

Abstract
As a component of chromatin-modifying complexes with histone acetyltransferase (HAT) activity, TRRAP has been shown to be involved in various cellular processes including gene transcription and oncogenic transformation. Inactivation of Trrap, the murine ortholog of TRRAP, in mice revealed its function in development and cell cycle progression. However, the underlying mechanism is unknown. Here, we show that the loss of Trrap in mammalian cells leads to chromosome missegregation, mitotic exit failure and compromised mitotic checkpoint. These mitotic checkpoint defects are caused by defective Trrap-mediated transcription of the mitotic checkpoint proteins Mad1 and Mad2. The mode of regulation by Trrap involves acetylation of histones H4 and H3 at the gene promoter of these mitotic players. Trrap associated with the HAT Tip60 and PCAF at the Mad1 and Mad2 promoters in a cell cycle-dependent manner and Trrap depletion abolished recruitment of these HATs. Finally, ectopic expression of Mad1 and Mad2 fully restores the mitotic checkpoint in Trrap-deficient cells. These results demonstrate that Trrap controls the mitotic checkpoint integrity by specifically regulating Mad1 and Mad2 genes.
Keywords: chromatin acetylation, Mad1 and 2, mitotic checkpoint, transcription regulation, Trrap
Introduction
The mitotic checkpoint (also known as the spindle assembly checkpoint) maintains genomic stability by ensuring correct segregation of chromosomes during cell division. This surveillance mechanism prevents anaphase from occurring until all chromosomes have attached properly to the spindle. When chromosomes are not attached properly to the spindle, the mitotic checkpoint prevents the onset of anaphase by maintaining sister-chromatid cohesion. The chief players in the mitotic checkpoint are Mad and Bub proteins that block the activity of a protein complex termed the anaphase-promoting complex (APC) (Musacchio and Hardwick, 2002; Peters, 2002). Inhibition of the ubiquitination activity of APC prevents the degradation of securin and thereby delays sister-chromatid separation and anaphase onset. In this way, the mitotic checkpoint prevents the occurrence of aneuploidy, which may lead to cancer. Inhibition of or mutants in molecules involved in the mitotic checkpoint and chromosome segregation, including securin (Jallepalli et al, 2001), cohesin Scc1 (Hauf et al, 2001), separase (Waizenegger et al, 2002) and polo-like kinase Plk1 (Simizu and Osada, 2000), exhibit defects in sister-chromatin separation and aberrant chromosome segregation. Defects in the mitotic checkpoint have been found frequently in human cancers (Lengauer et al, 1998). Studies on yeast and mammalian cells indicated that the levels of the mitotic checkpoint proteins are critical for their function (Musacchio and Hardwick, 2002) and a reduced expression and aberrant transcription were often detected in human cancers (Li and Benezra, 1996; Cahill et al, 1998; Takahashi et al, 1999; Wang et al, 2000; 2002; Lin et al, 2002).
Previous studies have revealed important crosstalk between proteins regulating the cell cycle regulatory apparatus, including cell cycle checkpoints, and histone acetylation (Chen et al, 2001; Wang et al, 2001). Covalent modification of histone tails by acetylation of conserved lysine residues by histone acetyltransferase (HAT) enzymes is believed to be an important mechanism by which cells regulate the function of chromatin DNA including accessibility to transcription factors (Brown et al, 2000; Cheung et al, 2000; Carrozza et al, 2003). Therefore, chromatin-remodeling activity plays an essential role in transcription regulation. TRRAP has been identified as a common component of several chromatin-modifying complexes with HAT activity, such as GCN5/PCAF (p300/CBP-associated factor) and Tip60/NuA4 (nucleosomal acetyltransferase of histone H4) (Grant et al, 1998; Saleh et al, 1998; Vassilev et al, 1998; Allard et al, 1999; Ikura et al, 2000; McMahon et al, 2000; Brown et al, 2001). Biochemical studies have established that TRRAP interacts with the transcription factors c-Myc and E2F (McMahon et al, 1998; 2000; Deleu et al, 2001). TRRAP is recruited to the transcription factor target site in chromatin, thereby mediating local histone acetylation and facilitating gene transcription (Bouchard et al, 2001; Frank et al, 2001).
Genetic studies in yeast showed that Tra1p, the yeast ortholog of TRRAP, is essential for cell viability (Saleh et al, 1998) and gene-specific transcription (Brown et al, 2001). In mammalian cells, expression of a TRRAP fragment that competes with the endogenous protein can suppress c-Myc- and E1A-mediated cellular transformation (McMahon et al, 1998; Deleu et al, 2001). Recently, we have shown that disruption of Trrap (the murine ortholog of TRRAP) blocks cell proliferation, resulting in loss of cell viability and embryonic lethality in mice (Herceg et al, 2001). We also demonstrated that Trrap regulates expression of genes by acetylation of histone H3 or H4 (Herceg et al, 2003). However, the biological targets of Trrap and Trrap-containing HATs have not been identified. In the present study, we investigated the molecular mechanism by which Trrap is involved in mitotic progression and identified Mad1 and Mad2 as critical targets by which Trrap regulates the mitotic checkpoint.
Results
Loss of Trrap in both immortalized and primary cells results in defective mitotic checkpoint in response to spindle poisons
Since loss of Trrap in immortalized mouse embryonic fibroblasts (MEFs) results in failure to sustain mitotic arrest in response to nocodazole (Herceg et al, 2001), we first tested the need for Trrap in mitotic checkpoint function in response to different spindle-damaging drugs. Immortalized MEF cell lines with inducible deletion of Trrap (Trrap ‘conditional' knockout cells, CER9) permit us to conditionally inactivate Trrap (Herceg et al, 2001). On addition of the inducer 4-hydroxy-tamoxifen (OHT), the Cre-ER fusion protein is translocated into the cell nucleus, resulting in the deletion of the Trrap gene by Cre recombinase (Herceg et al, 2001). We treated CER9 cells with nocodazole, colcemid or taxol in the presence (+OHT) or absence (−OHT) of the inducer and measured the mitotic index (defined as the percentage of mitotic cells) using fluorescence microscopy. We found that Trrap-containing cells (−OHT) showed a large increase in mitotic index (∼40–50%) in response to all three spindle poisons (Supplementary Figure S1), indicative of an active mitotic checkpoint. In contrast, only ∼10% of Trrap-depleted cells (+OHT) were arrested in metaphase in the presence of any of the spindle-damaging drugs, suggesting that their mitotic checkpoint was defective. Consistent with this observation, phase-contrast microscopy revealed that much fewer Trrap-deficient cells were round, indicative of failed mitotic arrest, in the presence of spindle poison (data not shown).
To rule out any effect that the immortalization process might have on mitotic progression, we repeated the experiment using primary MEFs (PMEFs) freshly isolated from Trrapfloxed/Δ (Trrapf/Δ) embryos. To induce Trrap deletion, PMEF cells were infected with Cre-containing adenovirus (Ad-Cre) and the inducibility of Trrap deletion was confirmed by PCR (Supplementary Figure S1). Analysis of the cells by phase-contrast microscopy (Supplementary Figure S1) and flow cytometry following staining by phospho-histone H3 antibody (Supplementary Figure S1) showed that primary Trrap-depleted cells (Ad-Cre) failed to accumulate in mitosis, suggesting that the mitotic checkpoint was defective in these cells. Consistent with the inability of Trrap-deficient cells to maintain the mitotic checkpoint response, depletion of Trrap resulted in degradation of securin, an inhibitor of chromatid separation (Supplementary Figure S1), and of cyclin B (Supplementary Figure S1), in the presence of spindle poison. While Trrap-containing cells accumulated and maintained high levels of securin and cyclin B, Trrap-deficient cells failed to maintain high levels of securin and cyclin B (Supplementary Figure S1), most likely due to activation of APC and exit from mitosis.
Introduction of nondegradable cyclin B restores mitotic arrest in Trrap-deficient cells
To rule out that low mitotic index seen in cells lacking Trrap (Supplementary Figure S1) is simply a result of Trrap-deficient cells failing to enter mitosis, we introduced nondegradable cyclin B into Trrap-deficient and Trrap-containing cells and followed their accumulation in mitosis. Since a nondegradable mutant of cyclin B arrests cells at mitosis with condensed chromosomes (Gallant and Nigg, 1992; Holloway et al, 1993), this approach allowed us to visualize the capacity of Trrap-deficient cells to enter mitosis. Cells were either mock-transfected or transfected with wild-type cyclin B-green fluorescent protein (CLB-GFP) or a nondegradable mutant of cyclin B-GFP (CLB-DM-GFP), incubated in the presence of nocodazole and scored for GFP-positive cells with condensed chromosomes (mitotic cells) and those with decondensed chromatin (interphase cells; Supplementary Figure S2). Trrap-containing cells either mock-transfected or CLB-GFP-transfected arrested in mitosis in the presence of nocodazole, as judged by a high mitotic index (Supplementary Figure S2), indicative of a proficient mitotic checkpoint. Neither mock transfection nor CLB-GFP transfection caused mitotic arrest in Trrap-deficient cells in the presence of nocodazole (Supplementary Figure S2). However, the nondegradable cyclin B (CLB-DM-GFP)-transfected Trrap-deficient cells showed a mitotic accumulation similar to that of Trrap-containing cells (Supplementary Figure S2). These results indicate that Trrap-deficient cells entered mitosis and initiated mitotic exit at similar kinetics as Trrap-containing cells. They also suggest that the checkpoint effectors downstream of cyclin B degradation are intact in cells lacking Trrap.
Trrap-deficient cells are defective in detecting chromosome missegregation
To follow mitotic progression in detail and to examine mitotic exit in cells lacking Trrap, we monitored cells in real time by observing chromosome segregation in living cells. To this end, we constructed Trrapfloxed/targeted (Trrapf/t) MEFs (Herceg et al, 2001) coexpressing Cre-ER recombinase and the histone H2B gene fused to the GFP gene (H2B-GFP) that allows labeling of mitotic chromosomes. Stable integrants coexpressing Cre-ER and H2B-GFP were identified by PCR and fluorescent microscopy, respectively (Supplementary Figure S3). Time-lapse microscopy of Trrap-containing cells (−OHT) revealed that the aligned chromosomes rapidly progressed into anaphase, characterized by separation and bipolar segregation of sister chromatids into two daughter cells (Supplementary Figure S3). In contrast, a large fraction of Trrap-deficient cells (+OHT) failed to separate their chromosomes adequately. The chromosomal material appeared either nonsegregated or stretched between two chromosome complements, giving rise to a micronucleus and interconnecting nuclear ‘bridge', respectively (Supplementary Figure S3). Although decondensation of chromatin appeared to be unaffected in the absence of Trrap, chromatin ‘bridges' were often observed (Supplementary Figure S3). Quantitative summary of time-lapse images showed a significant increase in chromosome segregation defects (both micronucleus and chromatin ‘bridge' formation) in Trrap-deficient cells in comparison to Trrap-containing cells (Supplementary Figure S3). These results demonstrate that Trrap-deficient cells, although capable of entering mitosis and initiating chromosome segregation, triggered anaphase onset in the presence of unattached chromosomes or inappropriately attached chromosomes, resulting in chromosome missegregation and aberrant mitotic exit. These data suggest that some machinery to detect and/or signal chromosome misattachment or missegregation is defective in the absence of Trrap.
Loss of Trrap leads to reduced levels of the mitotic checkpoint proteins Mad1 and Mad2
Because Mad and Bub proteins are major players in the mitotic checkpoint, we analyzed the expression of the mitotic checkpoint regulators Mad1, Mad2 and Bub3. Trrap-containing cells (CER9 cells, without OHT) and Trrap-deficient cells (CER9, +OHT) were synchronized by serum starvation and similar cell cycle progression was confirmed by flow cytometry (Figure 1A) and determination of the mitotic index (Figure 1B). In Trrap-containing cells, Mad2 protein levels were unchanged during cell cycle progression, while Trrap-deficient cells exhibited a significant reduction in Mad2 protein levels throughout the cell cycle (Figure 1C). Mad2 levels were also significantly reduced in unsynchronized cells lacking Trrap and when Trrap-deficient populations were released into the cell cycle from serum starvation in the absence of nocodazole (data not shown). Similarly, Trrap-deficient cells showed a marked reduction in the Mad1 level, whereas protein levels of Bub3 remained largely unchanged (Figure 1C). Consistent with these results, deletion of Trrap in primary MEF cells also resulted in downregulation of Mad2 and Mad1 but not Bub3 (Figure 1D). As a control, empty vector (pSG9) transfected cells PSG1 (in the presence or absence of OHT) showed no change in Mad2, Mad1 and Bub3 levels (Figure 1E).
Figure 1.
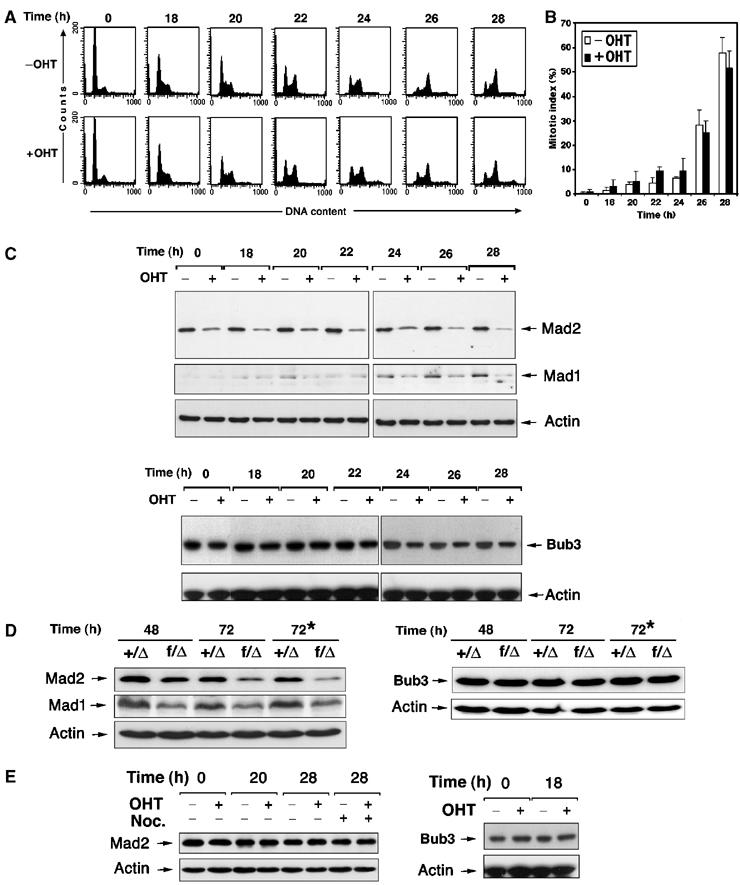
Loss of Trrap results in downregulation of Mad2 and Mad1 proteins. (A) Synchronization of Trrap-containing and Trrap-deficient cells. CER9 cells were synchronized at G0/G1 phase by serum starvation for 24 h with or without OHT pretreatment for 48 h. At indicated times after serum stimulation (i.e. 0 h corresponds to 48 h after addition of OHT), cells were stained with propidium iodide and analyzed by flow cytometry. (B) Cells were synchronized as in (A) and, after DAPI staining, the mitotic index was determined by scoring at least 100 cells. (C–E) Western blot analysis of mitotic checkpoint players in cells lacking Trrap. (C) CER9 cells were synchronized at G0/G1 phase by serum starvation for 24 h with or without OHT pretreatment for 48 h. Indicated times correspond to time after serum stimulation (i.e. 0 h corresponds to 48 h after addition of OHT). Protein extracts were prepared from samples taken at the indicated time points after serum stimulation and hybridized with anti-Mad2, anti-Mad1 and anti Bub3. Equal loading was verified by anti-actin antibody. (D) Primary MEFs of indicated genotypes were infected with adenovirus expressing Cre recombinase and at indicated time points after infection protein levels were determined by Western blotting. The asterisk (*) indicates that the cell cultures were incubated in the presence of nocodazole. (E) Western blot analysis of mitotic players in empty vector-transfected cells PSG1. Cell samples were taken at indicated time points after serum stimulation and protein levels of Mad2 and Bub3 were analyzed by Western blotting. Equal loading was verified by anti-actin antibody.
Loss of Trrap compromises transcription of Mad1 and Mad2 and not their protein stability
In order to establish if low levels of Mad proteins in cells lacking Trrap are due to increased protein degradation or to decreased gene expression, we first tested the effects of proteasome function on the accumulation of Mad proteins. Cells were synchronized by serum starvation and at 0 and 16 h after serum stimulation, the proteasome inhibitor N-acetyl-L-leucyl-L-leucyl-L-norleucinal (LLnL) was added to the cell culture. At 20 h after serum stimulation, protein lysates were analyzed by immunoblotting with anti-Mad2, anti-Mad1 and anti-Bub3 antibodies (Figure 2A). The addition of LLnL did not affect the protein levels of Mad2, Mad1 or Bub3 in either Trrap-deficient or Trrap-containing cells (Figure 2A), suggesting that the low levels of Mad proteins were not due to increased proteolysis in cells lacking Trrap.
Figure 2.
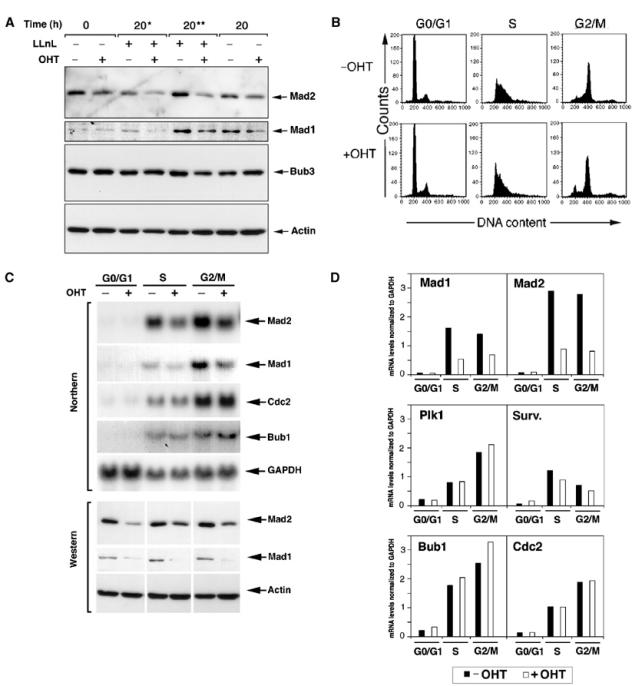
Loss of Trrap compromises transcription of Mad2 and Mad1 genes and not their protein stability. (A) Reduced protein levels of Mad players in cells lacking Trrap are not due to increased protein degradation. CER9 cells were synchronized at G0/G1 phase by serum starvation for 24 h with or without OHT pretreatment for 48 h and released into the serum-containing medium in the presence or absence of the proteasome inhibitor LLnL (50 μM). An asterisk (*) and two asterisks (**) indicate that LLnL was added at the 0 and 16 h time point, respectively, after addition of serum. Cell lysates prepared from cells harvested at the indicated time points were analyzed by Western blotting. (B–D) Mad mRNA levels were downregulated in Trrap-deficient cells. (B) CER9 cells were grown in the presence or absence of OHT for 48 h and then synchronized at G0/G1 phase by serum starvation for 24 h. Synchronized S and G2/M populations were obtained by collecting cells at 18 and 28 h in the presence of nocodazole, respectively, after release from serum starvation, as evidenced by flow cytometry analysis. These populations were used for Northern blotting, Western blotting and ChIP assay (see below). (C) CER9 cells were synchronized as in (B), and total RNA and protein lysates were analyzed by Northern and Western blotting, respectively. RNA loading was controlled by GAPDH probe and protein loading was verified by anti-actin antibody. (D) Quantification of mRNA expression levels of the mitotic players. mRNA levels of indicated genes were analyzed densitometrically and normalized to GAPDH.
We next examined the transcription levels of mitotic players over the cell cycle phases (Figure 2B, see Materials and methods) by Northern blotting. In Trrap-containing cells (−OHT), Mad2 mRNA accumulated at S phase reaching a peak at G2/M phase (Figure 2C and data not shown). In contrast, Trrap-deficient cells (+OHT) failed to accumulate Mad2 mRNA. Defective Mad2 and Mad1 mRNA accumulation was observed in Trrap-deficient cells in the presence or absence of nocodazole (data not shown). Similarly, Mad1 mRNA levels were reduced in the absence of Trrap, whereas mRNAs of Bub1, Cdc2, Plk1 and survivin remained unchanged (Figure 2C and D), suggesting that Mad1 and Mad2, but not Bub1, Cdc2, Plk1 or survivin, are regulated by Trrap. Therefore, low protein levels of Mad2 and Mad1 in Trrap-deficient cells (Figures 2C and 1C) are caused by compromised transcription of these players in the absence of Trrap.
Depletion of Trrap alters histones H4 and H3 acetylation at the Mad1 and Mad2 gene promoters
To elucidate the mechanism by which Trrap regulates the expression of Mad1 and Mad2 genes, we next used chromatin immunoprecipitation (ChIP) to examine the status of histone acetylation within the Mad1 and Mad2 gene promoters (Figure 3A) over the cell cycle phases (Figure 2B). In Trrap-containing cells, histone H4 acetylation levels were significantly increased (hyperacetylation) at G2/M phase compared to G0/G1 and S phases (‘basal' acetylation) (Figure 3B and C). In contrast, Trrap-deficient cells failed to show an increased histone H4 acetylation at G2/M phase (Figure 3B and C), demonstrating that Trrap is needed for G2/M-phase-specific hyperacetylation of histone H4, which correlates with an increased transcription of Mad2 gene at this stage (Figure 2C). In addition, acetylation levels of histone H4 appeared to be also slightly lower in Trrap-deficient cells at G0/G1 and S phases (Figure 3B and C).
Figure 3.
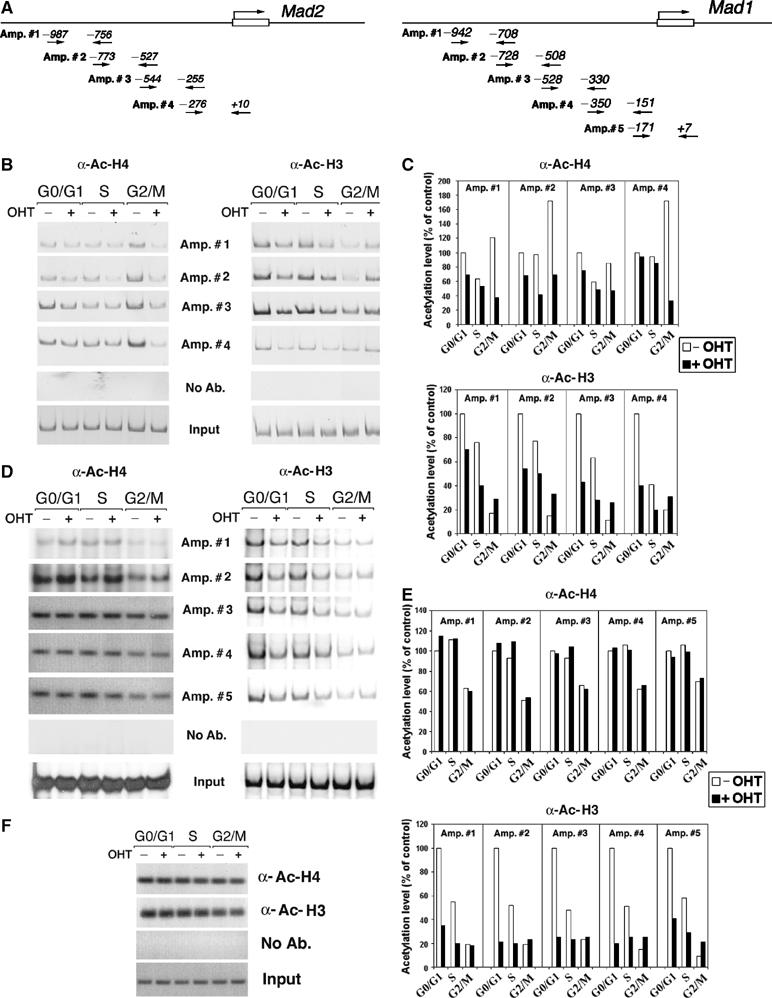
Effect of Trrap deletion on histone acetylation at the Mad gene promoters. (A) Schematic representation of the mouse Mad2 and Mad1 promoter. The arrows indicate the primers used in PCR amplification reactions and the numbers denote the position in relation to the start site. (B) ChIP analysis of histone acetylation at the Mad2 promoter. CER9 cells were synchronized as in Figure 2B and DNAs immunoprecipitated either by anti-acetyl-histone H4 or anti-acetyl-histone H3 were analyzed by PCR using primers recognizing the Mad2 promoter. (C) Quantification of acetylation levels at the Mad2 promoter in (B) by densitometric analysis and normalization versus input. (D) ChIP analysis of histone acetylation at the Mad1 promoter. Samples prepared as in (B) were analyzed by PCR using primers recognizing the Mad1 promoter. (E) Quantification of acetylation levels at the Mad1 promoter was carried out as in (D). (F) ChIP analysis of histone acetylation at the GAPDH promoter. Samples prepared as in (B) were analyzed by using primers recognizing the GAPDH promoter. As a control, mock immunoprecipitations without addition of antibodies (No Ab.) were included. Input corresponds to PCR reactions containing 1/150 of the total amount of chromatin used in immunoprecipitation reactions.
Furthermore, the examination of the acetylation status of histone H3 at the Mad2 promoter revealed that acetylation levels at G0/G1 and S phases were markedly reduced in Trrap-deficient cells (+OHT) compared to Trrap-containing cells (−OHT) (Figure 3B and C). At the G2/M phase, histone H3 acetylation levels in Trrap-containing cells were reduced in comparison to G0/G1 and S phases (Figure 3B and C), consistent with previous observations that histone H3 is hypoacetylated in mitotic cells (Krebs et al, 1999; Kruhlak et al, 2001). It seems that Trrap depletion caused a moderate increase in acetylation of histone H3 at G2/M phase (Figure 3C).
Analysis of acetylation status with different primer sets spanning the Mad1 promoter (Figure 3A) revealed that loss of Trrap specifically compromised acetylation of histone H3 at the Mad1 promoter at G0/G1 and S phases, as was observed for the Mad2 promoter (Figure 3D and E). However, acetylation levels of histone H4 at the Mad1 promoter were not affected in Trrap-deficient cells (Figure 3D and E). As a control, we analyzed acetylation levels of histones H4 and H3 at the GAPDH promoter and found that the depletion of Trrap did not alter acetylation levels of histones H3 and H4 at this promoter (Figure 3F). Together, these results demonstrate that Trrap is involved differentially in the acetylation of histones H4 and H3 at the Mad1 and Mad2 gene promoters during cell cycle progression.
Ectopic expression of Mad1 and Mad2 restores mitotic checkpoint in cells lacking Trrap
If Mad1 and Mad2 are specific targets for Trrap in mitotic checkpoint control, it should be possible to correct the mitotic checkpoint defects in cells lacking Trrap by restoration of Mad1 and Mad2 levels. To this end, Trrap-deficient and Trrap-containing cells were transiently transfected with Mad1 and Mad2 expression vectors together with H2B-GFP (as a fluorescent marker) and expression of exogenous genes was verified by Western blotting (Figure 4A). The transfected cells were incubated in the presence of nocodazole and the mitotic index was determined by counting H2B-GFP-positive cells with condensed chromosomes. As shown in Figure 4B, forced expression of Mad2 alone partially restored the mitotic checkpoint in cells lacking Trrap, whereas expression of Mad1 alone appeared to have no effect. Interestingly, coexpression of Mad1 and Mad2 almost completely restored the mitotic checkpoint in Trrap-deficient cells (Figure 4B). As a control, we transfected Trrap-deficient and Trrap-containing cells with the Mad1 and Mad2 expression vectors in the absence of nocodazole and found no increase in the mitotic index (Figure 4C), ruling out the possibility that Mad1 and Mad2 spontaneously activate the mitotic checkpoint and arrest cells in mitosis. Therefore, the Mad2 protein level appears to be the critical factor for mitotic checkpoint response, although both Mad1 and Mad2 are required for a fully functional mitotic checkpoint.
Figure 4.
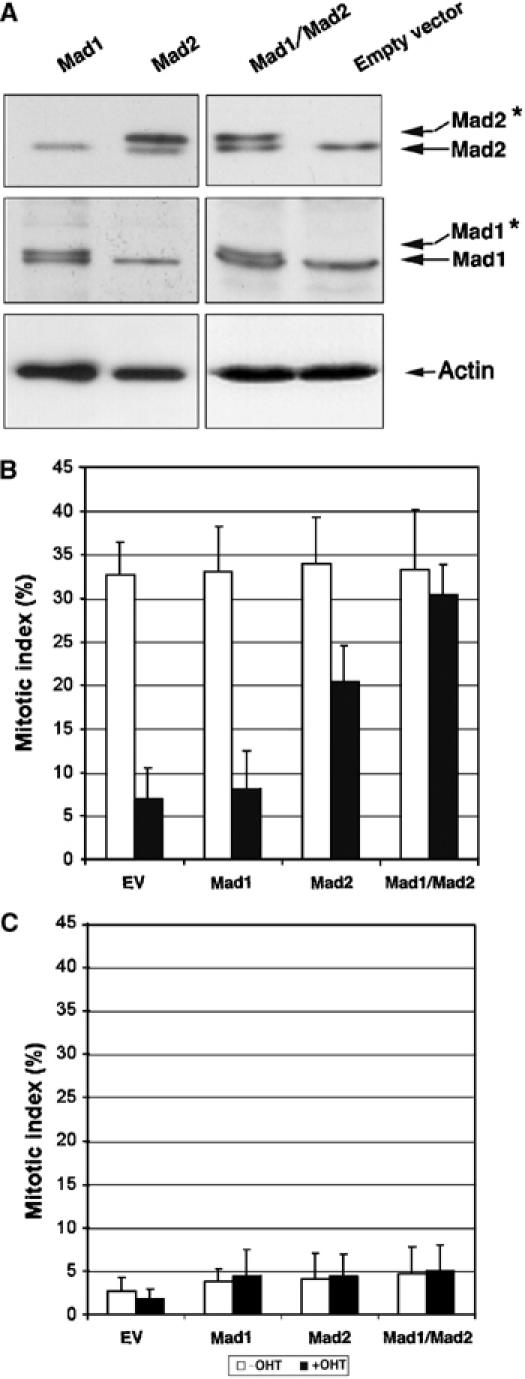
Ectopic expression of Mad1 and Mad2 in Trrap-deficient cells restores mitotic checkpoint. (A) Western blot analysis of protein expression of exogenous Mad1 and Mad2. CER9 cells were grown in the presence or absence of OHT for 24 h and then transfected by expression vector containing either Mad1 or Mad2 alone or Mad1 and Mad2 together. Note that exogenous human Mad1 and Mad2 proteins migrate at a slightly higher position on the gel (asterisk) in comparison to the corresponding endogenous mouse proteins. Actin was used as a loading control. (B, C) Ectopic expression of Mad proteins restores mitotic checkpoint. CER9 cells were grown in the presence or absence of OHT for 24 h and then cotransfected with an empty vector (EV) or indicated expression vectors and a vector expressing H2B-GFP. At 72 h after transfection, cells were incubated for further 10 h in the presence (B) or absence (C) of nocodazole. The mitotic index was determined by scoring GFP-positive cells with condensed chromosomes.
Trrap-dependent recruitment of HATs to the Mad1 and Mad2 promoters over the cell cycle
In order to gain better insight into the mechanism of transcriptional regulation of Mad1 and Mad2 by histone acetylation, we used ChIP assay with antibodies against Trrap, Tip60 and PCAF to examine the binding of these molecules to chromatin at the Mad1/2 promoters over cell cycle phases. As shown in Figure 5A and B, in Trrap-containing cells, Trrap association with the Mad2 and Mad1 promoters was correlated with Tip60 association; however, the correlation between Trrap and PCAF association with these promoters was found at G0/G1 and S phases but not at G2/M phase. Interestingly, patterns of HAT association with the Mad2 and Mad1 promoters (Figure 5A and B) were consistent with the histone acetylation at these promoters (Figure 3B and D). As expected, in Trrap-deficient cells, anti-Trrap antibody immunoprecipitated only residual amounts of chromatin (Figure 5A and B). It is to be noted that in contrast to Tip60, association of PCAF at the Mad2 and Mad1 promoters at G2/M phase was significantly reduced in Trrap-containing cells. Loss of Trrap was accompanied by a dramatic loss of both Tip60 and PCAF binding at the Mad2 and Mad1 promoters (Figure 5A and B), suggesting that Trrap is essential for the recruitment of these HATs to the Mad promoters.
Figure 5.
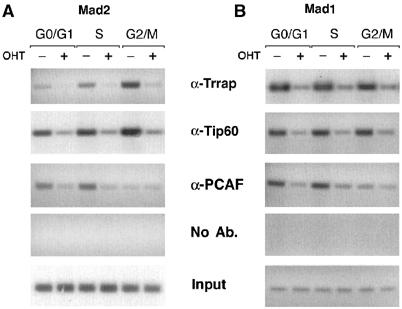
Cell cycle-dependent association of Trrap and HATs with the Mad1 and Mad2 promoters. Trrap-containing and Trrap-deficient cells were synchronized at indicated cell cycle phases as in Figure 2B, and analyzed by ChIP assay using anti-Trrap, anti-Tip60 and anti-PCAF antibodies. Immunoprecipitated DNA was analyzed by PCR using primers recognizing the Mad2 promoter (A) and the Mad1 promoter (B). Mock immunoprecipitation without addition of antibodies (No Ab.) was also included as a control. Input corresponds to PCR reactions containing 1/3750 of the total amount of chromatin used in immunoprecipitation reactions.
Discussion
Trrap is a component of several HAT complexes and is believed to regulate expression of diverse genes that are important for many cellular processes. In the present study, we focus on dissecting the mechanism underlying Trrap's involvement in the mitotic checkpoint and demonstrate that Trrap participates in the mitotic checkpoint by regulation of Mad1 and Mad2 expression and that the mode of regulation by Trrap involves acetylation of histones H3 and H4 at the gene promoters. Consistent with this notion, a recent study showed that deregulation of E2F1, a transcription factor known to interact with TRRAP (McMahon et al, 1998; Lang et al, 2001; Taubert et al, 2004), results in Mad2 overexpression and mitotic defects (Hernando et al, 2004). Although the target genes regulated by Trrap-mediated HATs are distinct from those of E2F1 with only partial overlap (Ren et al, 2002; Herceg et al, 2003), we cannot rule out that Trrap is involved in Mad2 gene expression through promotion of histone acetylation at the E2F1 binding sites in chromatin.
Although MAD and BUB family members are important for the mitotic checkpoint (Musacchio and Hardwick, 2002; Peters, 2002), we found that loss of Trrap compromises the transcription of Mad1 and Mad2, but not five other mitotic players analyzed including Bub1 and Bub3. In addition, ectopic expression of Mad proteins restores the mitotic checkpoint response in Trrap-deficient cells, indicating that Trrap controls the mitotic checkpoint function through specific regulation of Mad genes. It is interesting to note that expression of Mad1 alone in Trrap-deficient cells failed to restore the mitotic checkpoint response, whereas expression of Mad2 alone and Mad1/Mad2 led to partial and complete restoration, respectively, suggesting a differential dependence of the mitotic checkpoint on the Mad1 and Mad2 proteins. Although we cannot rule out that expression of exogenous Mad1 may not undergo normal post-transcriptional and post-translational regulation, two layers of regulation in addition to the transcriptional level shown to be operational in the control of Mad1 expression (Jin et al, 1998), it seems that mitotic checkpoint response is more sensitive to suboptimal levels of Mad2. In agreement with this observation, disruption of one Mad2 allele in mice causes compromised mitotic function and abnormal chromosomal segregation (Michel et al, 2001). However, this is not unexpected in view of the findings that Mad2 can form distinct complexes with Cdc20, a target molecule of the Mad proteins, independent of Mad1 (Sironi et al, 2001). Another possibility is that a reduced level of the Mad1 protein in cells lacking Trrap may still be sufficient to mediate Mad2 localization to kinetochores (Sironi et al, 2001; Chung and Chen, 2002). Another interesting observation is that in Trrap-containing cells, Mad2 mRNA expression levels increase in S and G2/M phases of the cycle, whereas the Mad2 protein levels are stable throughout the cell cycle (Figure 2C). These observations indicate that Mad2 transcription occurs mainly at later stages of the cell cycle, whereas its translation may not be confined to any cell cycle phase, although biological significance of the uncoupled transcription and translation of Mad2 is unknown.
Global histone acetylation dramatically changes during cell cycle progression, correlating well with general transcription (Krebs et al, 1999; Kruhlak et al, 2001). For example, general transcription of the eukaryotic genome, which is highly active in interphase, is abruptly silenced when cells enter mitosis (Gottesfeld and Forbes, 1997). However, promoter-specific and histone-specific acetylation provides a more delicate mechanism to coordinate specific gene activity through cell cycle phases. Indeed, this study shows that, in normal cells, the acetylation states of histones H3 and H4 differentially change through cell cycle at the Mad1 and Mad2 promoters. While histone H3 at the Mad2 promoter was hypoacetylated at G2/M cells, consistent with a mitosis-specific deacetylation of histone H3 (Kruhlak et al, 2001), histone H4 hyperacetylation was observed at G2/M at the Mad2 promoter but not at the Mad1 promoter. Interestingly, different patterns of histone acetylation at the Mad1 and Mad2 promoters were associated with different recruitment of HATs and the loss of Trrap affected histone acetylation patterns at these promoters in a different manner. These data suggest that the expression of Mad1 and Mad2 may be regulated by distinct histone acetylation patterns. This is consistent with the hypothesis commonly known as the ‘histone code' where different patterns of covalent histone modifications provide an epigenetic marker for gene expression patterns (Strahl and Allis, 2000). Consistent with this idea, we found that acetylation patterns depend on the cell cycle stage and the gene promoter.
Loss of Trrap abolished hyperacetylation of histone H4 at G2/M phase, concomitant with failure to accumulate Mad2 mRNA. In addition, acetylation levels of histone H4 at the Mad1/2 promoters at the G0/G1 and S phases were also low in Trrap-deficient cells. These results are in agreement with previous reports that Trrap recruits HAT complexes with affinity toward histone H4 (Allard et al, 1999; Ikura et al, 2000; Bouchard et al, 2001; Brown et al, 2001; Frank et al, 2001; 2003). However, Trrap is dispensable for acetylation of histone H4 at the Mad1 promoter, as Trrap depletion did not affect histone H4 acetylation at this promoter. Interestingly, loss of Trrap resulted in hypoacetylation of histone H3 at both Mad1 and Mad2 promoters at G0/G1 and S phases with a stronger effect on the Mad1 promoter. This effect may be explained by Trrap's role in several chromatin-remodeling complexes with histone H3 specificity (Grant et al, 1998; Vassilev et al, 1998; McMahon et al, 2000; Bouchard et al, 2001; Brown et al, 2001; Frank et al, 2001; 2003). These results suggest that Trrap is required for coordinated recruitment of specific HAT activities dependent on cell cycle stage and gene promoters. Surprisingly, we found that loss of Trrap resulted in an increased acetylation of histone H3 at the Mad2 promoter in the mitotic population (Figure 3B). Although the precise mechanism underlying this is not known, it raises an interesting possibility that crosstalk may exist between histones H3 and H4 acetylation during mitosis. A dynamic equilibrium exists between HAT and HDAC activities during mitosis (Kruhlak et al, 2001; Shin et al, 2003), and it is possible that loss of histone H4 acetylation caused by Trrap deficiency may compromise recruitment of HDAC with histone H3 specificity during mitosis.
In agreement with Trrap's function as a component of HAT complexes containing Tip60, which preferentially acetylates H4, and PCAF, which preferentially acetylates H3, we found colocalization of Trrap with Tip60 and PCAF at the Mad1 and Mad2 promoters. Trrap depletion abolished recruitment of these HATs to chromatin, consistent with an essential role for Trrap in the recruitment of HATs with H4 and H3 specificity at the Mad promoters (Carrozza et al, 2003). Interestingly, association patterns of Trrap and HATs with the Mad2 promoter were different from those with the Mad1 promoter, suggesting distinct patterns of recruitment of Trrap and HATs at these two promoters. In Trrap-containing cells, patterns of Tip60 and PCAF association with Mad1/2 promoters (Figure 5A and B) correspond to the patterns of histones H4 and H3 acetylation (Figure 3B and D), respectively, suggesting that these HATs are largely responsible for histone acetylation at the Mad1/2 promoters. We, however, note that loss of Trrap results in the absence of Tip60 loading at the Mad1 promoter (Figure 5A), although H4 acetylation levels at this promoter remain unchanged (Figure 3D). This suggests that the recruitment of a histone H4-acetylating HAT other than Tip60 to the Mad1 promoter occurs independently of Trrap.
The majority of human cancers exhibit genetic instability, caused in many cases by altered expression of mitotic checkpoint genes (Lengauer et al, 1998). For example, decreased expression of hMAD2 has been documented in human breast cancers, while haploinsufficiency of this gene causes chromosomal instability in both mouse and human cells (Michel et al, 2001). The essential nature of the Trrap gene in embryonic development represents an obstacle in testing the effects of the disruption of Trrap and histone acetylation in vivo (Herceg et al, 2001). Therefore, an animal model in which Trrap is inactivated in an inducible manner in tissues would allow the function of Trrap in chromosome instability and tumor development to be defined. Nevertheless, the present study demonstrates that Trrap, or Trrap-associated HAT, regulates the mitotic checkpoint response in a delicate manner through chromatin-remodeling activity. These findings highlight the HAT/HDAC complexes as a potential pharmaceutical target in prevention of chromosomal instability and suppression of tumor development.
Materials and methods
Cell culture
Immortalized MEFs that are amenable to inducible deletion of Trrap by Cre-ER were described previously (Herceg et al, 2001). The cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), penicillin/streptomycin and glutamine. Deletion of Trrap was induced with 1 μM OHT (Sigma, St Louis, MO), as described previously (Herceg et al, 2001). To obtain the cell populations in G0/G1 phase (quiescent cells), Trrap-deficient and Trrap-containing cells were serum starved for 24 h. To obtain the S-phase (S) and G2-phase/mitotic (G2/M) populations, cells were harvested at 18 and 28 h in the presence of nocodazole, respectively, after release from G0/G1 block. Cell cycle synchronization was verified by flow cytometry as described previously (Herceg et al, 2001). Primary MEFs were cultured under the same conditions as immortalized cells, except that β-mercaptoethanol (5 × 10−5 M) was added to the DMEM medium. For the experiments on primary MEFs, cells at early passages (2–3) were used.
Isolation of primary Trrap mutant MEF cells and deletion of Trrap by adenovirus expressing Cre recombinase
To generate primary Trrap ‘conditional' knockout cells, we first generated Trrap ‘floxed' (Trrapf/+) mice by blastocyst injection of Trrapf/+ embryonic stem cells established previously (Herceg et al, 2001). After crossing Trrapf/+ mice with Trrap+/Δ mice (Herceg et al, 2001), we obtained Trrapf/Δ offspring. Trrapf/Δ mice were then crossed with Trrap+/Δ mice, and primary MEFs of different genotypes were isolated from E13.5 fetuses. Trrap ‘floxed' allele was deleted by infection of adenovirus containing Cre recombinase under the control of the hCMV promoter (Akagi et al, 1997) for 48 h at a multiplicity of infection (MOI) of 100.
Establishment of Trrap ‘conditional' knockout MEFs expressing H2B-GFP
We cotransfected the human histone H2B gene fused to the gene encoding GFP (Kanda et al, 1998) and plasmid expressing Cre-ER recombinase (pCre-ER) into Trrapf/t MEFs (TA2; Herceg et al, 2001). Following blasticidine selection (2 μg ml−1), stable lines constitutively expressing H2B-GFP and Cre-ER were identified by green fluorescence and PCR, respectively.
Time-lapse microscopy of living cells
To visualize mitotic chromosomes in living cells, cells coexpressing H2B-GFP and Cre-ER (TA-5-8) were grown in 35-mm glass-bottom culture dishes in normal culture medium supplemented with 20 mM HEPES. The dishes were then mounted on an inverted confocal microscope (Axiovert 200M, Zeiss, Jena, Germany) equipped with an incubator to keep constant temperature, humidity and CO2 atmosphere. Images were acquired using Laser Scanning Microscope LSM 510 software (Zeiss).
Plasmids and transfections
Human cyclin B1 and a D-box mutant thereof subcloned into the mammalian expression vector pEFT7MCS (Pepperkok et al, 1999) were a gift of M Brandeis (Hebrew University, Jerusalem, Israel). Expression vectors containing human Mad2 cDNA under the control of CMV promoter and human Mad1 cDNA under the control of the SV40 promoter were a gift of Andrea Musacchio (European Institute of Oncology, Milan, Italy) and Kuan-Teh Jeang (NIH, Bethesda, MD, USA), respectively. All transfections were carried out using Lipofectamine reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. In all experiments, 1.5 μg of plasmid was used with the exception of the plasmid containing a selection marker, where 0.2 μg of plasmid was used.
Determination of mitotic index by fluorescent microscopy
Cells were grown in the presence and absence of OHT for 2 days and the microtubule-damaging drugs (100 ng ml−1 nocodazole, 10 ng ml−1 colcemid, 2 μM taxol; all from Sigma, St Louis, MO) were added to the culture and incubated for a further 10 h. Following fixation and staining with DAPI, the mitotic index was determined by scoring the percentage of cells with condensed chromosomes, as described previously (Herceg et al, 2001).
Determination of mitotic index in PMEFs by flow cytometry
Quantification of mitotic index in primary MEFs was performed by staining of chromosomal DNA by propidium iodide combined with immunofluorescent staining of phospho-histone H3 with anti-phospho-histone H3 antibodies (#06-570, Upstate Biotechnology, Charlottesville, VA) using a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA) as described previously (Xu et al, 2001).
Western blot analysis
Western blotting was carried out essentially as described previously (Herceg et al, 2001). Proteins were resolved by electrophoresis on 10% SDS–PAGE gels and the gels were blotted onto the nitrocellulose membrane (Bio-Rad, Hercules, CA). Primary antibodies used were as follows: rabbit α-Mad2 (1:1000; BD Transduction Laboratories, Franklin Lakes, NJ), rabbit α-Mad1 (1:1000; donated by K-T Jeang, NIH, Bethesda, MD, USA), rabbit α-securin (1:500; donated by JM Peters, Research Institute of Molecular Pathology, Vienna, Austria), rabbit α-Bub3 (1:1000; BD Transduction Laboratories, Franklin Lakes, NJ) and mouse α-actin (1:10 000; ICN Biomedicals Inc., Irvine, CA).
Northern blot analysis
RNA probes were prepared by reverse transcription–PCR (RT–PCR) on total RNA prepared from MEFs as described previously (Herceg et al, 2003). Total RNA (10 μg) was electrophoresed using a 0.9% agarose gel, in the presence of 6.7% formaldehyde as described previously (Herceg et al, 2003). 32P-labeled probes were made from 20–50 ng of cDNA using random priming by the Prime-IT RmT kit according to the manufacturer's instructions (Stratagene, La Jolla, CA). The conditions for probe hybridization were as instructed by the membrane manufacturer.
Chromatin immunoprecipitation assay
Cells cultured in 15-cm plates in the presence or absence of OHT for 48 h and synchronized by serum starvation for 24 h were crosslinked with formaldehyde. ChIPs were performed as previously described (Herceg et al, 2003), using polyclonal antibodies specific for acetylated histone H3 (#06-599), acetylated histone H4 (#06-866), Tip60, PCAF (all from Upstate Biotechnology, Charlottesville, VA) and TRRAP (Santa Cruz Biotechnology, Santa Cruz, CA). The ChIP primers used to analyze the Mad2 promoter were as follows: for amplicon #1 5′GCAGTCTGAGTACCAGAGTTA (Mad2.1.For) and 5′GATAGAGACACTTGATTTCC (Mad2.4.Rev); for amplicon #2 5′GGAAATCAAGTGTCTCTATC (Mad2.4.For) and 5′AGCATGGTCTCAGCTTCGAG (Mad2.5.Rev); for amplicon #3 5′CTCGAAGCTGAGACCATGCT (Mad2.5.For) and 5′CAACATTCCGCTCCACATCG (Mad2.6.Rev); for amplicon #4 5′CGATGTGGAGCGGAATGTTG (Mad2.6.For) and 5′GCTGTGCCATCACTAGATCG (Mad2.3.REV). The ChIP primers for the Mad1 promoter were as follows: for amplicon #1 5′TGTGTGCCATGGAGGTTAGG (Mad1.1.FOR) and 5′CTGTCCTTCACTCACACCAGG (Mad1.1.REV); for amplicon #2 5′CCTGGTGTGAGTGAAGGACAG (Mad1.2.FOR) and 5′CCAGGCTAATAACTGCCTGC (Mad1.2.REV); for amplicon #3 5′GCAGGCAGTTATTAGCCTGG (Mad1.3.FOR) and 5′TCACACAGAGGCAGGAAGAG (Mad1.3.REV); for amplicon #4 5′CTCTTCCTGCCTCTGTGTGA (Mad1.4.FOR) and 5′GGAAATGAGGCCCAAAGCAC (Mad1.4.REV); for amplicon #5 5′GTGCTTTGGGCCTCATTTCC (Mad1.5.FOR) and 5′CTTCCATGGTCAGCCTCCTT (Mad1.5.REV). The ChIP primers for the GAPDH promoter were 5′CCGCATCTTCTTGTGCAGTG (MmGAPDH-1.FOR) and 5′CATTCTCGGCCTTGACTGTG (MmGAPDH-1.REV).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Acknowledgments
We acknowledge the generous gifts of reagents from Andrea Musacchio (Mad1 and Mad2 vectors), Kuan-Teh Jeang (α-Mad1 antibodies and Mad1 and Mad2 vectors), Jan-Michael Peters (α-securin antibodies) and Michael Brandeis (CLB-GFP and CLB-DM-GFP vectors). We thank Joanna Loizou, Alain Puisieux, Bakary Sylla and Massimo Tommasino for helpful suggestions and critical reading of the manuscript. We also thank John Cheney and Fiona Corry for editing the manuscript. This work was supported by the Association for International Cancer Research (AICR), UK, and the Association pour la Recherche sur le Cancer (ARC), France.
References
- Akagi K, Sandig V, Vooijs M, Van der Valk M, Giovannini M, Strauss M, Berns A (1997) Cre-mediated somatic site-specific recombination in mice. Nucleic Acids Res 25: 1766–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard S, Utley RT, Savard J, Clarke A, Grant P, Brandl CJ, Pillus L, Workman JL, Cote J (1999) NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J 18: 5108–5119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Dittrich O, Kiermaier A, Dohmann K, Menkel A, Eilers M, Luscher B (2001) Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev 15: 2042–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Howe L, Sousa K, Alley SC, Carrozza MJ, Tan S, Workman JL (2001) Recruitment of HAT complexes by direct activator interactions with the ATM-related Tra1 subunit. Science 292: 2333–2337 [DOI] [PubMed] [Google Scholar]
- Brown CE, Lechner T, Howe L, Workman JL (2000) The many HATs of transcription coactivators. Trends Biochem Sci 25: 15–19 [DOI] [PubMed] [Google Scholar]
- Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B (1998) Mutations of mitotic checkpoint genes in human cancers. Nature 392: 300–303 [DOI] [PubMed] [Google Scholar]
- Carrozza MJ, Utley RT, Workman JL, Cote J (2003) The diverse functions of histone acetyltransferase complexes. Trends Genet 19: 321–329 [DOI] [PubMed] [Google Scholar]
- Chen H, Tini M, Evans RM (2001) HATs on and beyond chromatin. Curr Opin Cell Biol 13: 218–224 [DOI] [PubMed] [Google Scholar]
- Cheung P, Allis CD, Sassone-Corsi P (2000) Signaling to chromatin through histone modifications. Cell 103: 263–271 [DOI] [PubMed] [Google Scholar]
- Chung E, Chen RH (2002) Spindle checkpoint requires Mad1-bound and Mad1-free Mad2. Mol Biol Cell 13: 1501–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleu L, Shellard S, Alevizopoulos K, Amati B, Land H (2001) Recruitment of TRRAP required for oncogenic transformation by E1A. Oncogene 20: 8270–8275 [DOI] [PubMed] [Google Scholar]
- Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B (2003) MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep 4: 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B (2001) Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev 15: 2069–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant P, Nigg EA (1992) Cyclin B2 undergoes cell cycle-dependent nuclear translocation and, when expressed as a non-destructible mutant, causes mitotic arrest in HeLa cells. J Cell Biol 117: 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesfeld JM, Forbes DJ (1997) Mitotic repression of the transcriptional machinery. Trends Biochem Sci 22: 197–202 [DOI] [PubMed] [Google Scholar]
- Grant PA, Schieltz D, Pray-Grant MG, Yates JR III, Workman JL (1998) The ATM-related cofactor Tra1 is a component of the purified SAGA complex. Mol Cell 2: 863–867 [DOI] [PubMed] [Google Scholar]
- Hauf S, Waizenegger IC, Peters JM (2001) Cohesin cleavage by separase required for anaphase and cytokinesis in human cells. Science 293: 1320–1323 [DOI] [PubMed] [Google Scholar]
- Herceg Z, Hulla W, Gell D, Cuenin C, Lleonart M, Jackson S, Wang ZQ (2001) Disruption of Trrap causes early embryonic lethality and defects in cell cycle progression. Nat Genet 29: 206–211 [DOI] [PubMed] [Google Scholar]
- Herceg Z, Li H, Cuenin C, Shukla V, Radolf M, Steinlein P, Wang ZQ (2003) Genome-wide analysis of gene expression regulated by the HAT cofactor Trrap in conditional knockout cells. Nucleic Acids Res 31: 7011–7023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, Lowe SW, Cordon-Cardo C (2004) Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 430: 797–802 [DOI] [PubMed] [Google Scholar]
- Holloway SL, Glotzer M, King RW, Murray AW (1993) Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell 73: 1393–1402 [DOI] [PubMed] [Google Scholar]
- Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y (2000) Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102: 463–473 [DOI] [PubMed] [Google Scholar]
- Jallepalli PV, Waizenegger IC, Bunz F, Langer S, Speicher MR, Peters JM, Kinzler KW, Vogelstein B, Lengauer C (2001) Securin is required for chromosomal stability in human cells. Cell 105: 445–457 [DOI] [PubMed] [Google Scholar]
- Jin DY, Spencer F, Jeang KT (1998) Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell 93: 81–91 [DOI] [PubMed] [Google Scholar]
- Kanda T, Sullivan KF, Wahl GM (1998) Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 8: 377–385 [DOI] [PubMed] [Google Scholar]
- Krebs JE, Kuo MH, Allis CD, Peterson CL (1999) Cell cycle-regulated histone acetylation required for expression of the yeast HO gene. Genes Dev 13: 1412–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak MJ, Hendzel MJ, Fischle W, Bertos NR, Hameed S, Yang XJ, Verdin E, Bazett-Jones DP (2001) Regulation of global acetylation in mitosis through loss of histone acetyltransferases and deacetylases from chromatin. J Biol Chem 276: 38307–38319 [DOI] [PubMed] [Google Scholar]
- Lang SE, McMahon SB, Cole MD, Hearing P (2001) E2F transcriptional activation requires TRRAP and GCN5 cofactors. J Biol Chem 276: 32627–32634 [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B (1998) Genetic instabilities in human cancers. Nature 396: 643–649 [DOI] [PubMed] [Google Scholar]
- Li Y, Benezra R (1996) Identification of a human mitotic checkpoint gene: hsMAD2. Science 274: 246–248 [DOI] [PubMed] [Google Scholar]
- Lin SF, Lin PM, Yang MC, Liu TC, Chang JG, Sue YC, Chen TP (2002) Expression of hBUB1 in acute myeloid leukemia. Leuk Lymphoma 43: 385–391 [DOI] [PubMed] [Google Scholar]
- McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD (1998) The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell 94: 363–374 [DOI] [PubMed] [Google Scholar]
- McMahon SB, Wood MA, Cole MD (2000) The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol 20: 556–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R (2001) MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 409: 355–359 [DOI] [PubMed] [Google Scholar]
- Musacchio A, Hardwick KG (2002) The spindle checkpoint: structural insights into dynamic signalling. Nat Rev Mol Cell Biol 3: 731–741 [DOI] [PubMed] [Google Scholar]
- Pepperkok R, Squire A, Geley S, Bastiaens PI (1999) Simultaneous detection of multiple green fluorescent proteins in live cells by fluorescence lifetime imaging microscopy. Curr Biol 9: 269–272 [DOI] [PubMed] [Google Scholar]
- Peters JM (2002) The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol Cell 9: 931–943 [DOI] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 16: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh A, Schieltz D, Ting N, McMahon SB, Litchfield DW, Yates JR III, Lees-Miller SP, Cole MD, Brandl CJ (1998) Tra1p is a component of the yeast Ada.Spt transcriptional regulatory complexes. J Biol Chem 273: 26559–26565 [DOI] [PubMed] [Google Scholar]
- Shin HJ, Baek KH, Jeon AH, Kim SJ, Jang KL, Sung YC, Kim and CM, Lee CW (2003) Inhibition of histone deacetylase activity increases chromosomal instability by the aberrant regulation of mitotic checkpoint activation. Oncogene 22: 3853–3858 [DOI] [PubMed] [Google Scholar]
- Simizu S, Osada H (2000) Mutations in the Plk gene lead to instability of Plk protein in human tumour cell lines. Nat Cell Biol 2: 852–854 [DOI] [PubMed] [Google Scholar]
- Sironi L, Melixetian M, Faretta M, Prosperini E, Helin K, Musacchio A (2001) Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for the spindle checkpoint. EMBO J 20: 6371–6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41–45 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Haruki N, Nomoto S, Masuda A, Saji S, Osada H (1999) Identification of frequent impairment of the mitotic checkpoint and molecular analysis of the mitotic checkpoint genes, hsMAD2 and p55CDC, in human lung cancers. Oncogene 18: 4295–4300 [DOI] [PubMed] [Google Scholar]
- Taubert S, Gorrini C, Frank SR, Parisi T, Fuchs M, Chan HM, Livingston DM, Amati B (2004) E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol Cell Biol 24: 4546–4556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev A, Yamauchi J, Kotani T, Prives C, Avantaggiati ML, Qin J, Nakatani Y (1998) The 400 kDa subunit of the PCAF histone acetylase complex belongs to the ATM superfamily. Mol Cell 2: 869–875 [DOI] [PubMed] [Google Scholar]
- Waizenegger I, Gimenez-Abian JF, Wernic D, Peters JM (2002) Regulation of human separase by securin binding and autocleavage. Curr Biol 12: 1368–1378 [DOI] [PubMed] [Google Scholar]
- Wang C, Fu M, Mani S, Wadler S, Senderowicz AM, Pestell RG (2001) Histone acetylation and the cell-cycle in cancer. Front Biosci 6: D610–D629 [DOI] [PubMed] [Google Scholar]
- Wang X, Jin DY, Ng RW, Feng H, Wong YC, Cheung AL, Tsao SW (2002) Significance of MAD2 expression to mitotic checkpoint control in ovarian cancer cells. Cancer Res 62: 1662–1668 [PubMed] [Google Scholar]
- Wang X, Jin DY, Wong YC, Cheung AL, Chun AC, Lo AK, Liu Y, Tsao SW (2000) Correlation of defective mitotic checkpoint with aberrantly reduced expression of MAD2 protein in nasopharyngeal carcinoma cells. Carcinogenesis 21: 2293–2297 [DOI] [PubMed] [Google Scholar]
- Xu B, Kim S, Kastan MB (2001) Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol 21: 3445–3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3