO4. Upon treatment of cells with H2O2, the small GTPase Ral is activated and this results in a JNK-dependent phosphorylation of FOXO4 on threonine 447 and threonine 451. This Ral-mediated, JNK-dependent phosphorylation is involved in the nuclear translocation and transcriptional activation of FOXO4 after H2O2 treatment. In addition, we show that this signalling pathway is also employed by tumor necrosis factor α to activate FOXO4 transcriptional activity. FOXO members have been implicated in cellular protection against oxidative stress via the transcriptional regulation of manganese superoxide dismutase and catalase gene expression. The results reported here, therefore, outline a homeostasis mechanism for sustaining cellular reactive oxygen species that is controlled by signalling pathways that can convey both negative (PI-3K/PKB) and positive (Ras/Ral) inputs.
Keywords: FOXO, JNK, oxidative stress, Ral
Introduction
Reactive oxygen species (ROS) are oxygen free radicals that are highly reactive toward cellular constituents including protein, lipid and DNA. Formation of ROS can be caused by exogenous sources such as UV or ionizing radiation or by endogenous sources such as normal aerobic metabolism or by pathological conditions such as ischemia. Also in normal cell signalling, ROS are generated by growth factor-induced activation of enzyme complexes such as NADH oxidase (reviewed in Finkel, 2000). Furthermore, cellular levels of ROS fluctuate throughout the cell cycle and in fact ROS are required for cell proliferation (Clopton and Saltman, 1995; Shackelford et al, 2001). Nevertheless, cells have developed numerous antioxidant systems to prevent excess generation of ROS as the highly reactive nature of ROS will easily result in ROS-induced adverse modifications of protein, lipid or DNA. Normally, ROS-induced modification or damage is either repaired, as in the case of chromosomal DNA damage, or removed by degradation and subsequent resynthesis. In the case of excessive damage or ineffective repair, cell death (apoptosis) is triggered. Thus, cells require both a stringent homeostasis mechanism for ROS and efficient repair to tolerate levels of ROS required for normal cell function that will otherwise result in certain cell death.
Consistent with its role in normal growth factor-induced signalling, ROS generation by exogenous sources such as H2O2 treatment has been shown to trigger most of the signalling pathways downstream of growth factor receptors (reviewed in Finkel, 2000). For example, ROS have been shown to activate members of the JNK/p38 stress kinase family, MAPKs, PI-3K signalling, NF-κB and many more. Activation by ROS of some of these signalling cascades has been implicated primarily in the induction of apoptosis (JNK/p38), whereas others have been implicated in cell survival (PI-3K).
Protein kinase B (PKB/c-Akt) mediates many of the antiapoptotic effects of PI-3K signalling. A large number of PKB substrates have been implicated in the regulation of cellular survival (reviewed in Lawlor and Alessi, 2001). However, little is known as to how PI-3K/PKB signalling may regulate the cellular level of ROS. Recently, we and others have shown that the PKB-regulated Forkhead transcription factor FOXO3a can reduce the level of cellular oxidative stress by directly increasing mRNA and protein levels of manganese superoxide dismutase (MnSOD) and catalase (reviewed in Burgering and Medema, 2003). PKB-mediated phosphorylation of FOXO results in translocation of FOXO from the nucleus to the cytosol. Consequently, PKB activation decreases MnSOD and catalase levels and this is likely to contribute to an increase in cellular ROS. As cell cycle progression requires increased ROS level, this is in agreement with the role of PI-3K signalling in stimulating cell proliferation.
Because of the inverse relationship between PI-3K/PKB signalling and FOXO activity, we were interested whether an increase in ROS could regulate FOXO activity and oppose the effect of PI-3K/PKB signalling. Here, we show that oxidative stress induced by treatment of cells with H2O2 results in the activation of the small GTPase Ral. Activation of Ral results in the phosphorylation and activation of JNK and JNK-mediated phosphorylation of FOXO4 on T447 and T451. Phosphorylation of these residues is critical to FOXO4 transcriptional activity. Thus, H2O2 can induce FOXO4 transcriptional activity and this is further confirmed by the observation that H2O2 treatment results in translocation of FOXO4 from the cytosol to the nucleus. In addition, we show that tumor necrosis factor α (TNFα), a ligand known to increase cellular H2O2 levels, also activates FOXO4 transcriptional activity and that this involves cellular ROS, Ral and JNK. These results indicate that FOXOs can function in a negative feedback loop to control the cellular level of oxidative stress in a cell and therefore these results start to outline a novel homeostasis mechanism of ROS control.
Results
To investigate whether FOXO could function in a feedback mechanism to control cellular redox, we first analyzed the possibility that, similar to insulin signalling, cellular oxidative stress generated by H2O2 treatment of cells could induce FOXO phosphorylation. Cells transiently expressing HA-FOXO4 were labelled with [32P]orthophosphate and treated with various concentrations of H2O2. At the lowest concentration tested (20 μM), H2O2 treatment induced phosphorylation of FOXO4 (Figure 1A). Phosphorylation by H2O2 did not correlate with H2O2-induced PKB activation, as increased PKB phosphorylation in these cells was only observed at the highest concentration of H2O2 (200 μM; Figure 1A). By mutational analysis, we have previously defined two residues within FOXO4, threonine 447 (T447) and threonine 451 (T451), that can be phosphorylated independently of PKB activation (De Ruiter et al, 2001) and recently, we confirmed phosphorylation of these residues on FOXO4 by mass spectrometry (data not shown). To study regulation of T447/451 phosphorylation, we obtained phosphospecific antibodies against both phosphorylated T451 (T451P) and T447 (T447P). H2O2 treatment induced both T451 and T447 phosphorylation (Figure 1B). The T451P antibody did not recognize HA-FOXO4-T451A and the T447P antibody did not recognize HA-FOXO4-T447A isolated both from untreated and H2O2-treated cells, indicating their specificity. The T447P antibody also did not recognize HA-FOXO4-T451A, suggesting that T451 is an essential part of the epitope for the T447P antibody. Moreover, we analyzed phosphorylation of endogenous FOXO4. Mouse C2C12 cells expressing endogenous FOXO4 were treated with H2O2 and displayed increased T447 phosphorylation (Figure 1C). As T451 is not conserved between human and mouse FOXO4, we could not test endogenous T451 phosphorylation in these cells. These results show that in vivo FOXO4 becomes phosphorylated at T447 and T451 following treatment of cells with H2O2. The T451P antibody is of better quality compared to the T447P antibody. Therefore, the results with the T451P antibody are shown in the following figures, and similar results were obtained using the T447P antibody.
Figure 1.
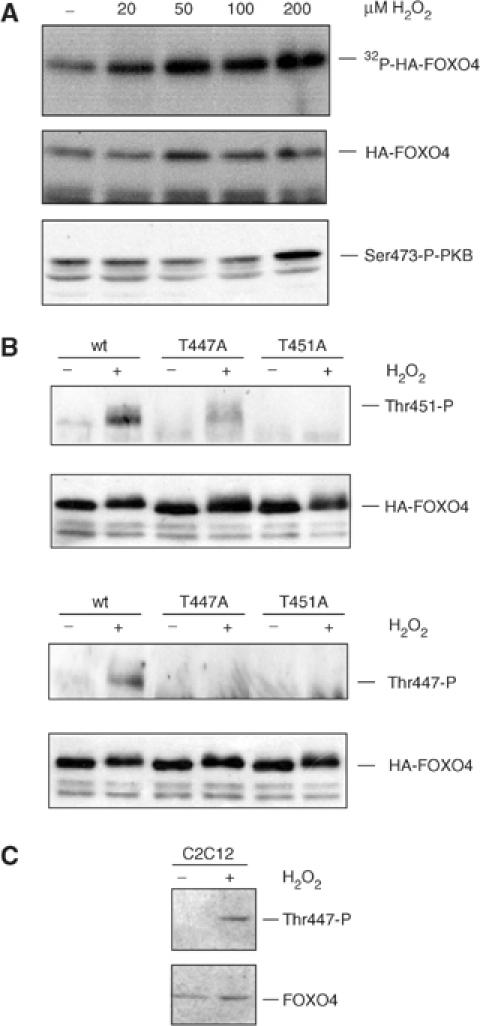
H2O2 induced phosphorylation of FOXO4 on T447 and T451. (A) A14 cells, transfected with HA-FOXO4, were labelled with [32P]orthophosphate for 3 h and left untreated or treated for 60 min with indicated H2O2 concentrations. Cells were lysed and HA-FOXO4 was immunoprecipitated. Following exposure to the film, the blot was probed with 12CA5 monoclonal antibody to ensure equal expression of HA-FOXO4 in each lane. H2O2 treatment induced a 2.5-fold increase in phosphorylation of FOXO4. In parallel, samples were analyzed on Western blot for Ser473 phosphorylation of PKB (lower panel). (B) 293T cells, transfected with HA-FOXO4, HA-FOXO4-T447A or HA-FOXO4-T451A, were left untreated or treated with 100 μM H2O2 for 60 min. HA-FOXO4 were immunoprecipitated and analyzed on Western blot for Thr447 or Thr451 phosphorylation. Same results were obtained with 200 and 400 μM H2O2. (C) Mouse C2C12 cells were left untreated or treated with 100 μM of H2O2 for 60 min. Endogenous FOXO4 was analyzed on blot for T447 phosphorylation. Same results were obtained using 200 or 400 μM H2O2.
In insulin signalling, phosphorylation of T447 and T451 occurs in a Ral-dependent manner (De Ruiter et al, 2001). Thus, we analyzed whether H2O2-induced phosphorylation of T447 and T451 was dependent on activation of the small GTPase Ral. Therefore, we first analyzed whether H2O2 could induce the activation of Ral. Cells treated for various periods of time with H2O2 were lysed and the level of active Ral (Ral-GTP) was determined by a pull-down assay (Wolthuis et al, 1998). H2O2 treatment induced a rapid and time-dependent increase in RalGTP levels (Figure 2A). To determine whether this H2O2-induced Ral activation mediated the phosphorylation of T451, we expressed HA-FOXO4 in the presence or absence of dominant-negative Ral (RalN28). Expression of dominant-negative Ral completely blocked phosphorylation of T451 (Figure 2B), indicating the involvement of Ral in H2O2-induced phosphorylation. Subsequently, we examined whether activation of endogenous Ral could mediate T451 phosphorylation. Activation of endogenous Ral, both through the expression of active Ras (RasV12) and through the expression of active Ral guanine nucleotide exchange factors (RlfCAAX and RalGEF2), but not the expression of a control in which the catalytic domain was mutated (RlfCAAXΔGEF), resulted in increased T451 phosphorylation (Figure 2C). Taken together, these data demonstrate that H2O2 treatment of cells results in the activation of the small GTPase Ral, which is necessary and sufficient to induce phosphorylation of T451 on FOXO4.
Figure 2.
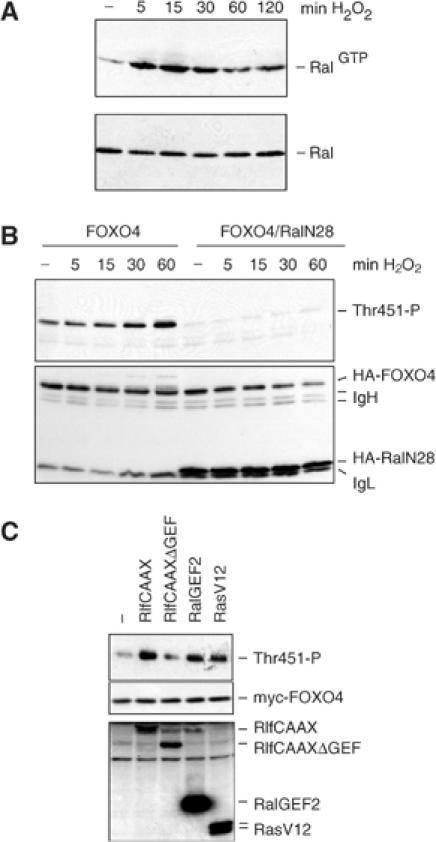
H2O2 induces Ral activation and Ral activation is necessary and sufficient for H2O2-induced T451 phosphorylation. (A) A14 cells were treated with 100 μM of H2O2 for the indicated time, and Ral GTP levels were analyzed on Western blot using a Ral pull-down assay (upper panel). The lower panel shows endogenous Ral protein levels. Same results were obtained using 200 and 400 μM H2O2. (B) 293T cells, transfected with HA-FOXO4 and HA-RalN28 or a control construct, were treated with 100 μM H2O2 for the indicated time and T451 phosphorylation was analyzed on Western blot. Same results were obtained using 200 or 400 μM H2O2. (C) 293T cells were transfected with myc-FOXO4 together with the indicated constructs. T451 phosphorylation was analyzed.
To investigate which kinase could mediate Ral-induced phosphorylation of T451, we treated cells with a variety of kinase inhibitors prior to H2O2 treatment. The MEK inhibitor PD98059, the PI-3K inhibitor LY294002 or the p38 inhibitor SB203580 could not inhibit H2O2-induced phosphorylation of T451 and T447 on FOXO4 (data not shown). These results indicate that T451 phosphorylation is not mediated by PI-3K, MAPK or p38. To study a potential involvement of JNK, we used immortalized mouse embryo fibroblasts (MEFs) derived from JNK1,2−/− mice, since there is no specific JNK inhibitor available. As these MEFs also do not express JNK3, they lack any JNK activity (Sabapathy et al, 1999). H2O2 treatment of JNK1,2−/− MEFs did not induce phosphorylation of T451, whereas H2O2 treatment of control MEFs (wild-type (wt) MEFs) did, strongly indicating that in vivo JNK mediates T451 phosphorylation (Figure 3A). To further confirm this, we rescued JNK expression in JNK1,2−/− MEF cells by coexpression of either JNK1 or JNK3. This restored H2O2-induced JNK activity and the induction of T451 phosphorylation (Figure 3B). JNK is often observed bound to its potential substrates. We therefore analyzed the binding between JNK and FOXO4. Treatment of cells with increasing concentrations of H2O2 induced the binding of JNK1 (data not shown) and JNK3 to FOXO4 (Figure 3C). Consistent with the in vivo data, active JNK1, but not p38α, could efficiently phosphorylate T451/447 of FOXO4 in vitro (Figure 3D). Thus, we conclude that JNK phosphorylates FOXO4 in vitro and in vivo at T451 and that this phosphorylation can be induced by H2O2 treatment.
Figure 3.
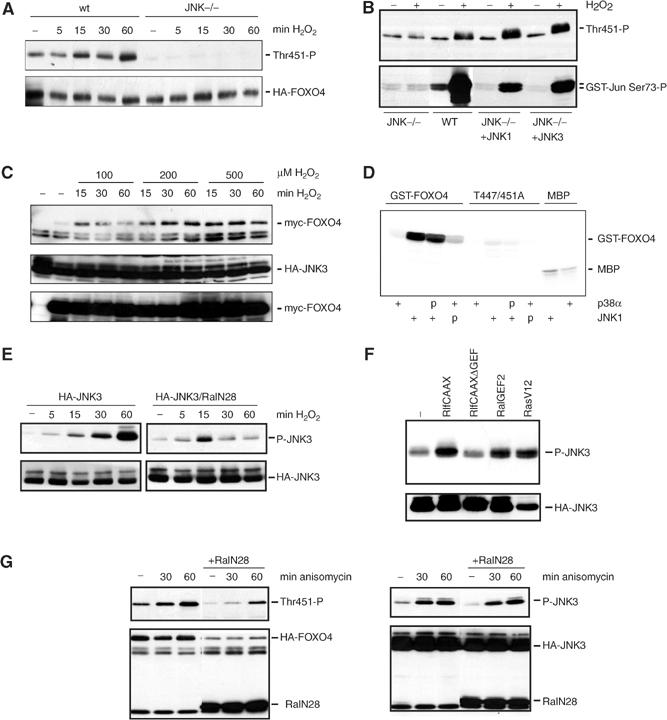
JNK is involved in the H2O2-induced Ral-mediated phosphorylation of T451 and T447 on FOXO4. (A) JNK1,2−/− MEFs, transfected with HA-FOXO4 together with JNK1, JNK3 or an empty vector, were treated with 100 μM H2O2 for the indicated time, and T451 phosphorylation was analyzed on Western blot. wt MEFs were included as control. Similar results were obtained using 200 or 400 μM H2O2. (B) JNK1,2−/− MEFs, wt MEFs and JNK−/− cotransfected with either JNK1 or JNK3, transfected with HA-FOXO4, were left untreated or treated with 100 μM of H2O2 for 60 min. T451 phosphorylation was analyzed. In parallel, a GST-Jun pull-down was performed to measure JNK activity (lower panel). Same results were obtained using 200 or 400 μM H2O2. (C) 293T, transfected with myc-FOXO4 and HA-JNK3, were treated with different concentrations of H2O2 for indicated times. HA-JNK3 was immunoprecipitated and binding of myc-FOXO4 to HA-JNK3 was analyzed on Western blot (upper panel). The lower panels show expression of the constructs. (D) Purified bacterially expressed GST-FOXO4(C) and GST-FOXO4-T447/451A(C) were incubated in the presence (+) or absence of active JNK or active p38α. P indicates pretreatment with either active JNK or p38α in the presence of unlabelled rATP. Prephosphorylation by JNK or p38α did not enhance the ability of p38α or JNK to subsequently phosphorylate GST-FOXO4. MBP substrate was included as control for the activity of active JNK and p38α. (E) 293T cells transfected with HA-JNK3 with or without HA-RalN28 were treated with 100 μM of H2O2 for increasing periods of time. JNK phosphorylation was analyzed on western blot. Similar results were obtained using 200 or 400 μM of H2O2. (F) 293T cells were transfected with HA-JNK3 together with indicated constructs. JNK phosphorylation was analyzed on Western blot. (G) 293T cells transfected with HA-FOXO4 or HA-JNK3 with or without HA-RalN28 were untreated or treated with 10 μg/ml anisomycin for 30 or 60 min. FOXO4-T451 and JNK phosphorylations were analyzed on Western blot. The 60 min treatment of anisomycin induced a three-fold increase in FOXO4-T451 phosphorylation, both in the presence and absence of RalN28.
Our results thus far suggest a role for Ral in mediating H2O2-induced JNK activation in vivo. To test this directly, we expressed HA-JNK1 (not shown) or HA-JNK3 either in the absence or the presence of dominant-negative RalN28 and stimulated JNK activity by H2O2 treatment. Dominant-negative Ral inhibited, especially at later time points, the induction of JNK phosphorylation and activation by H2O2 (Figure 3E). Again, we tested whether activation of endogenous Ral would be sufficient to increase JNK activity. Indeed, as was shown for T451 phosphorylation, coexpression of active RalGEFs but not that of the inactive GEF increased JNK activity (Figure 3F). To analyze the specificity of the involvement of Ral in H2O2-induced JNK activation, we also tested whether Ral is involved in anisomycin-induced JNK activation. Consistent with a role for JNK in mediating T447/451 phosphorylation of FOXO4, anisomycin treatment also induced T451 phosphorylation. However, RalN28 did not block the anisomycin-induced phosphorylation of both JNK and T451 (Figure 3G). Therefore, we conclude that FOXO4 is phosphorylated by JNK at T447 and T451 and that JNK is differentially regulated following cellular stress: JNK activation following oxidative stress as generated by H2O2 treatment is mediated by the small GTPase Ral, whereas JNK activation following ER stress as generated by anisomycin treatment occurs independently of Ral.
We and others have previously shown that insulin signalling results in the translocation of FOXO from the nucleus to the cytosol (Biggs et al, 1999; Brownawell et al, 2001). Thus, we analyzed the effect of increased oxidative stress on FOXO4 localization and transcriptional activity. As was shown by others (Brunet et al, 2004), treatment of cells with H2O2 cultured in the presence of serum, when FOXO4 is predominantly localized in the cytosol, induced relocalization of FOXO4 from the cytosol to the nucleus (Figure 4A). Translocation induced by H2O2 appeared to be stochastic. For reasons that are not clear, cells appear to respond in an all-or-none fashion. An example of this is shown in Figure 4A (middle panel). To analyze the effect on transcriptional activity, we performed reporter assays using FOXO responsive promoters. Interestingly, we only observed a small but reproducible increase of FOXO transcriptional activity at low H2O2 concentrations, as measured by an increase in activity on the MnSOD promoter construct (Figure 4B) (Kim et al, 1999). Similar results were obtained using the 6xDBE or p27 promoter constructs, both of which are FOXO responsive promoters (Furuyama et al, 2000; Medema et al, 2000; data not shown). The decrease in FOXO transcriptional activity following overnight H2O2 treatment at higher concentrations is often accompanied by a decrease in HA-FOXO4 expression (Figure 4B, loading control). Whether this decrease in FOXO transcriptional activity observed at higher H2O2 concentrations is also due to PKB/c-Akt signalling, which is switched on at high concentrations of H2O2, or that other H2O2-induced modifications inhibit FOXO activity and/or expression in a dominant fashion, is at present unknown.
Figure 4.
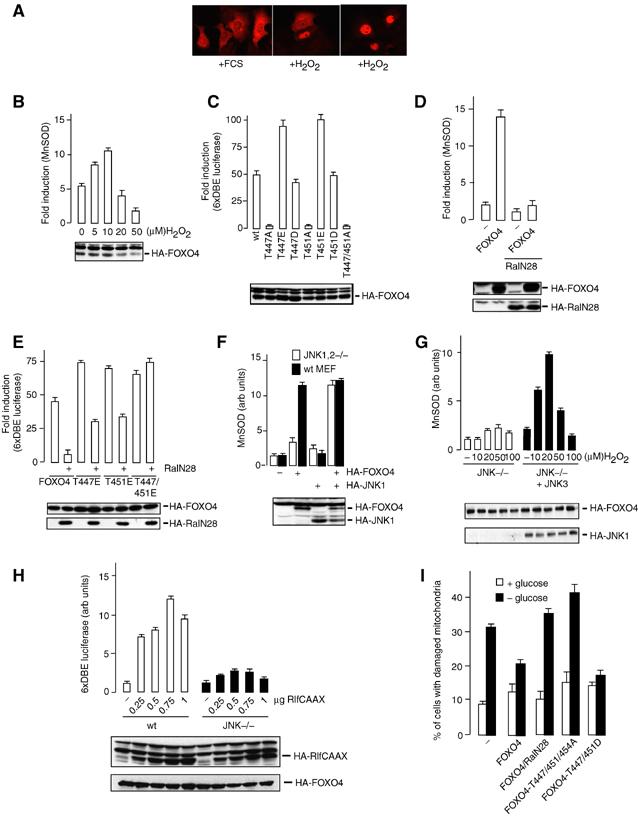
H2O2 induces nuclear translocation and activation of FOXO4. (A) DLD1 cells, transfected with HA-FOXO4, were maintained in the presence of serum and left untreated (left panel) or treated with 100 μM of H2O2 for 60 min (middle and right panels). Cells were fixed and HA-FOXO4 was stained. The middle panel shows differential response to oxidative stress with respect to localization. Same results were obtained using 200 or 400 μM of H2O2. (B) DLD1 cells, transfected with pSODLUC-3340 in the absence or presence of HA-FOXO4, were treated with indicated concentrations of H2O2 for 16 h and subjected to luciferase assays. Equal expression was tested by Western blot. Data represent the average of three independent experiments. (C) DLD1 cells were transfected with 6xDBE-luciferase together with the indicated constructs, and subjected to luciferase assays as described in panel B. (D) DLD-1 cells were transfected with pSODLUC-3340 in the absence or presence of HA-FOXO4 and absence or presence of dominant-negative Ral (RalN28), and subjected to luciferase assays as described in panel B. (E) A14 cells were transfected with 6xDBE-luciferase in the presence of HA-FOXO4 or the indicated mutant constructs and either in the presence or absence of dominant-negative Ral (RalN28), and subjected to luciferase assays as described in panel B. (F) JNK1,2−/− MEFs and wt MEFs were transfected with pSODLUC-3340 in the absence or presence of HA-FOXO4, HA-JNK1 or both, and subjected to luciferase assays as described in panel B. (G) JNK1,2−/− MEFs, transfected with pSODLUC-3340 with or without JNK3, were treated with indicated concentrations of H2O2 for 16 h, and subjected to luciferase assays as described in panel B. (H) wt MEFs and JNK1,2−/− were transfected with 6xDBE-luciferase together with indicated amounts of HA-RlfCAAX, and subjected to luciferase assays as described in panel B. (I) A14 cells were transfected with the indicated constructs and a puromycin selection vector. At 36 h after transfection, cells were put on 2 μg/ml puromycin and either cultured in medium containing FCS with glucose or medium with FCS lacking glucose. After 48 h of glucose deprivation, cells were harvested, stained with rhodamine-1,2,3 and analyzed for mitochondrial membrane stability.
To further demonstrate that phosphorylation of either T447 or T451 results in activation of FOXO4, we tested a series of T447 and T451 mutants that can no longer be phosphorylated at these sites or could mimic phosphorylation (Figure 4C). Mutating either T447 or T451 to alanine was already sufficient to almost completely block transcriptional activity. Importantly, both the phospho-mimicking T447E and the T451E mutant displayed enhanced transcriptional activity compared to wt FOXO4. Thus, these data suggest that phosphorylation of either T447 or T451 is sufficient to activate FOXO4 transcriptional activity. In agreement with a role for Ral, introduction of the dominant-negative RalN28 completely blocks FOXO4 activity (Figure 4D), whereas the phospho-mimicking mutants were either partially (T447E and T451E) or not inhibited by RalN28 coexpression (T447/451E; Figure 4E). These data indicate that the effect of Ral on FOXO4 transcriptional activity is entirely through the regulation of T447 and T451 phosphorylation. To confirm the role of JNK in transcriptional activation via T447/451 phosphorylation, we analyzed FOXO transcriptional activity in JNK1,2−/− cells. In these cells, FOXO activity was lower compared to wt MEFs, and reintroducing JNK, clearly enhanced FOXO4 transcriptional activity (Figure 4F). H2O2-induced activation of FOXO activity was also reduced in JNK1,2−/− MEFs. However, reintroduction of JNK3 in these cells restored stress-induced FOXO4 activity (Figure 4G). Previously, we reported that the effect of RlfCAAX on FOXO4-mediated transcription was sensitive to the amount of RlfCAAX used (De Ruiter et al, 2001). Here, we show that the effect of RlfCAAX on FOXO activity is also dependent on JNK, since RlfCAAX-induced FOXO activity is lowered in JNK−/− cells (Figure 4H). The dose-dependent effect of RlfCAAX shows similarities with the dose-dependent effect of H2O2 treatment.
Next, we tested whether the loss of transcriptional activity as a result of the T447/451A mutation in FOXO4 as measured by the reporter assays resulted in a change in FOXO function. Previously, we have shown that FOXOs can protect cells from glucose deprivation-induced mitochondrial membrane instability (Kops et al, 2002) and thus we tested whether the FOXO4 mutants were compromised in this respect. Inhibition of Ral signalling and expression of the T447/451A mutant reduced the ability of FOXO4 to protect cells from glucose deprivation, and consistent with this the phospho-mimicking T447/451D mutant displayed slightly enhanced protection (Figure 4I).
Thus, from these data, we conclude that H2O2 induces a translocation of FOXO4 from the cytosol to the nucleus and that this translocation is part of the mechanism whereby H2O2 induces transcriptional activation of FOXO4. Mutant analysis shows that this transcriptional activation involves T447 and T451 phosphorylation and is dependent on the presence of JNK.
Finally, we tested whether stimuli other than H2O2 that are known to influence intracellular ROS levels could activate FOXO transcriptional activity. TNFα is a cytokine that has been shown to increase intracellular H2O2 levels and likely concomitantly to increase cellular oxidative stress (Goossens et al, 1995). This increase in cellular ROS may mediate the cytotoxic action of TNFα, although the exact mechanism is largely unknown. For example, overexpression of antioxidants including catalase to reduce cellular H2O2 has been shown to enhance (Bai and Cederbaum, 2000) or reduce TNFα cytotoxicity (Wong et al, 1989). There are also reports that demonstrate unaltered TNFα cytotoxicity after overexpression of antioxidants (O'Donnell et al, 1995). Interestingly, TNFα has also been shown to increase the expression of MnSOD (Wong et al, 1989) and FOXOs can induce MnSOD expression (Kops et al, 2002). A14 cells are sensitive to TNFα, as measured by activation of NF-κB, but show no obvious signs of TNFα-induced cell death (data not shown). Treatment of A14 cells with increasing concentrations of TNFα induced a dose-dependent increase in FOXO4 transcriptional activity (Figure 5D). Similar to the H2O2-induced FOXO4 transcriptional activity, the TNFα-induced increase is mediated by Ral, as it is inhibited by expression of the dominant-negative RalN28. Moreover, it is mediated by JNK, as it is absent in JNK1,2−/− cells and can be restored by reintroducing JNK in JNK1,2−/− cells (Figure 5B). Importantly, TNFα-induced JNK activation involved an increase in cellular oxidative stress, as pretreatment with N-acetyl-L-cysteine (NAC), which enhances the scavenging of oxygen radicals, reduced TNFα-induced JNK activation (Figure 5C).
Figure 5.
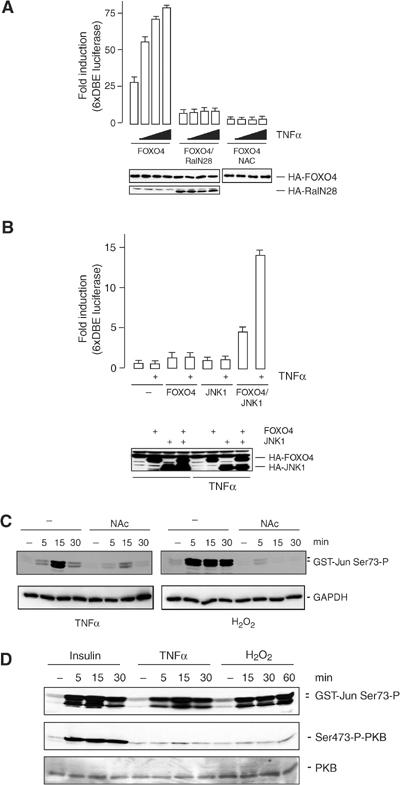
TNFα induces FOXO4 transcriptional activity. (A) A14 cells were transfected with the HA-FOXO4 with or without HA-RalN28, together with 6xDBE-luciferase and Tk-renilla. Cells were treated with increasing concentrations of TNFα (5, 10 and 20 ng/ml). Pretreatment with NAC was performed by adding 10 mM of NAC 16 h before treatment with TNFα. At 16 h after TNFα treatment, luciferase activity was measured. Data represent the average of three independent experiments. (B) JNK1,2−/− MEFs were transfected with HA-FOXO4 with or without HA-JNK1 together with 6xDBE-luciferase and Tk-renilla, and treated for 16 h with TNFα (20 ng/ml). Luciferase activity was measured as described in panel B. (C) A14 cells were treated with 10 mM NAC and the next day treated with TNFα (20 ng/ml) or H2O2 (100 μM) for the indicated time points. JNK activity was measured by a GST-Jun pull-down. (D) A14 cells were treated with TNFα (20 ng/ml), H2O2 (100 μM) or insulin (1 μg/ml) for the indicated time points. JNK activity was measured by means of a GST-Jun pull-down and PKB activity was monitored using the S473 phosphospecific antibody.
The ability to increase cellular oxidative stress is not unique to TNFα, and for several growth factors it has been suggested that a change in intracellular redox contributes to activation of downstream signalling pathways (Sundaresan et al, 1995; Bae et al, 1997). However, in general, these growth factors repress FOXO transcriptional activity through PKB-mediated nuclear exclusion. Thus, we compared the levels of PKB activation with the activation of JNK by the stimuli employed in this study. As already indicated by our initial experiments (Figure 1), H2O2 at relatively low concentrations induces T447/T451 phosphorylation, and consistently H2O2 induces considerable JNK activity, whereas little or no induction of PKB activity could be demonstrated (Figure 5D). Similar to H2O2, and consistent with the ability to induce FOXO transcriptional activity, TNFα induced considerable JNK activity, whereas only a small and transient increase in PKB activity was observed. In contrast, insulin induces a robust increase in PKB activity and represses FOXO transcriptional activity (Kops et al, 1999; data not shown), despite the induction of JNK activity comparable to TNFα and H2O2. This comparison therefore indicates that PKB activation can act dominantly over JNK activation and explains how TNFα and H2O2 result in FOXO activity.
A possible mechanism whereby the opposing effects of JNK- and PKB-mediated phosphorylation of FOXO4 can be integrated is through the regulation of binding of cofactors such as 14-3-3. Binding of 14-3-3 is suggested to function as cytoplasmic anchor for FOXO and thereby to inhibit FOXO transcriptional activity. Therefore, we tested whether phosphorylation of T447/451 would affect the ability of PKB-mediated 14-3-3 binding to FOXO4 (Figure 6A). We could not observe reproducible changes in insulin-induced 14-3-3 binding to FOXO4 and the FOXO4 mutants T447/451A and T447/451D. Also insulin-induced PKB-mediated phosphorylation of T28 and S193 did not appear significantly changed (Figure 6B). Thus, JNK-mediated phosphorylation of T447/451 appears to increase transcriptional activity, rather than relieve the inhibition by PKB imposed via 14-3-3 binding.
Figure 6.
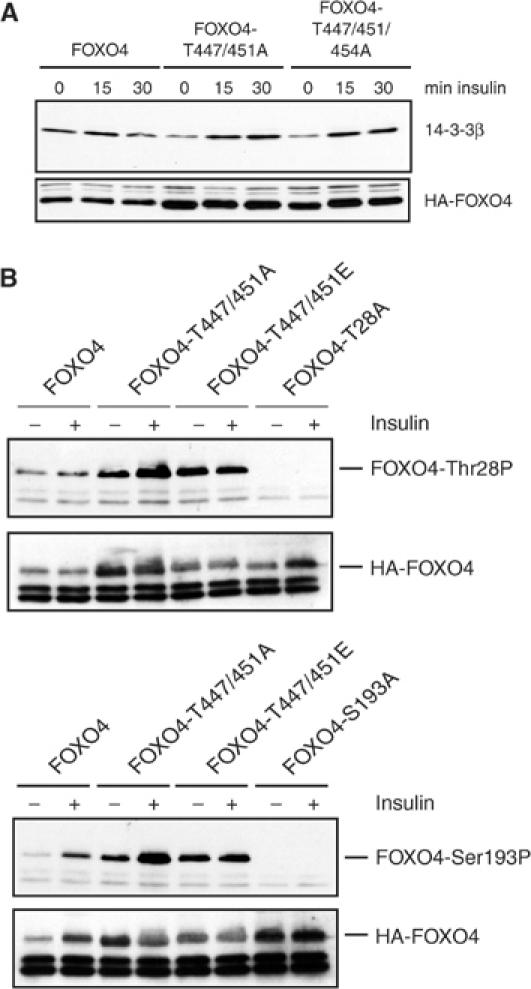
JNK phosphorylation does not affect 14-3-3 binding to FOXO. (A) A14 cells were transfected with the indicated constructs and stimulated with insulin (1 μg/ml) for the indicated time points. Binding of endogenous 14-3-3 to HA-FOXO4 and HA-FOXO4 mutants was assayed by immunoprecipitation of HA-FOXO4 followed by Western blotting using an antibody directed against 14-3-3β. (B) A14 cells were transfected with the indicated constructs and treated with insulin (1 μg/ml) for 30 min. Phosphorylation of T28 and S193 was assayed using phosphospecific antibodies. The specificity of these antibodies was demonstrated by including the FOXO4-Thr28A and FOXO4-S193A mutants respectively.
Discussion
Activation of PI-3K/PKB signalling decreases FOXO activity and thus the levels of FOXO target genes like MnSOD and catalase (Kops et al, 2002). MnSOD and catalase belong to a large and diverse family of antioxidant enzymes. Their regulation via PI-3K/PKB/FOXO signalling therefore implies that insulin, through this signalling cascade, may modulate the cellular ROS level. Consistent with this hypothesis, we have previously shown that FOXO-mediated upregulation of MnSOD expression results in considerable lowering of cellular ROS (Kops et al, 2002). Here, we demonstrate that an increase in ROS will enhance FOXO transcriptional activity, and thus functions as a feedback mechanism. An increase in ROS levels induces activation of the small GTPase Ral, which will in turn lead to the phosphorylation and activation of the stress kinase JNK. Active JNK induces the phosphorylation of T447 and T451 on FOXO4. Phosphorylation of these residues is essential for FOXO4 transcriptional activity as shown by mutational analysis. Consistent with this, H2O2 treatment increases FOXO transcriptional activity and translocation of FOXO4 from the cytoplasm to the nucleus and activation of the transcription factor. Activation of FOXO4 through T447/451 phosphorylation can now induce transcription of MnSOD and catalase, leading to a decrease in ROS levels. Thus, activation of FOXO4 by oxidative stress is part of a negative feedback loop to reduce the levels of oxidative stress in a cell, preventing damage to DNA, lipids and proteins (Figure 7).
Figure 7.
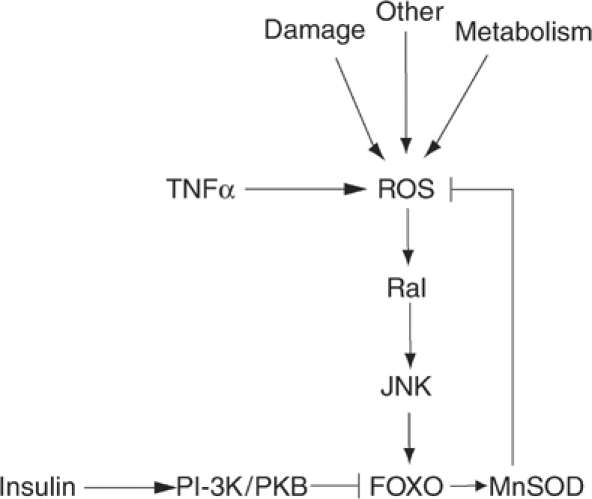
Model for FOXO-mediated control of cellular ROS levels. During periods of increased oxidative stress, FOXO4 is activated by Ral- and JNK-mediated phosphorylation on T447 and T451. Increased activity of FOXO4 will lead to induction of transcription of the antioxidant enzymes MnSOD and catalase, leading to a decrease of ROS levels. Thus, activation of FOXO4 by oxidative stress is part of a negative feedback loop to reduce the levels of ROS in a cell. Growth factors can balance cellular ROS through the activation of PKB or JNK, depending on the magnitude and kinetics of PKB and JNK activation. Cellular ROS can be increased through a variety of events including cellular metabolism and various forms of damage (DNA, protein).
The homeostasis mechanism for controlling ROS levels presented here is controlled by signalling pathways that can provide both negative (PI-3K/PKB) and positive (Ras/Ral/JNK) inputs on FOXO. Growth factors, including insulin, have the ability to regulate both pathways simultaneously. The question remains as to what would then determine the effective outcome of growth factor signalling of cells. H2O2 induces nuclear translocation in the presence of serum growth factors (Figure 4A), and we performed our transcriptional assays in the presence of serum growth factors. In the presence of serum, cells maintain a basal level of PKB activity and apparently H2O2 treatment even at relatively low concentrations can sufficiently activate Ral/JNK signalling in order to activate FOXO. Thus, when PKB activity is relatively low, for example as compared to insulin-induced PKB activity, the Ral/JNK signalling pathway acts opposite and dominant over PKB. At higher concentrations of H2O2, or for example following treatment of cells with insulin, PKB activity increases to a level at which it can act dominant over JNK activity. The importance of this differential activation of intracellular signalling pathways is further illustrated by our finding that TNFα, unlike insulin, increases FOXO transcriptional activity in A14 cells and this occurs with a concomitant strong activation of JNK and weak activation of PKB. If PKB activity outweighs the JNK activity, this will result in relocalization of FOXO to the cytosol. In addition, it has been suggested that PKB-mediated phosphorylation of FOXO also results in ubiquitination and consequent degradation of the FOXO proteins (Matsuzaki et al, 2003). This would be consistent with the reduced FOXO expression we frequently observe following long-term treatment with higher concentrations of H2O2.
Recent studies have also revealed another mechanism by which the activity of FOXOs can be regulated. In response to H2O2 treatment, FOXO3a and FOXO4 are substrates for acetylation by the acetylase transferases p300/CBP and subsequent deacetylation by SIRT1 and other deacetylases (Brunet et al, 2004; Motta et al, 2004; van der Horst et al, 2004). The results on the effect of acetylation and deacetylation on FOXO transcriptional activity are contradictory. Both negative (Brunet et al, 2004; van der Horst et al, 2004) and positive (Motta et al, 2004) regulations of FOXO transcriptional activity by acetylation have been reported. Taken together and consistent with our own data on the role of acetylation in regulating FOXO4 (van der Horst et al, 2004), the data presented here suggest that during periods of low oxidative stress, FOXOs are initially activated by JNK-mediated phosphorylation, but thereafter or at higher concentrations or longer periods of time inactivated by acetylation and/or ubiquitination. Deacetylation of FOXO by Sir2 can thus prolong the FOXO activity induced by H2O2 treatment in order to ensure full activation of the antioxidant targets of FOXO. To test this hypothesis, we are currently investigating the kinetics of H2O2-induced phosphorylation and acetylation of FOXO and the effect on FOXO transcriptional activity.
The human FOXO family of transcription factors consists of three different isoforms. Here we show the regulation of FOXO4 via T447/451 phosphorylation. Clustal W alignment of FOXO4 with the other isoforms, FOXO1 and FOXO3a, did not reveal conserved T447/451 phosphorylation sites in the C-terminal parts of the protein. However, for example for FOXO3a, it has been shown that several sites within the C-terminal part of the protein can be phosphorylated upon H2O2 treatment (Brunet et al, 2004). These phosphorylation sites resemble potential JNK phosphorylation sites. We are currently investigating whether JNK can induce a phosphorylation-dependent activation of FOXO1 and FOXO3a upon treatment with H2O2, and which of the phosphorylation sites are involved, and thus are functionally equivalent to T447/451.
The model that derives from the data presented here bears striking similarities to the proposed role and regulation of DAF-16, the Caenorhabditis elegans homolog of mammalian FOXO. In C. elegans, a variety of stresses including oxidative stress have been shown to induce translocation of DAF-16/GFP from the cytosol to the nucleus (Henderson and Johnson, 2001). Oxidative stress strongly induces the expression of SOD-3, the C. elegans homologue of MnSOD, in a DAF-16-dependent manner (Honda and Honda, 1999). Thus, both in C. elegans and mammalian cells, oxidative stress activates DAF-16/FOXO to act in a negative feedback control of ROS. In C. elegans, little is known as to what mediates stress-induced activation of DAF-16. Both Ral and JNK isoforms are present in C. elegans and it will be of interest to investigate whether these are involved in DAF-16 control by certain forms of oxidative stress. Interestingly, it has recently been demonstrated that in Drosophila, increased JNK signalling also increases oxidative stress resistance (Wang et al, 2003). Similarly, dFOXO has also been shown to increase oxidative stress resistance in Drosophila (Junger et al, 2003). In the regulation of cell shape in Drosophila, there is genetic evidence for a connection between dRal and dJNK (Sawamoto et al, 1999). However, here dRal appears to regulate cell shape changes through inhibition of the JNK pathway, which is in contrast to what we observe here. The genetic interaction between FOXO, Ral and JNK in regulating oxidative stress resistance has not been examined in Drosophila yet. However, the JNK-mediated FOXO activation as described here may likely be a mechanism whereby increased oxidative stress resistance is also achieved in Drosophila.
Studies in both C. elegans and Drosophila support a link between stress resistance and prolonged longevity. For example, in both organisms, removing oxidants by overexpressing superoxide dismutase and catalase resulted in animals with a prolonged lifespan (Honda and Honda, 1999; Taub et al, 1999). In C. elegans, prolonged lifespan, protection against oxidative stress and regulation of superoxide dismutase and catalase are all dependent on the status of DAF-16 (Murphy et al, 2003). In mammals, the pathways involved in the basic controls for aging will likely be more complex. Recently, FOXO3a has been linked to the important mammalian gerontogene p66shc. In cells lacking p66shc, the activity of FOXO3a is increased. H2O2 treatment of these p66shc−/− cells reduced the activation of PKB and consequent PKB-mediated FOXO3a inactivation, indicating that the redox-dependent FOXO inactivation is reduced (Nemoto and Finkel, 2002). Yet, a mechanism as to how p66Shc ablation could result in FOXO activation, rather than solely preventing its inhibition in an indirect manner, remains to be determined. Our results may provide such a possible mechanism. It has been shown that p66Shc inhibits the p46/p52Shc isoforms. The latter isoforms are involved in growth factor-induced Ras and MAPK activation. We have shown that insulin-induced Ras activation is mediated almost exclusively by p46/p52 Shc (Pronk et al, 1994) and insulin-induced Ral activation is mediated by Ras (Wolthuis et al, 1998). Thus, loss of p66Shc would be expected to specifically enhance the Ras/Ral signalling branch following insulin stimulation. A major consequence would be enhanced FOXO activity due to a shift from negative regulatory signalling (PI-3K/PKB) to positive regulatory signalling (Ras/Ral). This possibility is currently under investigation.
In summary, our data describe a mechanism by which FOXO activity is regulated by oxidative stress, resulting in prevention of cellular damage. They bring us one step closer to unravelling the mechanisms that control aging in mammalian cells.
Materials and methods
Cell culture, transfection and treatment
A14 cells (mouse NIH3T3 cells overexpressing the human insulin receptor) (Burgering and Coffer, 1995), wt MEFs, JNK9 MEFs (JNK1−/−, JNK2−/− MEFs) (Sabapathy et al, 1999), HEK293T cells and mouse C2C12 myoblast cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with L-glutamine, penicillin/streptomycin and 10% FCS. DLD1 human colon carcinoma cells were maintained in RPMI 1640 medium supplemented with L-glutamine, penicillin/streptomycin and 10% FCS. For luciferase assays, cells were cultured in six-well plates. For the GST-RalBD pull-down assay, cells were cultured in 90 mm dishes. For all other experiments, cells were cultured in 60 mm dishes. HEK293T cells, wt MEFs, JNK9 MEFs and DLD1 cells were transiently transfected using FuGENE6 reagent according to the manufacturer (Roche). A14 cells were transfected using the calcium phosphate method. Total amounts of transfected DNA were equalized using pBluescript KSII+. Anisomycin, EGF, insulin, TNFα and H2O2 were added at 10 μg/ml, 20 ng/ml, 1 μg/ml, 20 ng/ml and 20–500 μM, respectively, as indicated. For luciferase assays, H2O2 was added at 5–20 μM overnight.
Plasmids and recombinant proteins
The following plasmids have been described before: pMT2-HA-FOXO4 (Kops et al, 1999), pMT2-HA-FOXO4-T447A, pMT2-HA-FOXO4-T451A, pMT2-HA-FOXO4-T447/451/454A, pMT2-HA-FOXO4-T447D, pMT2-HA-FOXO4-T451D (De Ruiter et al, 2001), pMT2-HA-RalN28, pMT2-HA-Rlf-CAAX, pMT2-HA-Rlf-CAAXΔGEF, pSVE-RASV12 (Wolthuis et al, 1997), pMT2-HA-RalGEF2 (de Bruyn et al, 2000), pMT2-HA-JNK1, pMT2-HA-JNK3 (de Groot et al, 1997), 6xDBE-luciferase (Furuyama et al, 2000) and pSODLUC-3340 (Kim et al, 1999). pRL-Tk (Tk Renilla luciferase) was purchased from Promega.
pMT2-HA-FOXO4-T451E and pMT2-HA-FOXO4-T447E were generated using mutagenesis PCR. pcDNA3.1-myc-FOXO4 was created by ligating a Klenow blunted SalI/NotI fragment from pMT2-HA-FOXO4 into Klenow blunted BamHI/NotI-digested pcDNA3.1-myc.
GST-FOXO4(C) and GST-FOXO4-T447A/T451A(C) (Kops et al, 1999), GST-Jun (de Ruiter et al, 2000) and GST-RalBD (Wolthuis et al, 1998) have been described before.
Antibodies
Monoclonal 12CA5 and 9E10 antibodies were produced using hybridoma cell lines. Monoclonal antibody against Ral was obtained from Transduction Laboratories. Phosphospecific polyclonal antibodies recognizing PKB-S473, FOXO4-S193 and Jun-S73 were obtained from Cell Signaling. Phosphospecific polyclonal antibody against FOXO3a-T32 was obtained from Upstate Technology. Polyclonal HA antibody and polyclonal 14-3-3β (K19) antibody were obtained from Santa Cruz.
The phosphospecific antibody against the phosphorylated T447 was made by using the peptide KALGTpPVLTPPTEAC to immunize rabbits (Sigma). The phosphospecific antibody against the phosphorylated T451 was generously provided by Cell Signaling.
Immunoprecipitations and Western blots
Cells were lysed in RIPA buffer (50 mM Tris–HCl (pH 7.5), 0.1% NP-40, 0.5% deoxycholate, 10 mM EDTA, 150 mM NaCl, 50 mM NaF, 1 μM leupeptin and 0.1 μM aprotinin). Lysates were cleared for 10 min at 14 000 r.p.m. at 4°C, and incubated for 2 h at 4°C with either 1 μl 12CA5 (HA) or 1 μl 9E10 (myc) and 50 μl of prewashed protein A beads. Immunoprecipitations were washed four times in lysis buffer, cleared of all supernatant and 25 μl of 1 × Laemmli sample buffer was added. Samples were subjected to SDS–PAGE and transferred to PVDF (Perkin Elmer). Western blot analysis was performed under standard conditions, using indicated antibodies. For phosphospecific antibodies, membranes were blocked in 1% BSA and washed in TBS–Tween.
In vitro kinase assay
GST-FOXO4 and GST-FOXO4-T447/451A were precoupled to glutathione beads and washed twice with reaction buffer (50 mM HEPES pH 7.4, 15 mM MgCl2 and 200 μM sodium vanadate). For kinase reactions, the beads were incubated in kinase buffer (containing 100 μM ATP or 5 μM ATP and 10 μCi [γ-32P]ATP per reaction) at 30°C for 30 min, resuspended in sample buffer and analyzed by SDS–PAGE followed by autoradiography.
[32P]orthophosphate labelling
In vivo labelling of A14 cells transfected with HA-FOXO4 was performed as described previously (Burgering and Coffer, 1995).
Determination of Ral-GTP
Ral-GTP levels were determined using a Gst-RalBD pull-down assay as described previously (Wolthuis et al, 1998).
Immunofluorescence
DLD1 cells were cultured on coverslips, transfected with 1 μg of pMT2-HA-FOXO4 and fixed in 4% paraformaldehyde. Cells were permeabilized with 0.1% Triton X-100 in PBS, and nonspecific binding was blocked with 0.5% BSA in PBS for 45 min. Incubation with the HA polyclonal antibody for a period of 1 h was followed by 1 h incubation with anti-rabbit-CY3 secondary antibody. Coverslips were washed and mounted on glass slides using Immuno-Mount (Shandon, Pittsburgh, PA).
Luciferase assays
Cells were transfected with a reporter construct bearing six canonical FOXO binding sites (6xDBE-luciferase) or bearing a −3340 bp promoter fragment of the human SOD2 gene for MnSOD (pSODLUC-3340). Cells were cotransfected with indicated constructs. Transfections were performed in triplicate. Luciferase counts were normalized using Tk Renilla luciferase. After overnight treatment with H2O2 or TNFα, or 48 h post-transfection, cells were lysed in passive lysis buffer (PLB) and luciferase activity was analyzed using a luminometer and dual-luciferase assay kit according to the manufacturer (Promega).
Measurement of mitochondrial membrane integrity
Cells were glucose deprived for 48 h by adding DMEM medium lacking glucose and pyruvate, but supplemented with 8% FCS. Note that owing to the small amount of glucose in FCS, glucose amounts are estimated to be about 10- to 20-fold less in this medium than under normal culture conditions. Cells were digested with trypsin and incubated with 10 μg/ml rhodamine-1,2,3 (Scaduto and Grotyohann, 1999) at 37°C for 30 min. After two washes with PBS, mitochondrial membrane staining was measured by standard flow cytometry.
Acknowledgments
We thank Arjan Brenkman, Fried Zwartkruis, Armando van der Horst, Pieter van den Heuvel, Leo Price and Hans van Dam for discussions and critical reading of the manuscript. We thank Cell Signaling for providing samples of Thr451P antibody and Erwin Wagner for providing JNK−/− cells. This research was supported by grants from the Dutch Cancer Foundation (KWF) and the Netherlands Organization for Scientific Research (NWO).
References
- Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG (1997) Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem 272: 217–221 [PubMed] [Google Scholar]
- Bai J, Cederbaum AI (2000) Overexpression of catalase in the mitochondrial or cytosolic compartment increases sensitivity of HepG2 cells to tumor necrosis factor-alpha-induced apoptosis. J Biol Chem 275: 19241–19249 [DOI] [PubMed] [Google Scholar]
- Biggs WH III, Meisenhelder J, Hunter T, Cavenee WK, Arden KC (1999) Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA 96: 7421–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownawell AM, Kops GJ, Macara IG, Burgering BM (2001) Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol Cell Biol 21: 3534–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015 [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ (1995) Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 376: 599–602 [DOI] [PubMed] [Google Scholar]
- Burgering BM, Medema RH (2003) Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J Leukoc Biol 73: 689–701 [DOI] [PubMed] [Google Scholar]
- Clopton DA, Saltman P (1995) Low-level oxidative stress causes cell-cycle specific arrest in cultured cells. Biochem Biophys Res Commun 210: 189–196 [DOI] [PubMed] [Google Scholar]
- de Bruyn KM, de Rooij J, Wolthuis RM, Rehmann H, Wesenbeek J, Cool RH, Wittinghofer AH, Bos JL (2000) RalGEF2, a pleckstrin homology domain containing guanine nucleotide exchange factor for Ral. J Biol Chem 275: 29761–29766 [DOI] [PubMed] [Google Scholar]
- de Groot RP, van Dijk TB, Caldenhoven E, Coffer PJ, Raaijmakers JA, Lammers JW, Koenderman L (1997) Activation of 12-O-tetradecanoylphorbol-13-acetate response element- and dyad symmetry element-dependent transcription by interleukin-5 is mediated by Jun N-terminal kinase/stress-activated protein kinase kinases. J Biol Chem 272: 2319–2325 [DOI] [PubMed] [Google Scholar]
- De Ruiter ND, Burgering BM, Bos JL (2001) Regulation of the Forkhead transcription factor AFX by Ral-dependent phosphorylation of threonines 447 and 451. Mol Cell Biol 21: 8225–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ruiter ND, Wolthuis RM, van Dam H, Burgering BM, Bos JL (2000) Ras-dependent regulation of c-Jun phosphorylation is mediated by the Ral guanine nucleotide exchange factor–Ral pathway. Mol Cell Biol 20: 8480–8488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T (2000) Redox-dependent signal transduction. FEBS Lett 476: 52–54 [DOI] [PubMed] [Google Scholar]
- Furuyama T, Nakazawa T, Nakano I, Mori N (2000) Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J 349: 629–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens V, Grooten J, De Vos K, Fiers W (1995) Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci USA 92: 8115–8119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE (2001) daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol 11: 1975–1980 [DOI] [PubMed] [Google Scholar]
- Honda Y, Honda S (1999) The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J 13: 1385–1393 [PubMed] [Google Scholar]
- Junger MA, Rintelen F, Stocker H, Wasserman JD, Vegh M, Radimerski T, Greenberg ME, Hafen E (2003) The Drosophila Forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J Biol 2: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HP, Roe JH, Chock PB, Yim MB (1999) Transcriptional activation of the human manganese superoxide dismutase gene mediated by tetradecanoylphorbol acetate. J Biol Chem 274: 37455–37460 [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419: 316–321 [DOI] [PubMed] [Google Scholar]
- Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM (1999) Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature 398: 630–634 [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 114: 2903–2910 [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A (2003) Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci USA 100: 11285–11290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM (2000) AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404: 782–787 [DOI] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L (2004) Mammalian SIRT1 represses forkhead transcription factors. Cell 116: 551–563 [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424: 277–283 [DOI] [PubMed] [Google Scholar]
- Nemoto S, Finkel T (2002) Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 295: 2450–2452 [DOI] [PubMed] [Google Scholar]
- O'Donnell VB, Spycher S, Azzi A (1995) Involvement of oxidants and oxidant-generating enzyme(s) in tumour-necrosis-factor-alpha-mediated apoptosis: role for lipoxygenase pathway but not mitochondrial respiratory chain. Biochem J 310 (Part 1): 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk GJ, de Vries-Smits AM, Buday L, Downward J, Maassen JA, Medema RH, Bos JL (1994) Involvement of Shc in insulin- and epidermal growth factor-induced activation of p21ras. Mol Cell Biol 14: 1575–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF (1999) Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech Dev 89: 115–124 [DOI] [PubMed] [Google Scholar]
- Sawamoto K, Winge P, Koyama S, Hirota Y, Yamada C, Miyao S, Yoshikawa S, Jin MH, Kikuchi A, Okano H (1999) The Drosophila Ral GTPase regulates developmental cell shape changes through the Jun NH(2)-terminal kinase pathway. J Cell Biol 146: 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaduto RC Jr, Grotyohann LW (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 76: 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford RE, Innes CL, Sieber SO, Heinloth AN, Leadon SA, Paules RS (2001) The Ataxia telangiectasia gene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts. J Biol Chem 276: 21951–21959 [DOI] [PubMed] [Google Scholar]
- Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T (1995) Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270: 296–299 [DOI] [PubMed] [Google Scholar]
- Taub J, Lau JF, Ma C, Hahn JH, Hoque R, Rothblatt J, Chalfie M (1999) A cytosolic catalase is needed to extend adult lifespan in C. elegans daf-C and clk-1 mutants. Nature 399: 162–166 [DOI] [PubMed] [Google Scholar]
- van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM (2004) FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J Biol Chem 279: 28873–28879 [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H (2003) JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev Cell 5: 811–816 [DOI] [PubMed] [Google Scholar]
- Wolthuis RM, de Ruiter ND, Cool RH, Bos JL (1997) Stimulation of gene induction and cell growth by the Ras effector Rlf. EMBO J 16: 6748–6761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolthuis RM, Zwartkruis F, Moen TC, Bos JL (1998) Ras-dependent activation of the small GTPase Ral. Curr Biol 8: 471–474 [DOI] [PubMed] [Google Scholar]
- Wong GH, Elwell JH, Oberley LW, Goeddel DV (1989) Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell 58: 923–931 [DOI] [PubMed] [Google Scholar]