

Abstract
Butyrate response factor (BRF1) belongs to the Tis11 family of CCCH zinc-finger proteins, which bind to mRNAs containing an AU-rich element (ARE) in their 3′ untranslated region and promote their deadenylation and rapid degradation. Independent signal transduction pathways have been reported to stabilize ARE-containing transcripts by a process thought to involve phosphorylation of ARE-binding proteins. Here we report that protein kinase B (PKB/Akt) stabilizes ARE transcripts by phosphorylating BRF1 at serine 92 (S92). Recombinant BRF1 promoted in vitro decay of ARE-containing mRNA (ARE-mRNA), yet phosphorylation by PKB impaired this activity. S92 phosphorylation of BRF1 did not impair ARE binding, but induced complex formation with the scaffold protein 14-3-3. In vivo and in vitro data support a model where PKB causes ARE-mRNA stabilization by inactivating BRF1 through binding to 14-3-3.
Keywords: exosome, insulin, mRNA turnover, PKB, zinc-finger protein
Introduction
The stability of mRNA varies considerably among different species of transcripts and is in many cases regulated in a complex fashion in response to external stimuli. Short-lived transcripts carry cis elements that appear to regulate the access to the mRNA decay machinery. A major element is the so-called AU-rich element (ARE), which is located in the 3′ untranslated region (3′UTR) of many short-lived transcripts from cytokines, proto-oncogenes, growth factors or cell cycle regulators (Shaw and Kamen, 1986; Chen and Shyu, 1995; Bakheet et al, 2001). AREs promote deadenylation and decapping (Gao et al, 2001), followed by degradation of the mRNA body (Shyu et al, 1991; Xu et al, 1997). Mammalian ARE-containing mRNA (ARE-mRNA) is thought to be degraded mainly by the exosome, a multiprotein complex containing 3′–5′ exonucleases and helicases (Chen et al, 2001; Mukherjee et al, 2002). Stabilization of short-lived ARE-containing transcripts by exogenous signals leads to a rapid accumulation of mRNA, with a consequent increase of protein levels. Physiological examples are found in T-cells activated by immune stimulation (Lindsten et al, 1989), mast cells responding to IgE-linked allergens (Wodnar-Filipowicz et al, 1989; Wodnar-Filipowicz and Moroni, 1990), or macrophages stimulated by IL-1/TNFα (Huang et al, 2000), where increased cytokine production occurs. Deregulated ARE-dependent mRNA turnover can contribute to oncogenic transformation (Schuler and Cole, 1988; Nair et al, 1994; Stoecklin et al, 2003), inflammation (Carballo et al, 1998) and immunopathology (Kontoyiannis et al, 1999), underlying the physiological relevance of this process. Also, stress stimuli, including UV exposure (Gorospe et al, 1998; Wang et al, 2000), heat shock (Laroia et al, 1999) and hypoxia (Paulding and Czyzyk-Krzeska, 2000), can lead to stabilization of ARE-mRNA. In addition to mRNA turnover, the ARE has been reported to regulate translation (Kontoyiannis et al, 1999; Piecyk et al, 2000).
Several signal transduction pathways have been implicated in mRNA decay control. Activation of stress-induced c-jun N-terminal kinase (JNK) (Chen et al, 1998; Ming et al, 1998), p38 mitogen-activated protein kinase (MAPK) (Dean et al, 1999; Winzen et al, 1999; Brook et al, 2000; Clark et al, 2003; Frevel et al, 2003), MAPKAP kinase 2 (MK2) (Neininger et al, 2002), phosphatidylinositol 3-kinase (PI3-K) (Ming et al, 2001) and wnt-β-catenin pathway (Briata et al, 2003) has been shown to trigger stabilization of various transcripts.
Several ARE-binding proteins (AUBPs) affecting mRNA turnover have been identified. HuR, a member of the embryonic-lethal abnormal vision (ELAV in Drosophila melanogaster) family of RNA-binding proteins (Ma et al, 1996), stabilizes ARE-containing transcripts (Fan and Steitz, 1998). AUF1 (hnRNPD) exerts a stabilizing as well as a destabilizing function depending on cell type (Zhang et al, 1993; Chen et al, 2002) and isoform (Raineri et al, 2004). The Tis11 protein family members tristetraprolin (TTP) and butyrate response factors (BRF1 and BRF2) share a highly conserved CCCH tandem zinc-finger (Zn-finger) motif that binds RNA (Lai et al, 1999). TTP was initially identified by the Blackshear group as a positive regulator of TNFα mRNA decay (Carballo et al, 1998) by studying the autoimmune-like phenotype of TTP knockout (k.o.) mice. Subsequently, it was found to act by targeting ARE-mRNA to the exosome (Chen et al, 2001). BRF1 was recently identified by a functional genetic screen aimed at finding genes responsible for ARE-dependent decay (Stoecklin et al, 2002). Interestingly, BRF1 has been found to be a circadian gene (Storch et al, 2002) and its k.o. is lethal at embryonic day 11 (Stumpo et al, 2004). Both TTP and BRF1 can stimulate deadenylation (Lai et al, 2003). TIA/TIAR are RNA recognition motif (RRM)-containing proteins that regulate translational silencing of TNFα mRNA and recruitment into stress granules (Gueydan et al, 1999; Piecyk et al, 2000). Lastly, KSRP, a K homology (KH)-motif RNA-binding protein originally described as a neuronal specific splicing enhancer of c-src (Min et al, 1997), has been recently demonstrated to be an ARE-mRNA-specific destabilizing factor both in vitro and in vivo (Gherzi et al, 2004). An ARE-binding helicase has been described by Tran et al (2004).
AUBPs are obvious candidate targets for signaling pathways regulating mRNA turnover. HuR has been shown to act in concert with p38 in stabilizing IL-3 mRNA (Ming et al, 2001), although direct phosphorylation of HuR was not detected. TTP was found to be phosphorylated by p38, which inhibited its ARE-binding activity (Carballo et al, 2001; Zhu et al, 2001). In addition, the extracellular signal-related kinase 2 (ERK2) (Taylor et al, 1995) and MK2 (Mahtani et al, 2001) are able to phosphorylate TTP in vitro. Phosphorylation of TTP by MK2 leads to 14-3-3 binding, inhibition of TTP activity and exclusion of TTP from stress granules (Johnson et al, 2002; Chrestensen et al, 2003; Stoecklin et al, 2004). 14-3-3 may also interact with BRF1, as suggested by results from a yeast two-hybrid screening (Bustin and McKay, 1999) and from GST-pulldown experiments (Johnson et al, 2002).
Considerable progress in the analysis of mRNA turnover has been made by the development of in vitro decay systems. As control can be exercised over the choice of reagents to be added/omitted, different aspects of post-transcriptional regulation, including deadenylation, decapping and degradation of ARE-containing RNA, can be studied (Ford et al, 1999; Chen et al, 2000; Gao et al, 2001). In vitro AREs destabilize RNA through binding of AUBPs such as KSRP and TTP, which in turn recruit both the exosome and the deadenylase, thus targeting the RNA for exosomal decay (Chen et al, 2001; Lai et al, 2003; Gherzi et al, 2004). In vitro systems therefore provide a powerful tool to study the regulation of ARE-dependent decay.
Here we show that BRF1 is able to promote ARE-dependent decay in an in vitro system, and that this activity is strongly reduced by phosphorylation of BRF1 by protein kinase B (PKB) at serine 92 (S92). Using a phospho-specific antipeptide antibody, we present evidence that BRF1 phosphorylation at S92 mediates insulin-induced mRNA stabilization in vivo. Furthermore, BRF1 phosphorylation was found to provide a docking site to 14-3-3, suggesting that this protein may sequester BRF1 from the cellular decay-promoting machinery.
Results
Activated PKB stabilizes ARE-containing reporter mRNA
Previously, we have shown that activated PI3-K is able to stabilize ARE-containing reporter mRNA upon transfection into NIH 3T3 cells (Ming et al, 2001). We tested whether PKB, a downstream effector of PI3-K, mediates this effect. An activated form of PKB (m/pPKB) (Andjelkovic et al, 1997) was transfected together with a tetracycline-sensitive β-globin reporter construct containing the ARE of IL-3 (Tet-β-globin-IL3UTR) (Stoecklin et al, 2002) into NIH 3T3 B2A2-23 mouse fibroblasts (Xu et al, 1998). These cells stably express the Tet-responsive transcriptional activator tTA. When transcription of the reporter was inhibited by the addition of tetracycline, rapid degradation of the message with a half-life of less than 1 h was observed, while the reporter remained stable in the presence of activated PKB (Figure 1A and B). Activation of PKB was therefore sufficient to stabilize the ARE reporter transcript.
Figure 1.

ARE-mRNA stabilization by PKB. (A) The Tet-β-globin-IL3UTR reporter gene was transfected alone (lanes 1–3) or in combination with constitutively activated m/pPKB (lanes 4–6). After 24 h, transcription was stopped by addition of doxycycline. Cytoplasmic RNA was isolated at the indicated time points and processed for northern blotting. (B) The graph shows the quantifications of five independent decay assays normalized to the actin signal. Standard errors are shown unless too small to be represented.
BRF1 is phosphorylated at S92 in vitro
Inspection of the coding sequence of BRF1 (SwissProt database; accession number Q07352) revealed the presence of two overlapping RXRXXS motifs, including S90 and S92, corresponding to PKB consensus phosphorylation sites (Alessi et al, 1996). Interestingly, this PKB recognition motif is highly conserved in human, mouse and Xenopus leavis BRF1 (Figure 2A), but it is not present in TTP, although these closely related proteins show considerable homology throughout other regions.
Figure 2.

PKB phosphorylates BRF1 on S92. (A) Alignment of BRF1 sequences from human, mouse, rat and X. leavis. S90 and S92 are indicated. (B) In vitro phosphorylation reactions were performed for the times indicated using 40 ng of activated recombinant PKB and 20 ng of either full length rBRF1-wt, a N-terminal fragment (aa 3–110), the Zn-finger domain (aa 111–179) or a C-terminal fragment (aa 180–338). The asterisk denotes the full-length BRF1 protein, lower bands are degradation products. (C) In vitro phosphorylation reactions were performed for the times indicated, using 40 ng of activated recombinant PKB and 20 ng of either rBRF1-wt, rBRF1-S90A or rBRF1-S92A.
To address the question whether BRF1 is indeed a substrate of PKB, we first performed in vitro phosphorylation experiments. Either full-length recombinant BRF1 or three fragments consisting of the N-terminus (aa 3–110), the central Zn-finger domain (aa 111–179) or the C-terminus of BRF1 (aa 180–338) were tested as substrates for PKBβ. Only the full-length rBRF1 and the N-terminal fragment containing S90 and S92 proved to be efficiently phosphorylated (Figure 2B, lanes 5–12).
We used the N-terminal fragment (aa 3–110) of BRF1 to identify the site phosphorylated by PKB via mass spectrometry (see Materials and methods). This revealed that BRF1 is preferentially phosphorylated at S92. Accordingly, a peptide containing the S92 residue (DSRFRDRSFSEG) was phosphorylated in vitro by purified activated PKBα and β at 60% of the rate of the GSK3 peptide (RPRTSSFAEG), which is the best-characterized substrate of this kinase (data not shown). To investigate whether S92 is indeed the major PKB phosphorylation site, we mutated S90 or S92 to alanine (S90A, S92A), and tested the recombinant proteins for substrate activity. In line with the mass spectrometry data, BRF1-wt and BRF1-S90A were equally phosphorylated by PKB (Figure 2C, lanes 5–12), whereas phosphorylation was considerably reduced by the S92A mutation (Figure 2C, lanes 13–16). It appears that in the absence of the S92 phosphorylation site PKB can phosphorylate S90, which represents an alternative consensus site.
BRF1 S92 phosphorylation regulates ARE-dependent RNA decay in vitro
We were curious to see whether BRF1 may show similar in vitro decay activity as reported for its close homolog TTP (Chen et al, 2001), as this would allow us to study the nature of the S92 phosphorylation under controlled in vitro conditions. We co-incubated S100 extracts from slowC cells, a line lacking BRF1 expression as the result of frameshift mutations in both alleles (Stoecklin et al, 2002), with a radio-labeled RNA containing the 59 nt long ARE from IL-3 (AREIL3). While AREIL3 was stable in the presence of slowC extracts (Figure 3A, lanes 1–4), it decayed rapidly when 5 ng or more of rBRF1 was added (Figure 3A, lanes 5–20). As a control for both ARE specificity of the observed decay and equal loading, a 250 nt RNA containing the IL-3 3′UTR lacking the ARE (ΔARE) was added at the onset of the reaction. No degradation of the ΔARE control could be observed under all conditions tested. We concluded that rBRF1 is able to promote rapid RNA decay in this ARE-dependent in vitro system.
Figure 3.
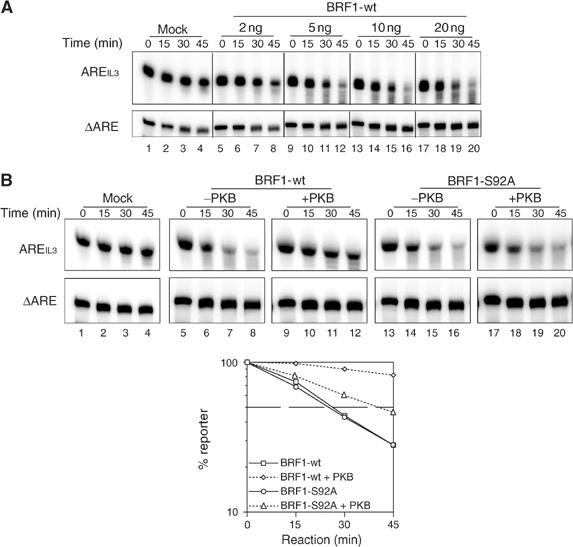
BRF1 S92 phosphorylation regulates ARE-dependent RNA decay in vitro. (A) Radioactively labeled AREIL3 and ΔARE transcripts were incubated together in S100 extract from slowC cells. Decay reactions were performed at 37°C in the presence of increasing amounts of rBRF1-wt, stopped at the indicated time points and resolved on a 10% urea-polyacrylamide gel. ΔARE transcripts served as specificity and loading control. (B) Decay reactions were carried out in the absence (lanes 1–4) or in the presence of 20 ng of rBRF1-wt (lanes 5–12) or rBRF1-S92A (lanes 13–20) after pretreatment with buffer (lanes 5–8/13–16) or activated PKB (lanes 9–12/17–20). The graph below shows quantification of at least four independent experiments after normalizing to the ΔARE signal. Standard error bars were too small to be represented.
We next investigated whether phosphorylation of rBRF1 by PKB would affect the in vitro decay rate. Unphosphorylated rBRF1-wt was compared to rBRF1-wt pretreated with activated PKB for 30 min (Figure 3B, lanes 5–12). A striking reduction of AREIL3 decay was observed with phospho-rBRF1-wt (p-rBRF1-wt) as compared to unphosphorylated rBRF1-wt. To check whether this effect was due to phosphorylation of BRF1 at S92, we performed the same experiment using the rBRF1-S92A mutant. Again, rBRF1-S92A promoted degradation of AREIL3 (Figure 3B, lanes 13–16), yet the mutant appeared refractory to inhibition by PKB (Figure 3B, lanes 17–20). Quantification (Figure 3B, lower panel) revealed an intermediate decay pattern, which may result from alternative phosphorylation at S90 (see also Figure 2C). From these data, we concluded that, in vitro, the decay-promoting activity of BRF1 is inhibited by phosphorylation at S92.
BRF1 is phosphorylated at S92 in vivo
To extend this study to endogenous BRF1, we first raised an antibody directed to a trisdekapeptide containing phosphorylated S92 (FRDRSFpSEGGERL). Affinity-purified antibody recognized rBRF1 phosphorylated by PKB with a high specificity (Figure 4A). As an additional control, we preincubated either the antibody described above or an antibody recognizing the C-terminus of BRF1 (Raineri et al, 2004) with the S92-containing phosphopeptide (PP; see above). In the case of the phospho-antibody, the western signal could specifically be competed away, while detection of unphosphorylated BRF1 was not impaired (Figure 4B, compare lanes 3 and 6).
Figure 4.
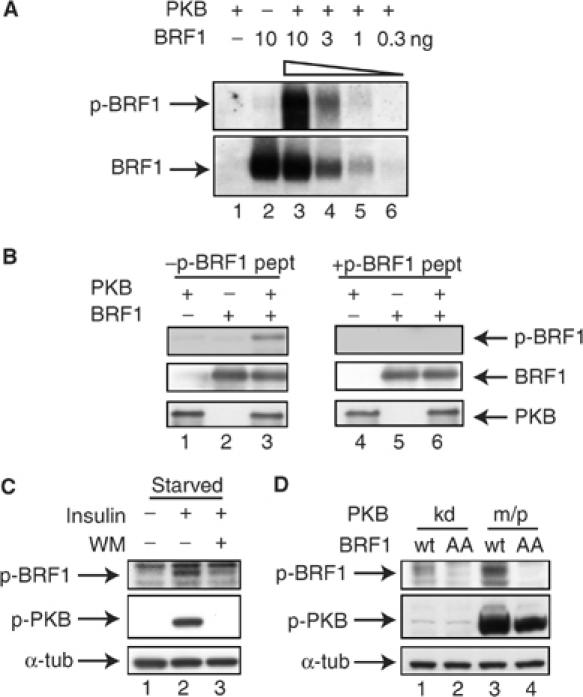
BRF1 is phosphorylated in vivo at S92. (A) Indicated amounts of rBRF1-wt were incubated with buffer alone or activated PKB for 30′ at 30°C and processed for western blotting. Phosphorylated (upper panel) or unphosphorylated BRF1 (lower panel) were detected by corresponding antibodies directed against a p-S92 containing or a C-terminal peptide (see Materials and methods). (B) rBRF1 phosphorylated as in (A) was processed for western blotting. Antibodies recognizing phosphorylated BRF1 (upper panel), BRF1 (middle panel) or PKB (lower panel) were pre-incubated on ice for 30′ with or without 50 μg of the PP used for immunization before adding to the blot. (C) HIRc-B cells were serum starved overnight, treated with WM (200 nM) for 30′ where indicated before stimulation with insulin (20 μg/ml) for 15′ (lanes 1–3). Whole-cell extracts were analyzed by western blot, detecting p-BRF1, phospho-PKB and α-tubulin as a loading control. (D) NIH3T3 B2A2-23 cells were transfected with m/pPKB or kinase-dead PKB (PKBkd) and BRF1-wt or BRF1S90A/S92A (BRF1AA). Whole-cell extracts were analyzed by western blot, detecting p-BRF1, phospho-PKB and α-tubulin as a loading control.
We then examined whether BRF1 becomes phosphorylated at S92 in vivo. Insulin, a known activator of PKB (Burgering and Coffer, 1995) was used to stimulate HIRc-B rat fibroblasts, which express the human insulin receptor and are widely used to investigate insulin signaling (McClain et al, 1987). To reduce PKB background activity, we starved the cells overnight in serum-free medium. After stimulation with insulin for 15 min, PKB was strongly activated (Figure 4C, lane 2, middle panel) and we monitored BRF1 phosphorylation by western blotting using the p-BRF1 antibody. Phosphorylation of BRF1 at S92 could be detected under conditions of PKB activation (Figure 4C, lane 2, upper panel). BRF1 levels did not change in response to insulin stimulation (data not shown). This phosphorylation was partially inhibited by pretreating the cells with the PI3-K inhibitor wortmannin (WM), which completely abolishes PKB activation (Figure 4C, lanes 2 and 3). As WM treatment was not able to block BRF1 phosphorylation completely, we hypothesize that, in addition to PKB, another unidentified kinase may also phosphorylate BRF1 at S92. Phosphorylation of a single serine by more than one kinase is known to occur (Johannessen et al, 2004). To obtain in vivo evidence that indeed PKB is phosphorylating BRF1, we transfected BRF1-wt or the BRF1-S90A/S92A double mutant construct together with m/pPKB or a kinase-dead variant of PKB (PKBkd) into 3T3 cells. Strong phosphorylation of BRF1-wt, but not of the mutant, was observed in m/pPKB-transfected cells (Figure 4D). Together, these data indicate that PKB does phosphorylate BRF1 in vivo at S92.
ARE-mRNA is stabilized under conditions that phosphorylate BRF1
We then wanted to know whether phosphorylation of BRF1 impairs its decay-promoting activity in vivo. We co-transfected the Tet-β-globin-IL3UTR reporter together with the tTA transactivator (pTetOff, Clontech) into HIRc-B cells. When transcription of the reporter was turned off by adding doxycycline, we observed rapid decay of the reporter mRNA with a half-life of approximately 1 h (Figure 5A, lanes 1–3). After pretreatment of cells with insulin for 15 min, the mRNA was more stable (lanes 4–6), and this effect was sensitive to WM (lanes 7–9). We concluded that in vivo ARE-mRNAs become stabilized under conditions where phosphorylation of BRF1 at S92 occurs. Quantification from three parallel experiments indicated that the observed changes are statistically significant (Figure 5B).
Figure 5.
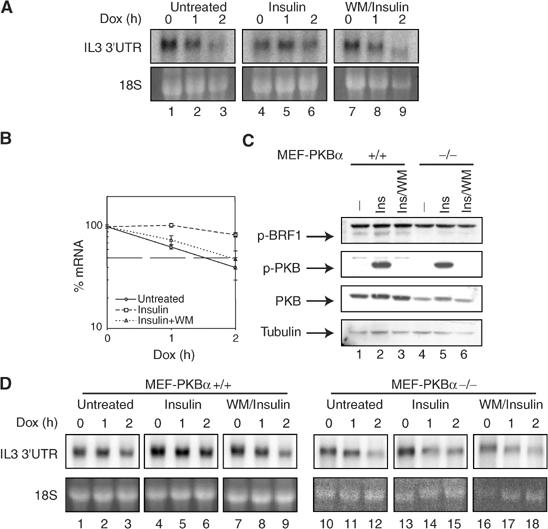
Effect of insulin on reporter mRNA decay. (A) At 24 h after co-transfection of the Tet-β-globin-IL3UTR reporter gene and the Tet-responsive transcriptional activator tTA (pTET-on), HIRc-B cells were split into fresh medium for 16 h. Where indicated cells were pretreated with WM (200 nM) for 30′ before addition of insulin (20 μg/ml) for another 15′. Then doxycycline was added (2 μg/ml) to stop transcription, cytoplasmic RNA was isolated at the indicated time points and processed for northern blotting. (B) Quantification of three independent experiments is shown normalized to the 18S rRNA signal. (C) MEF-PKBα−/− and +/+ cells were pretreated with WM for 30′ prior to insulin (Ins) stimulation for 15′ as indicated. p-BRF1, phospho-PKB, PKB and tubulin protein levels were analyzed by western blot. (D) MEF-PKBα−/− and +/+ cells were treated as described for HIRc-B in (A). Reporter RNA was analyzed by northern blot. One representative experiment is shown.
To further evaluate the involvement of PKB in stabilizing ARE-mRNA, we made use of mouse embryonic fibroblasts with k.o. of PKBα (MEF-PKBα−/−) (Yang et al, 2003). When MEF-PKBα−/− and +/+ cells were compared by western blotting, we noticed strong PKB phosphorylation upon insulin stimulation even in −/− cells, apparently via the PKBβ and γ isoforms. In unstimulated MEF-PKBα+/+ cells, there was background phosphorylation of BRF1 (Figure 5C), presumably by a kinase other than PKB (see also Figure 4C). Insulin stimulation increased this signal and stabilized the reporter mRNA in a WM-sensitive fashion. In MEF-PKBα cells, the p-BRF1 signal was low with no response to insulin. When mRNA stability of the reporter was examined, decay was generally more rapid in −/− cells and insulin failed to induce stabilization (5D). These data support an in vivo role for PKBα in stabilization of ARE-mRNA.
BRF1 phosphorylation leads to 14-3-3 binding
To elucidate the mechanism how PKB inhibits BRF1, we first asked whether BRF1 phosphorylation affects ARE binding, as it was found for TTP (Carballo et al, 2001). By electrophoretic mobility shift assay, we compared increasing amounts of unphosphorylated rBRF1 to rBRF1 pretreated with activated PKB for its ability to bind a radio-labeled AREIL3 probe. Phosphorylation did not impair ARE binding (Figure 6A). The inhibitory effect of S92 phosphorylation on BRF1-destabilizing activity could therefore not be explained by a change in the RNA-binding capacity of BRF1. An alternative mechanism responsible could be binding of phosphorylated BRF1 to an inhibitory factor. A candidate binding partner was 14-3-3, as various isoforms of this protein bind to targets phosphorylated by PKB (Tzivion and Avruch, 2002) and, in addition, 14-3-3 has been found to interact with BRF1 (Bustin and McKay, 1999; Johnson et al, 2002). To test this possibility, His-tagged rBRF1, either untreated or phosphorylated by PKB, was added to slowC cell extracts, purified by Ni-NTA beads and processed for western blotting. Phosphorylated but not unphosphorylated BRF1 pulled down 14-3-3 (Figure 6B, lanes 1 and 2). Furthermore, addition of the PP containing phosphorylated S92 antagonized 14-3-3 binding in a dose-dependent fashion (lanes 3–5), whereas the analogous peptide containing the S92A substitution (AP) was unable to interfere with 14-3-3 binding (lanes 6–8).
Figure 6.
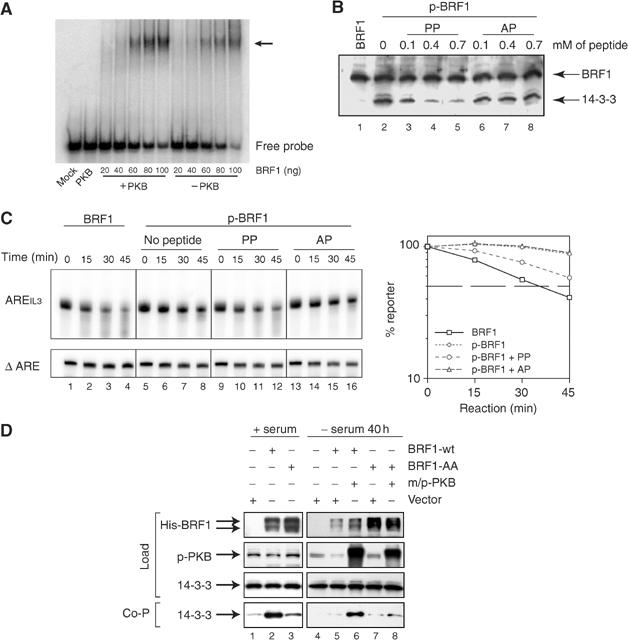
S92 phosphorylation of BRF1 induces complex formation with 14-3-3. (A) Electrophoretic mobility shift assay. Indicated amounts of rBRF1 were pre-incubated with or without activated PKB, mixed with radio-labeled AREIL3 RNA and resolved on 4% nondenaturing PAGE. (B) His-tagged rBRF1 pretreated with buffer (lane 1) or activated PKB (lanes 2–11) was mixed with S100 extract from slowC cells. Increasing concentrations of the phosphorylated peptide surrounding S92 (aa 86–98) of BRF1 (PP) or the corresponding S92A mutant peptide (AP) were added. rBRF1 was purified using Ni-NTA beads, and 14-3-3 and BRF1 were detected with the corresponding antibodies. The lower band represents 14-3-3, the upper His-tagged rBRF1. (C) In vitro decay reactions were carried out as described in Figure 3 in the presence of 0.4 mM of PP (lanes 9–12) or AP (lanes 13–16). The graph on the right shows quantification of four independent experiments after normalizing to the ΔARE signal. Standard error bars were too small to be represented. (D) COS7 cells were transfected with vector, bsdHisBRF1 wt, bsdHisBRF1 aa, either alone (lanes 1–3) or in combination with m/pPKB (lanes 5–8), as indicated. In the experiment shown on the right (lanes 4–8), FCS was withdrawn for 40 h. Expression of His-tagged BRF1, phospho-PKB and 14-3-3 in the cell extracts was determined by western blotting (load). After purification of BRF1 by Ni-NTA beads, co-precipitation of 14-3-3 was tested by western blotting (Co-P).
If interaction with 14-3-3 was responsible for BRF1 inhibition, we would expect the PP to affect binding and hence BRF1 activity in the in vitro decay assay. Indeed, addition of PP could restore decay activity of phosphorylated BRF1 (Figure 6C, lanes 9–12), whereas the AP peptide was ineffective (Figure 6C, lanes 13–16). Taken together, these data indicate that PKB-induced inhibition of BRF1 occurs through binding of 14-3-3.
Finally, in vivo experiments were performed to verify that PKB regulates the interaction between BRF1 and 14-3-3. For this experiment we chose COS7 cells, as complex formation between phosphorylated TTP and 14-3-3 was recently demonstrated in this line (Stoecklin et al, 2004). Cells were transiently transfected with wt or mutant BRF1 constructs carrying a His-tag. BRF1 was purified from the cell extracts using Ni-NTA beads, and western blotting revealed co-precipitation of 14-3-3 with BRF1-wt (Figure 6D, lane 2). Mutation of BRF1 at S90/92 strongly prevented the interaction with 14-3-3 (lane 3). In order to see whether activation of the PKB pathway would stimulate 14-3-3 binding, cells were co-transfected with m/pPKB and serum-starved to reduce endogenous PKB activity. m/pPKB strongly induced binding of 14-3-3 to BRF1-wt (lane 6). Notably, induced binding of 14-3-3 was not observed with the S90A/S92A double mutant (BRF1AA, lane 8). Together, these data suggest that S92 phosphorylation by PKB leads to interaction of 14-3-3 with BRF1. Whereas this interaction does not impair RNA binding, it may inhibit recruitment of the BRF1–RNA complex to the exosomal decay machinery and thereby lead to stabilization of ARE-mRNA.
Discussion
In this report, we provide evidence that phosphorylation of BRF1 by PKB stabilizes ARE-mRNA. As initially reported for PI3-K (Ming et al, 2001), its downstream target PKB efficiently stabilized ARE-containing reporter transcripts in transfection experiments (Figure 1). BRF1 contains two overlapping consensus sequences for PKB phosphorylation at S90 and S92, both of which are conserved between mammals and Xenopus (Figure 2). Mass spectrometry analysis of a recombinant BRF1 N-terminal peptide phosphorylated by PKB revealed that phosphorylation occurs exclusively at S92. In vitro kinase assays with full-length wt protein and S90A/S92A mutants as substrates confirmed S92 as the major phosphorylation site.
To explore the role of S92 phosphorylation, we took advantage of the fact that rBRF1, as previously reported for TTP, is able to promote degradation of ARE-RNA when added to an S100 extract. Interestingly, the decay-promoting activity of BRF1 (Figure 3), but not its ARE-binding activity (Figure 6A), was abrogated by PKB phosphorylation. The rBRF1-S92A mutant was competent in promoting decay, but insensitive to PKB inhibition, establishing S92 as a regulatory site. To our knowledge, this is the first example where specific phosphorylation was shown to affect ARE-dependent mRNA decay in vitro.
To validate these findings in vivo, we analyzed endogenous BRF1 phosphorylation using a S92 phospho-specific antibody. In HIRc-B cells expressing the human insulin receptor, treatment with insulin triggered rapid phosphorylation of both PKB and BRF1 (Figure 4). The latter effect was only partially blocked by the PI3-K inhibitor WM, suggesting that a second kinase in addition to PKB may also phosphorylate S92. Support for this notion was found in MEF-PKBα+/+ cells, where in the absence of PKB activation background BRF1 phosphorylation was observed (Figure 5C). A single serine can indeed be phosphorylated by more than one kinase, as known from nuclear factor CREB, which becomes phosphorylated at position 133 not only by PKA, but also by PKB and other AGC kinases (Johannessen et al, 2004). That PKB can phosphorylate in vivo BRF1 at S92 was obvious from transfection experiments, where activated PKB phosphorylated BRF1-wt but not BRF1-S90A/S92A (Figure 4D).
When ARE reporter mRNA decay following insulin stimulation was examined, we observed WM-sensitive stabilization in HIRc-B (Figure 5A) and MEF-PKBα+/+ cells, but not in MEF-PKBα−/− cells. In the latter, the p-BRF1 signal was low under all conditions and correlated with rapid decay.
To explore the consequences of BRF1 phosphorylation, we turned our attention to the 14-3-3 protein. Members of the family of 14-3-3 proteins are known to bind a variety of target proteins phosphorylated by kinases belonging to the AGC kinase family, which includes PKB. The optimal binding site for 14-3-3 is RSXpSXP (Tzivion and Avruch, 2002), with preference for an aromatic or positively charged amino acid at position −1. The sequence surrounding S92 of BRF1 (RSFpSEG, see Figure 2) represents a 14-3-3 binding site only lacking a P at position +2 compared to the optimal consensus. However, BRF1 has been shown to interact with 14-3-3 in yeast two-hybrid assays and pulldown experiments (Bustin and McKay, 1999; Johnson et al, 2002). Our experiments demonstrate that PKB induces complex formation between BRF1 and 14-3-3 in the same extracts in which BRF1-dependent in vitro RNA decay is regulated by PKB. The S92-containing PP, which competitively dissociated the BRF1/14-3-3 complex, also specifically antagonized the inhibitory effect of PKB on BRF1 activity. As our BRF1 antibody was not able to pull down endogenous protein from HIRc-B cell (data not shown), we overexpressed His-tagged BRF1 in COS7 cells. 14-3-3 co-purified with BRF1-wt, but not with BRF1-S90A/S92A. In addition, co-transfection of activated PKB strongly stimulated binding of 14-3-3 to BRF1-wt, but not to mutant BRF1. Together, our results suggest that BRF1 phosphorylation at S92 leads to binding of 14-3-3, which inhibits the decay-promoting activity of BRF1. Interaction with 14-3-3 has been reported to have diverse functional consequences depending on the targets involved, including inhibition of apoptosis (BAD protein), increasing nuclear export rates (FKH transcription factor), enhancement of DNA binding (p53 protein), protection from proteolysis or by simply serving as a phosphorylation-dependent scaffold (for references, see Tzivion and Avruch, 2002). A reasonable assumption, therefore, is that 14-3-3 sequesters BRF1 in vivo and thereby restricts its access to the decay machinery. Interestingly, Stoecklin et al (2004) have recently reported that phosphorylation of TTP by MK2 at S178 and S52 leads to formation of a similar complex with 14-3-3, which reduces TTP activity and excludes TTP from the stress granules. While a homologous site to S178 in TTP is present in BRF1 (S201 or 203), the N-terminal S52 is not. On the other hand, TTP does not contain a homolog to S92 in BRF1. It will be interesting to see whether S201 or 203 in BRF1 is also phosphorylated by MK2 or another kinase, as this may allow cooperative regulation of BRF1 by two independent signal pathways. As 14-3-3 proteins generally bind as dimers to two phosphorylated residues on their target, dual phosphorylation of BRF1 may be required for full binding of 14-3-3 and robust inhibition of its activity.
An emerging model of regulated ARE-dependent mRNA decay has the following features: In resting cells, AUBPs such as BRF1, TTP, KSRP or AUF1 form part of a multiprotein complex that binds the ARE and facilitates access of the RNA to the decay machinery. More than one AUBP may act on a given species of mRNA, as shown for KSRP and BRF1 in HT1080 cells (Gherzi et al, 2004), and the relative contributions of AUBPs may depend on the cell type. The two Zn-finger domains of the Tis11 gene family proteins recognize the UUAUUUAUU nonamer, the hallmark of class-II AREs, by binding to two adjacent 5′-UAUU-3′ subsites (Hudson et al, 2004). Thus, an ARE containing more than one nonamer is likely to bind several AUBPs. Cell activation or stress leads to a change in the phosphorylation status of TTP, BRF1 or AUF1 (Wilson et al, 2003a; 2003b), changing their interacting partners and thereby altering the composition of the multiprotein complex targeting the ARE. In such a situation, the stabilizing HuR protein may be able to bind preferentially to the ARE, while the poly(A)-ribonuclease (PARN), the exosome, the decapping machinery and the 5′ to 3′ exonucleases lose access to the ARE-mRNA. In addition, mRNA has not only the binary choice of being degraded or delivered to the translation machinery, but also may alternatively be stored in stress granules for later translation. Elucidation of the interplay between these complex and dynamic processes requires the identification of the major proteins involved, as well as their post-translational modification and their functional significance.
Materials and methods
Plasmids
The plasmids bsdHisBRF1S90A and bsdHisBRF1S92A were generated from bsd-HisBRF1wt (Stoecklin et al, 2002) by site-directed mutagenesis, using as upstream primers TV43 (5′-CCGCGCCTTCTCGGAAGGGGGCGAG-3′) and M2521 (5′-GGAGCGGTCTCGGAAGCGGC-3′), respectively, and as downstream primers TV44 (5′-TCTCGGAAGCGGCTGTCTCGCGAGC-3′) and M2518 (5′-TTCGCCGAAGGGGGCGAGCG-3′), respectively. bsd-HisBRF1S90A served as template to construct bsd-HisBRF1AA using M2518 and M2522 (5′-GGCGCGGTCTCGGAAGCGGC). For plasmid SP6-ARE, a 59 nt fragment spanning the ARE of IL-3, including six AUUUA pentamers, was amplified from murine IL-3 cDNA by PCR using primers M1977 (5′-ATGGATCCTTCCATTAAGGC-3′) and M1978 (5′-ATAGATCTTCACAGAAGGC-3′). The amplicon was digested with BamHI and BglII, and ligated into the BglII site of pSP73 (Promega). Plasmids (m/p)-HA-PKBα and T7-ARE− have been described previously (Andjelkovic et al, 1997; Stoecklin et al, 2002). For recombinant BRF1, the constructs used were pQE-30-BRF1S90A, pQE-30-BRF1S92A and pQE-30-BRF1AA, which were constructed in the same way as pQE-30-BRF1wt (Stoecklin et al, 2002), using bsd-HisBRF1S90A, bsd-HisBRF1S92A and bsd-HisBRF1AA, respectively. To generate pQE-30-N-term., pQE-30-BRF1wt was digested with HindIII and HincII, blunt-ended and religated. For pQE-30-C-term., the SapI (blunt)–XhoI fragment of bsd-HisBRF1wt was inserted into the SmaI–SalI sites of pQE-30 (Qiagen). For pQE-30-Zn-finger, the HincII(blunt)–SapI(blunt) fragment of pQE-30-BRF1wt was inserted into the SalI (blunt) site of pQE-30. Recombinant proteins were produced as described (Stoecklin et al, 2002).
Cell culture and transfection
SlowC (Stoecklin et al, 2002), NIH 3T3 B2A2-23 (Xu et al, 1998) and COS7 cells were grown in Iscove's modified Dulbecco medium (IMDM) supplemented with 10% fetal calf serum (FCS), 50 μM 2-mercaptoethanol, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. HIRc-B cells (McClain et al, 1987), MEF-PKBα−/− and +/+ cells (Yang et al, 2003) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with FCS, 2-mercaptoethanol, glutamine, penicillin and streptomycin as described above. To reduce the background PKB activity, cells were starved overnight in serum-free medium before stimulation with insulin (20 μg/ml). Transfection was performed using Lipofectamine 2000 reagent (Life Technologies) following the manufacturer's protocol.
In vitro phosphorylation
The reaction was performed in 30 mM Tris–HCl (pH 7.5), 5mM MgCl2, 1 mM DTT, 0.2 mM ATPγS (unlabeled or 32P-labeled) with 4 ng/μl rBRF1 and 10 ng/μl activated PKB for 30′ at 30°C (Yang et al, 2002).
Electrophoretic mobility shift assay
The electrophoretic mobility shift assay was performed as described (Stoecklin et al, 2002), with the exception that AREIL3 RNA transcribed from SP6-ARE was used as probe.
ARE-mRNA decay analysis
In vivo measurement of RNA decay in NIH 3T3 B2A2-23 and HIRc-B cells using the tetracycline-sensitive Tet-β-globin-IL3UTR reporter gene was performed as described previously (Stoecklin et al, 2002). For in vitro analysis of RNA decay, S100 fractions were prepared from slowC cells and RNA decay reactions were performed as described previously (Chen et al, 2001), with the addition of 100 nM of okadaic acid. 32P-labeled transcripts from SP6-ARE and T7-ARE− (Stoecklin et al, 2002) were synthesized in vitro using SP6 or T7 RNA polymerase (Promega), respectively. Both transcripts were co-incubated in all reactions.
Northern blotting analysis
RNA extraction and northern blotting analysis have been described previously (Stoecklin et al, 2000).
Western blot analysis
The following antibodies were used for Western blotting: sc-629 rabbit anti 14-3-3β (Santa Cruz), sc-7270 mouse monoclonal anti-omni probe (Santa Cruz), phospho-Akt (Ser 473) 587F11 Monoclonal Antibody (Cell Signaling Technology), rabbit polyclonal anti-AKT (Cell Signaling) and mouse monoclonal anti α-tubulin (236-10501) (Molecular Probes).
Anti-p-BRF1 and anti-BRF1 antibodies were generated by immunizing rabbits with the KLH-linked peptides F-R-D-R-S-F-S(PO3H2)-E-G-G-E-R-L (aa 86–98) and S-D-Q-E-G-Y-L-S-S-S-S-S-S-H-S-G-S-D-S-P-T (aa 300–320) (Neosystem). Sera were affinity-purified using the above-mentioned peptides linked to activated CH Sepharose 4B (Pharmacia) according to the manufacturer's protocol. Proteins were visualized using ECL Advance (Amersham) or CDP-star (Roche).
Pulldown assay
In vivo: At 24–48 h after transfection of COS7 cells using lipofectamine 2000, cells were lysed in buffer containing 1% NP-40, 150 mM NaCl, 50 mM Tris–HCl (pH 8.0), 1 mM MgCl2, 10% glycerol, 20 mM 2-mercaptoethanol, 10 mM imidazole, 1 mM Na-vanadate, 50 mM NaF, 20 nM okadaic acid and EDTA-free ‘complete' protease inhibitors (Roche). After incubation of the cytoplasmic fraction for 1 h at 4°C with 40 μl of Ni-NTA magnetic agarose beads (Qiagen), the beads were washed four times in lysis buffer containing 20 mM imidazole, eluted in SDS-sample buffer and resolved on 4–20% gradient polyacrylamide tris-glycine gels (Invitrogen).
In vitro: In all, 40 ng of phosphorylated or unphosphorylated rBRF1 and different amounts of the peptides PP (F-R-D-R-S-F-S(PO3H2)-E-G-G-E-R-L corresponding to aa 86–98) or AP (F-R-D-R-S-F-A-E-G-G-E-R-L; Neosystem) were incubated with 40 μg of S100 slowC extract in 100 mM Tris–HCl (pH 8.0), 1 mM Mg-acetate, 1.5 mM K-acetate, 150 mM NaCl, 10% glycerol, 10 mM imidazole and 100 nM okadaic acid in a total volume of 800 μl. After incubation at 37°C for 30 min, Ni-NTA affinity purification was performed as described above.
Mass spectrometry
The N-terminal BRF1 band (aa 3–110) was excised from the gel, reduced with 10 mM DDT, alkylated with 55 mM iodoacetamide and cleaved with 0.5 μg LysC (Achromobacter, Wako BioProducts) at 37°C overnight in 25 mM ammonium bicarbonate buffer (pH 8.0). After the LysC cleavage, the gel was dried and 0.5 μg of Asp-N (Roche; sequencing grade) was added in 40 μl of 50 mM sodium phosphate buffer (pH 8.0) at 37°C for 6 h. The extracted peptides were analyzed by capillary liquid chromatography tandem mass spectrometry (LC-MSMS) using a Magic C18 100 μm × 10 cm HPLC column (Spectronex) connected on-line to an iontrap Finnigan DecaXP (ThermoFinnigan). PPs were identified with the neutral loss function, searching for fragment ions formed by the loss of phosphoric acid, 32.3, 48.5 or 97 Da from the (M+3H)3+, (M+2H)2+ and (M+H)+ ions, respectively. The PP was isolated by LC-MSMS, followed by cleavage with 0.1 μg trypsin (Promega; sequencing grade) in 50 mM ammonium bicarbonate (pH 8.0) at 37°C for 1 h. The peptide mixture was then analyzed by LC-MSMS as described above and the PP was additionally sequenced by an MS3 experiment.
Acknowledgments
CM was supported by grant 31-57065.99 by the Schweizerische Nationalfonds zur Förderung der wissenschaftlichen Forschung, RG was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC). The Friedrich Miescher Institute is part of the Novartis Research Foundation. We thank Jing Yang and David Barford (Institute of Cancer Research, London) for producing some of the purified PKB preparations, Jianhua Feng for advice and purified protein and Dr Don Benjamin for discussion and helpful comments on the manuscript.
References
- Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P (1996) Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett 399: 333–338 [DOI] [PubMed] [Google Scholar]
- Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA (1997) Role of translocation in the activation and function of protein kinase B. J Biol Chem 272: 31515–31524 [DOI] [PubMed] [Google Scholar]
- Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS (2001) ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res 29: 246–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briata P, Ilengo C, Corte G, Moroni C, Rosenfeld MG, Chen CY, Gherzi R (2003) The Wnt/beta-catenin → Pitx2 pathway controls the turnover of Pitx2 and other unstable mRNAs. Mol Cell 12: 1201–1211 [DOI] [PubMed] [Google Scholar]
- Brook M, Sully G, Clark AR, Saklatvala J (2000) Regulation of tumour necrosis factor alpha mRNA stability by the mitogen-activated protein kinase p38 signalling cascade (in process citation). FEBS Lett 483: 57–61 [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ (1995) Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 376: 599–602 [DOI] [PubMed] [Google Scholar]
- Bustin SA, McKay IA (1999) The product of the primary response gene BRF1 inhibits the interaction between 14-3-3 proteins and cRaf-1 in the yeast trihybrid system. DNA Cell Biol 18: 653–661 [DOI] [PubMed] [Google Scholar]
- Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ (2001) Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J Biol Chem 6: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ (1998) Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281: 1001–1005 [DOI] [PubMed] [Google Scholar]
- Chen CY, Del Gatto-Konczak F, Wu Z, Karin M (1998) Stabilization of interleukin-2 mRNA by the c-Jun NH2-terminal kinase pathway. Science 280: 1945–1949 [DOI] [PubMed] [Google Scholar]
- Chen CY, Gherzi R, Andersen JS, Gaietta G, Jurchott K, Royer HD, Mann M, Karin M (2000) Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation. Genes Dev 14: 1236–1248 [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M (2001) AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 107: 451–464 [DOI] [PubMed] [Google Scholar]
- Chen CY, Shyu AB (1995) AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 20: 465–470 [DOI] [PubMed] [Google Scholar]
- Chen CY, Xu N, Shyu AB (2002) Highly selective actions of HuR in antagonizing AU-rich element-mediated mRNA destabilization. Mol Cell Biol 22: 7268–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW (2003) MK2 phosphorylates tristetraprolin on in vivo sites including S178, a site required for 14-3-3 binding. J Biol Chem 19: 19. [DOI] [PubMed] [Google Scholar]
- Clark AR, Dean JL, Saklatvala J (2003) Post-transcriptional regulation of gene expression by mitogen-activated protein kinase p38. FEBS Lett 546: 37–44 [DOI] [PubMed] [Google Scholar]
- Dean JL, Brook M, Clark AR, Saklatvala J (1999) p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem 274: 264–269 [DOI] [PubMed] [Google Scholar]
- Fan X, Steitz J (1998) Overexpression of HuR, a nuclear–cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J 17: 3448–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford LP, Watson J, Keene JD, Wilusz J (1999) ELAV proteins stabilize deadenylated intermediates in a novel in vitro mRNA deadenylation/degradation system. Genes Dev 13: 188–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frevel MA, Bakheet T, Silva AM, Hissong JG, Khabar KS, Williams BR (2003) p38 mitogen-activated protein kinase-dependent and -independent signaling of mRNA stability of AU-rich element-containing transcripts. Mol Cell Biol 23: 425–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Wilusz CJ, Peltz SW, Wilusz J (2001) A novel mRNA-decapping activity in HeLa cytoplasmic extracts is regulated by AU-rich elements. EMBO J 20: 1134–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherzi R, Lee KY, Briata P, Wegmuller D, Moroni C, Karin M, Chen CY (2004) A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol Cell 14: 571–583 [DOI] [PubMed] [Google Scholar]
- Gorospe M, Wang X, Holbrook NJ (1998) p53-dependent elevation of p21Waf1 expression by UV light is mediated through mRNA stabilization and involves a vanadate-sensitive regulatory system. Mol Cell Biol 18: 1400–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueydan C, Droogmans L, Chalon P, Huez G, Caput D, Kruys V (1999) Identification of TIAR as a protein binding to the translational regulatory AU-rich element of tumor necrosis factor alpha mRNA. J Biol Chem 274: 2322–2326 [DOI] [PubMed] [Google Scholar]
- Huang ZF, Massey JB, Via DP (2000) Differential regulation of cyclooxygenase-2 (COX-2) mRNA stability by interleukin-1 beta (IL-1 beta) and tumor necrosis factor-alpha (TNF-alpha) in human in vitro differentiated macrophages. Biochem Pharmacol 59: 187–194 [DOI] [PubMed] [Google Scholar]
- Hudson BP, Martinez-Yamout MA, Dyson HJ, Wright PE (2004) Recognition of the mRNA AU-rich element by the zinc finger domain of TIS11d. Nat Struct Mol Biol 11: 257–264 (Epub 2004 February 8) [DOI] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Moens U (2004) What turns CREB on? Cell Signal 16: 1211–1227 [DOI] [PubMed] [Google Scholar]
- Johnson BA, Stehn JR, Yaffe MB, Blackwell TK (2002) Cytoplasmic localization of Tristetraprolin involves 14-3-3-dependent and -independent mechanisms. J Biol Chem 8: 8. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10: 387–398 [DOI] [PubMed] [Google Scholar]
- Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ (1999) Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol 19: 4311–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WS, Kennington EA, Blackshear PJ (2003) Tristetraprolin and its family members can promote the cell-free deadenylation of AU-rich element-containing mRNAs by poly(A) ribonuclease. Mol Cell Biol 23: 3798–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroia G, Cuesta R, Brewer G, Schneider RJ (1999) Control of mRNA decay by heat shock–ubiquitin–proteasome pathway. Science 284: 499–502 [DOI] [PubMed] [Google Scholar]
- Lindsten T, June CH, Ledbetter JA, Stella G, Thompson CB (1989) Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science 244: 339–343 [DOI] [PubMed] [Google Scholar]
- Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H (1996) Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. Biol Chem 271: 8144–8151 [DOI] [PubMed] [Google Scholar]
- Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR (2001) Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol Cell Biol 21: 6461–6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain DA, Maegawa H, Lee J, Dull TJ, Ulrich A, Olefsky JM (1987) A mutant insulin receptor with defective tyrosine kinase displays no biologic activity and does not undergo endocytosis. J Biol Chem 262: 14663–14671 [PubMed] [Google Scholar]
- Min H, Turck CW, Nikolic JM, Black DL (1997) A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev 11: 1023–1036 [DOI] [PubMed] [Google Scholar]
- Ming XF, Kaiser M, Moroni C (1998) c-jun N-terminal kinase is involved in AUUUA-mediated interleukin-3 mRNA turnover in mast cells. EMBO J 17: 6039–6048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming XF, Stoecklin G, Lu M, Looser R, Moroni C (2001) Parallel and independent regulation of interleukin-3 mRNA turnover by phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase. Mol Cell Biol 21: 5778–5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee D, Gao M, O'Connor JP, Raijmakers R, Pruijn G, Lutz CS, Wilusz J (2002) The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J 21: 165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair AP, Hahn S, Banholzer R, Hirsch HH, Moroni C (1994) Cyclosporin A inhibits growth of autocrine tumour cell lines by destabilizing interleukin-3 mRNA. Nature 369: 239–242 [DOI] [PubMed] [Google Scholar]
- Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M (2002) MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem 277: 3065–3068 (Epub 2001 December 6) [DOI] [PubMed] [Google Scholar]
- Paulding WR, Czyzyk-Krzeska MF (2000) Hypoxia-induced regulation of mRNA stability. Adv Exp Med Biol 475: 111–121 [DOI] [PubMed] [Google Scholar]
- Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, Chen S, Gueydan C, Kruys V, Streuli M, Anderson P (2000) TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J 19: 4154–4163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raineri I, Wegmueller D, Gross B, Certa U, Moroni C (2004) Roles of AUF1 isoforms, HuR and BRF1 in ARE-dependent mRNA turnover studied by RNA interference. Nucleic Acids Res 32: 1279–1288 (print 2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler GD, Cole MD (1988) GM-CSF and oncogene mRNA stabilities are independently regulated in trans in a mouse monocytic tumor. Cell 55: 1115–1122 [DOI] [PubMed] [Google Scholar]
- Shaw G, Kamen R (1986) A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 46: 659–667 [DOI] [PubMed] [Google Scholar]
- Shyu AB, Belasco JG, Greenberg ME (1991) Two distinct destabilizing elements in the c-fos message trigger deadenylation as a first step in rapid mRNA decay. Genes Dev 5: 221–231 [DOI] [PubMed] [Google Scholar]
- Stoecklin G, Colombi M, Raineri I, Leuenberger S, Mallaun M, Schmidlin M, Gross B, Lu M, Kitamura T, Moroni C (2002) Functional cloning of BRF1, a regulator of ARE-dependent mRNA turnover. EMBO J 21: 4709–4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Gross B, Ming XF, Moroni C (2003) A novel mechanism of tumor suppression by destabilizing AU-rich growth factor mRNA. Oncogene 22: 3554–3561 [DOI] [PubMed] [Google Scholar]
- Stoecklin G, Ming XF, Looser R, Moroni C (2000) Somatic mRNA turnover mutants implicate tristetraprolin in the interleukin-3 mRNA degradation pathway. Mol Cell Biol 20: 3753–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P (2004) MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J 11: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ (2002) Extensive and divergent circadian gene expression in liver and heart. Nature 417: 78–83 [DOI] [PubMed] [Google Scholar]
- Stumpo DJ, Byrd NA, Phillips RS, Ghosh S, Maronpot RR, Castranio T, Meyers EN, Mishina Y, Blackshear PJ (2004) Chorioallantoic fusion defects and embryonic lethality resulting from disruption of Zfp36L1, a gene encoding a CCCH tandem zinc finger protein of the tristetraprolin family. Mol Cell Biol 24: 6445–6455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor GA, Thompson MJ, Lai WS, Blackshear PJ (1995) Phosphorylation of tristetraprolin, a potential zinc finger transcription factor, by mitogen stimulation in intact cells and by mitogen-activated protein kinase in vitro. J Biol Chem 270: 13341–13347 [DOI] [PubMed] [Google Scholar]
- Tran H, Schilling M, Wirbelauer C, Hess D, Nagamine Y (2004) Facilitation of mRNA deadenylation and decay by the exosome-bound, DExH protein RHAU. Mol Cell 13: 101–111 [DOI] [PubMed] [Google Scholar]
- Tzivion G, Avruch J (2002) 14-3-3 proteins: active cofactors in cellular regulation by serine/threonine phosphorylation. J Biol Chem 277: 3061–3064 [DOI] [PubMed] [Google Scholar]
- Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y, Holbrook N, Gorospe M (2000) HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol 20: 760–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GM, Lu J, Sutphen K, Suarez Y, Sinha S, Brewer B, Villanueva-Feliciano EC, Ysla RM, Charles S, Brewer G (2003a) Phosphorylation of p40AUF1 regulates binding to A+U-rich mRNA-destabilizing elements and protein-induced changes in ribonucleoprotein structure. J Biol Chem 278: 33039–33048 [DOI] [PubMed] [Google Scholar]
- Wilson GM, Lu J, Sutphen K, Sun Y, Huynh Y, Brewer G (2003b) Regulation of A+U-rich element-directed mRNA turnover involving reversible phosphorylation of AUF1. J Biol Chem 278: 33029–33038 [DOI] [PubMed] [Google Scholar]
- Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J 18: 4969–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodnar-Filipowicz A, Heusser CH, Moroni C (1989) Production of the haemopoietic growth factors GM-CSF and interleukin-3 by mast cells in response to IgE receptor-mediated activation. Nature 339: 150–152 [DOI] [PubMed] [Google Scholar]
- Wodnar-Filipowicz A, Moroni C (1990) Regulation of interleukin 3 mRNA expression in mast cells occurs at the posttranscriptional level and is mediated by calcium ions. Proc Natl Acad Sci USA 87: 777–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Chen CY, Shyu AB (1997) Modulation of the fate of cytoplasmic mRNA by AU-rich elements: key sequence features controlling mRNA deadenylation and decay. Mol Cell Biol 17: 4611–4621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Loflin P, Chen CY, Shyu AB (1998) A broader role for AU-rich element-mediated mRNA turnover revealed by a new transcriptional pulse strategy. Nucleic Acids Res 26: 558–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Cron P, Thompson V, Good VM, Hess D, Hemmings BA, Barford D (2002) Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell 9: 1227–1240 [DOI] [PubMed] [Google Scholar]
- Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, Hemmings BA (2003) Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J Biol Chem 278: 32124–32131 [DOI] [PubMed] [Google Scholar]
- Zhang W, Wagner BJ, Ehrenman K, Schaefer AW, DeMaria CT, Crater D, DeHaven K, Long L, Brewer G (1993) Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol Cell Biol 13: 7652–7665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Brauchle MA, Di Padova F, Gram H, New L, Ono K, Downey JS, Han J (2001) Gene suppression by tristetraprolin and release by the p38 pathway. Am J Physiol Lung Cell Mol Physiol 281: L499–L508 [DOI] [PubMed] [Google Scholar]