

Abstract
Using the proteomic tandem affinity purification (TAP) method, we have purified the Saccharomyces cerevisie U2 snRNP-associated splicing factors SF3a and SF3b. While SF3a purification revealed only the expected subunits Prp9p, Prp11p and Prp21p, yeast SF3b was found to contain only six subunits, including previously known components (Rse1p, Hsh155p, Cus1p, Hsh49p), the recently identified Rds3p factor and a new small essential protein (Ysf3p) encoded by an unpredicted split ORF in the yeast genome. Surprisingly, Snu17p, the proposed yeast orthologue of the seventh human SF3b subunit, p14, was not found in the yeast complex. TAP purification revealed that Snu17p, together with Bud13p and a newly identified factor, Pml1p/Ylr016c, form a novel trimeric complex. Subunits of this complex were not essential for viability. However, they are required for efficient splicing in vitro and in vivo. Furthermore, inactivation of this complex causes pre-mRNA leakage from the nucleus. The corresponding complex was named pre-mRNA REtention and Splicing (RES). The presence of RES subunit homologues in numerous eukaryotes suggests that its function is evolutionarily conserved.
Keywords: RES, SF3a, SF3b, tandem affinity purification (TAP), U2 snRNP
Introduction
Pre-mRNA splicing occurs by two sequential transesterification reactions that are catalysed by a large and dynamic ribonucleoprotein complex: the spliceosome. Every splicing reaction necessitates the stepwise assembly of a full spliceosome onto the pre-mRNA. Interestingly, two different but evolutionarily related spliceosomes co-exist in most eukaryotic cells. Formation of major spliceosomes involves the U1, U2, U4, U5 and U6 snRNPs, while that of the minor spliceosomes requires U11, U12, U4ATAC, U5 and U6ATAC snRNPs. For major spliceosomes, U1 snRNP binding to the intron 5′ splice site allows the formation of a commitment complex for yeast or complex E in mammals (Séraphin and Rosbash, 1989a; Michaud and Reed, 1991). This step is followed by pre-spliceosome formation through U2 snRNP addition. Finally, the U4/U6.U5 triple snRNP joining generates the complete spliceosome (Pikielny et al, 1986; Konarska and Sharp, 1987). After remodelling steps, during which the U1 and U4 snRNPs dissociate, the first splicing reaction occurs. A second remodelling step precedes the last transesterification reaction. Recently, an alternative scenario, suggesting that a pre-assembled complex of all snRNPs binds the pre-mRNA as a single unit, was proposed (Stevens et al, 2002). In this model, the spliceosome also must undergo remodelling before each catalytic step. In the cell, molecular mechanism must act in parallel to splicing to ensure that pre-mRNAs are not exported to the cytoplasm before their complete processing. This process is not well understood. Many factors involved in spliceosome assembly have associated pre-mRNA leakage and splicing inhibition phenotypes (Legrain and Rosbash, 1989). Pre-mRNA leakage is then likely to result indirectly from pre-mRNA accumulation, even though this is not always the case (Rutz and Séraphin, 2000). More recently, the new yeast factor, Mlp1, was directly implicated in nuclear pre-mRNA retention without affecting splicing itself (Galy et al, 2004).
U2 and U6 snRNPs have been implicated in catalysis of splicing in the active spliceosome (Valadkhan and Manley, 2001), while U5 snRNP is known to interact with exons (Newman and Norman, 1992; Wyatt et al, 1992). Spliceosome remodelling allows the U2 and U6 snRNAs to form a base pair (Brow, 2002). In addition, these snRNAs interact with the pre-mRNA: U2 snRNA is involved in branchpoint adenosine definition (Parker et al, 1987), while U6 snRNA selects the 5′ splice site (Kandels-Lewis and Séraphin, 1993; Lesser and Guthrie, 1993). The U2 snRNP is a large structure containing the U2 snRNA, seven Sm proteins and two U2-specific proteins: U2A′ and U2B″ (Lea1p and Msl1p in Saccharomyces cerevisiae) (Lührmann et al, 1990; Tang et al, 1996; Caspary and Séraphin, 1998). In addition, two multisubunit complexes associate with U2 snRNP: SF3a and SF3b (Brosi et al, 1993). SF3a is composed of three polypeptides (SF3a120, SF3a66, SAP130, in human; Prp9p, Prp11p and Prp21p in yeast; Krämer, 1996), while human SF3b was reported to contain seven proteins (SAP130, SAP155, SAP145, SF3b49, SF3b14b, p14 and SF3b10)(Gozani et al, 1996; Das et al, 1999; Will et al, 2002). Yeast homologues of five of these factors have been characterized (Wells et al, 1996; Igel et al, 1998; Caspary et al, 1999; Wang and Rymond, 2003). Interestingly, human SF3a acts exclusively during the splicing of major introns (Will et al, 1999), while SF3b is required for splicing of both types of introns by associating with U2 and U12 snRNPs, respectively (Das et al, 1999; Will et al, 1999). Thus, both factors are essential for splicing. Furthermore, many SF3b subunits can be crosslinked to the branchpoint region of the pre-mRNA (Staknis and Reed, 1994; Gozani et al, 1996; Query et al, 1996; McPheeters and Muhlenkamp, 2003). Interestingly, the human SF3b p14 subunit that crosslinks directly to the branchpoint lies in the centre of a shell made by the other SF3b subunits in a structural model of the complex obtained by cryo-electron microscopy (Golas et al, 2003).
We have used the TAP proteomic approach developed in our laboratory (Rigaut et al, 1999; Puig et al, 2001), to characterize the yeast U2 snRNP-associated complexes. Identification of the SF3a subunits by mass spectrometry revealed the presence of the three previously known factors: Prp9p, Prp11p and Prp21p, but no additional component. In-depth analysis of SF3b revealed six subunits, four of which had been identified previously, namely Rse1p, Hsh155p, Cus1p and Hsh49p (Wells et al, 1996; Igel et al, 1998; Caspary et al, 1999; Puig et al, 2001). We also found Rds3p that was independently identified while this work was in progress (Wang and Rymond, 2003). Interestingly, we identified a novel, unpredicted small yeast protein, as a new subunit of this complex. This subunit, encoded by a small split yeast ORF, is essential for yeast growth. Surprisingly, however, Snu17p, which was suggested to represent the yeast orthologue of the human p14 branchpoint-binding protein (Gottschalk et al, 2001; Wang and Rymond, 2003), was consistently absent from our purifications. TAP purification revealed that Snu17p is present in an independent trimeric complex. Subunits of this new complex, called pre-mRNA REtention and Splicing (RES), are dispensable for yeast cell viability. These proteins are, however, conserved in eukaryotes and required for efficient pre-mRNA splicing in vivo and in vitro. More importantly, inactivation of RES subunits induces nuclear pre-mRNA leakage. Overall, our proteomic analysis of the yeast SF3a and SF3b splicing factors established definitively their composition, revealing altogether the presence of unsuspected subunits. It also led to the identification and functional characterization of the new RES complex. These findings have implication for our understanding of the composition and organization of the mammalian splicing machinery.
Results
Yeast SF3a contains three subunits
The yeast SF3a subunits, Prp9p, Prp11p and Prp21p, were identified genetically and, based on their homology to human subunits and in vitro studies, suggested to be SF3a components (reviewed in Krämer, 1996). However, as yeast SF3a was not purified, it remained possible that more subunits were present and/or that these three proteins did not form the predicted complex. Thus, we purified SF3a using a Prp9p TAP fusion. Purified products were fractionated by SDS–PAGE. This consistently revealed three proteins present in apparent relative stoichiometric amount (Figure 1A). These factors were identified by mass spectrometry as Prp9p, Prp11p and Prp21p. We conclude that yeast SF3a contains only three subunits.
Figure 1.
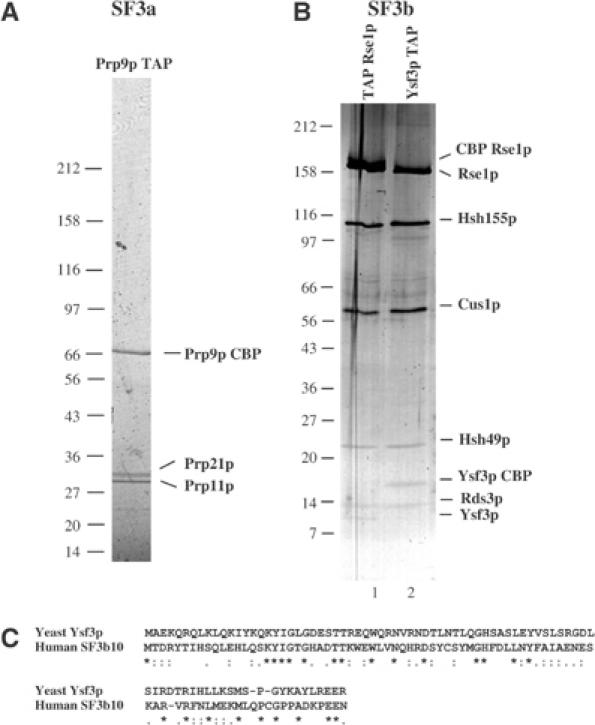
Characterization of yeast SF3a and SF3b complexes. (A) TAP purification of yeast SF3a using a PRP9 TAP fusion identifies three subunits. The TAP purified material was fractionated on a 5–20% gradient SDS–PAGE gel and stained with Coomassie Blue. Proteins were identified by MALDI. (B) TAP purification of yeast SF3b complex using TAP Rse1 or Ysf3 TAP fusions identifies six subunits. The purified material was fractionated on a 5–20% gradient SDS–PAGE gel, which was then stained with silver. Proteins were identified by MALDI. (C) Sequence alignment of human SF3b10 protein and its yeast homologue Ysf3p. Symbols indicate identity (*), high (:) or low (.) similarity.
Characterization of yeast SF3b reveals new subunits
TAP purification of the yeast U2 snRNP led us previously to identify and characterize Rse1p, a yeast homologue to human SF3b130 (Caspary et al, 1999). Hsh155p was also identified in the purified material (data not shown). We used a N-terminal TAP-tagged Rse1p to characterize yeast SF3b (Puig et al, 2001). Mass spectrometry analysis identified previously known components of yeast SF3b (Rse1p, Hsh155p, Cus1p and Hsh49p; Puig et al, 2001) as well as a new 14 kDa protein: Rds3p (data not shown). While this work was in progress, the presence of Rds3p in yeast SF3b was independently reported (Wang and Rymond, 2003). Additional smaller products were consistently seen in SF3b purifications (e.g., Figure 1B, lane 1), but we were unable to identify them in the yeast protein database through mass spectrometry (data not shown). The recent purification of human SF3b revealed the presence of two new small subunits of 14 and 10 kDa, namely SF3b14 and SF3b10 (Will et al, 2002). While the similarity between SF3b14 and Rds3p was noted, SF3b10 was reported to have no homologues in yeast. BLAST searches against the yeast genome database revealed, however, a sequence that, when translated, showed significant similarity to human SF3b10 (Figure 1C). The corresponding ORF had not been predicted because a putative intron disconnects the initiating ATG from the downstream coding sequence. To confirm that this new gene, yeast splicing factor 3b subunit (YSF3), was active, we performed a 5′ RACE analysis. Sequencing of the resulting product confirmed that this region was transcribed and that the putative intron was removed, thus generating the predicted coding sequence (data not shown). To confirm that the cognate polypeptide was made and that it was a subunit of yeast SF3b, we fused the TAP tag to the C-terminus of Ysf3p. Western blot analysis confirmed that Ysf3p was expressed and the profile of the six subunits recovered after TAP purification, gel fractionation and silver staining was identical to the one observed following purification of TAP Rse1p (Figure 1B, compare lanes 1 and 2; note the expected mobility change of Rse1p and Ysf3p due to the tag). Mass spectrometry identified unequivocally the six subunits consistently recovered in such purifications in apparent relative stoichiometric amounts as Rse1p, Hsh155p, Cus1p, Hsh49p, Rds3p and Ysf3p. To confirm this finding and to rule out the presence of an undetected co-migrating subunit, we TAP tagged the remaining subunits of the yeast SF3b complex, namely Hsh155p, Cus1p, Hsh49p and Rds3p. Parallel TAP purification with the six SF3b subunits revealed that the major proteins recovered co-migrated (except for the tagged subunit that was enlarged by the expected mass) and that no additional subunit could be detected (data not shown). Overall, these results demonstrated that yeast SF3b contains only six subunits, including the newly identified Rds3p and Ysf3p factors.
Snu17p/Ist3 belongs to a new evolutionally conserved trimeric complex: RES
While seven proteins have been reported to be present in human SF3b (Will et al, 2002), our analysis failed to detect the yeast homologue to the human p14 subunit (Table I). More importantly, we could not detect the Snu17p factor, which was suggested to represent the yeast p14 homologue (Gottschalk et al, 2001; Will et al, 2001; Wang and Rymond, 2003). To ascertain that Snu17p was not a yeast SF3b subunit, we TAP purified Snu17p and associated factors. Consistent with our findings, the protein profile was different from the pattern observed in SF3b purifications (compare Figures 1B and 2A). Three proteins were present in apparent relative stoichiometric amounts. This new complex was named RES (see below). Mass spectrometry identified its subunits as Snu17p, Bud13p and the product of the YLR016c ORF (pre-mRNA leakage 1 (Pml1p), see below), as well as low levels of contaminating ribosomal and heat shock proteins that were not reproducibly found (data not shown). None of the RES subunits were detected or identified in the six TAP purifications performed with tagged SF3b subunits (see above).
Table 1.
Comparison of yeast and human SF3a, SF3b and RES subunits
| Yeast | Human | Domains |
|---|---|---|
| SF3a | ||
| Prp9p | SF3a60 | 2 × Zn finger |
| Prp11p | SF3a66 | Zn finger |
| Prp21p | SF3a120 | Swap domain |
| SF3b | ||
| Rse1p | SAP130 | β-Propeller repeats |
| Hsh155p | SAP155 | HEAT repeats |
| Cus1p | SAP145 | |
| Hsh49p | SF3b49 | 2 × RRM |
| Rds3p | SF3b14b | Zn finger |
| — | p14 | RRM |
| Ysf3p | SF3b10 | |
| RES | ||
| Snu17p | CGI-79 | RRM |
| Bud13p | MGC13125 | |
| Pml1p | Snip | FHA domain |
Figure 2.
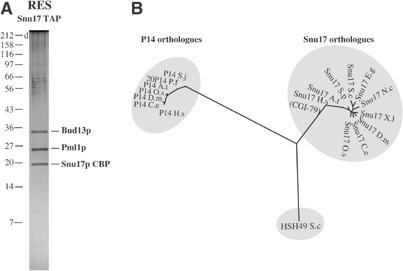
Identification of the new RES complex associated with Snu17p. (A) Proteins associated with Snu17p were purified by the TAP method. Purified material was fractioned on a 10% tricine PAGE gel and stained with Coomassie Blue. Proteins were identified by MALDI. In addition to Snu17p, Bud13p and Pml1p, we identified Ssa2p heat shock protein and some ribosomal proteins. These common contaminants were not reproducibly seen in other purifications (not shown). (B) Snu17 is not a P14 branchpoint-binding protein orthologue. A phylogenetic tree of Snu17 and human P14 homologue RNA-binding domains was built and rooted with the RNA-binding domain of Hsh49. Accession numbers of the sequences are as follows: P14 Plasmodium falciparum (Q8I5G8); P14 Arabidopsis thaliana (Q9FMP4); P14 Oryza sativa (Q7XZG6); P14 Schistosoma japonicum (Q86EF3); P14 Caenorhabditis elegans (Q8ITY4); P14 Drosophila melanogaster (Q9VRV7) P14 Homo sapiens (Q9Y3B4); Snu17 C. elegans (Q18318); Snu17 D. melanogaster (Q9VIS0); Snu17 Xenopus laevis (AAH56844) Snu17 Schizosaccharomyces pombe (O94290); Snu17 O. sativa (Q94GF0); Snu17 Mus musculus (Q8R0F5); Snu17 Neurospora crassa (CAE76416); Snu17/CGI-79 H. sapiens (Q9Y3I8); Snu17 Eremothecium gossypii (AAS51937); Snu17 S. cerevisiae (YIR005W); Hsh49 S. cerevisiae (YOR319W).
Biocomputing analyses revealed that RES subunits are conserved and that they contain various domains (Table I). Like human p14, Snu17p possesses an RNA-binding domain (RBD). Interestingly, BLAST searches revealed that Snu17p is more related to the human CGI-79 protein than to p14. Phylogenetic analysis of proteins similar to these factors, rooted using the RBD domain of Hsh49p, indicates that p14 and Snu17p belong to two separate lineages (Figure 2B). In each species, besides yeast, two factors are present, one from each subfamily. However, no clear p14 orthologue is found in the yeast genome. This result is consistent with our finding that human and yeast SF3b have different compositions and that Snu17p belongs to a new complex. Bud13p contains a unique, phylogenetically conserved, C-terminal region of unknown function, while the C-terminus of Pml1p contains a conserved FHA domain, implicated in phosphothreonine binding (Hammet et al, 2000). Interestingly, Pml1p was identified in complex containing all five snRNPs, supporting a role in splicing (Stevens et al, 2002).
SF3b subunits are essential for yeast growth, while inactivation of RES causes slow growth phenotypes
To understand the role of the newly identified factors, we disrupted the corresponding genes in yeast. Inactivation of RDS3 and YSF3 in a diploid strain followed by tetrad analysis revealed that they are, like all other SF3b subunits, essential for vegetative growth (data not shown). In contrast, all RES subunits were dispensable but their inactivation generated a slow growth phenotype that was exacerbated at 37°C (Figure 3). Interestingly, this phenotype was stronger for BUD13 and SNU17 inactivation than for PML1. Overall, the genetic analysis was consistent with the biochemical data indicating the presence of two functionally different complexes.
Figure 3.
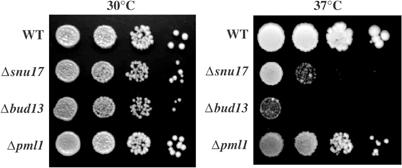
RES subunits are required for wild-type growth. Serial dilutions of strains harbouring deletion of the RES subunits (Δsnu17, Δbud13, or Δpml1) and an isogenic wild-type strain control were placed on YPDA plates and incubated at 30 and 37°C for 2 and 3 days, respectively.
RES is not associated with snRNAs
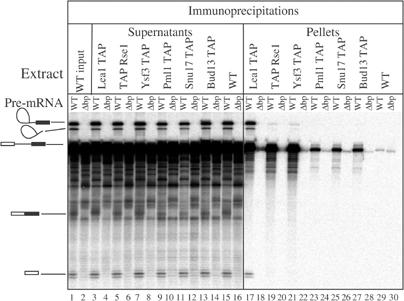
SF3b subunits have been shown to co-precipitate the U2 snRNA in yeast and human cells, while Snu17p was also proposed to associate with this species (Gottschalk et al, 2001).We performed coimmunoprecipitation experiments to assess whether RES subunits were associated with the U2 snRNP. Extracts from strains expressing either Snu17p, Bud13p, Pml1p, or as positive controls the U2 snRNP protein Lea1p (U2A′), or SF3b subunits Rse1p and Ysf3p fused to the TAP tag were precipitated with IgG-coated beads and the presence of snRNAs in the pellets was analysed by primer extension. A wild-type extract served as a negative control. Surprisingly, for tagged Snu17p, Bud13p and Pml1p, like for the wild-type extract, we did not detect snRNA co-precipitation (Figure 4). In contrast, under these conditions, Lea1p, Rse1p and Ysf3p co-precipitated efficiently the U2 snRNA as well as small amounts of U5 and U6 snRNAs. Western blotting revealed that more than 50% of all tagged proteins were immunoprecipitated (data not shown). Thus, lack of snRNA co-precipitation by RES subunits does not result from inefficient pull-down. These data confirm that Snu17 is not an SF3b subunit, and indicates further that RES is not stably associated with an snRNP.
Figure 4.

In contrast to SF3b, the RES complex is not stably associated with snRNAs. Extracts from a control wild-type yeast strain (WT) and strains harbouring TAP-tagged Lea1p, Rse1p, Ysf3p, Snu17p, Bud13p or Pml1p were immunoprecipitated with IgG-coated beads. RNA extracted from input and pellet was analysed by primer extension with primers specific for the U1, U2, U4, U5 and U6 snRNAs. The positions of the corresponding signals are shown on the left. RNA from a four times more extract was used for the pellets relative to the input fractions.
RES associates with pre-mRNA in vitro
In the absence of snRNA pull-down, we tested for a link between RES and splicing by performing in vitro splicing reactions and assaying its association with pre-mRNA, splicing intermediates and/or products. Extracts from a wild-type strain and strains expressing TAP-tagged Snu17p, Bud13p, Pml1p and Lea1p, Rse1p, Ysf3p were incubated either with radioactively labelled wild-type pre-mRNA or mutant pre-mRNA containing a branchpoint deletion as a negative control. After precipitation with IgG-coated beads, RNAs were extracted from pellets and detected by autoradiography following denaturing acrylamide gels electrophoresis. Supernatants and one input were analysed in parallel to control for precipitation efficiency and absence of RNA degradation. As expected (Figure 5), Lea1p, Rse1p and Ysf3 precipitated significant amount of pre-mRNA over the background observed with wild-type extract or with the mutant pre-mRNA lacking a branchpoint (Séraphin and Rosbash, 1991). In addition, Rse1p and Ysf3p co-precipitated low but significant amount of lariat intermediate, while Lea1p efficiently pulled down splicing intermediates and the lariat intron. The pre-mRNA was also co-precipitated with the tagged RES, even though to a lower extent than with the other tagged factors. The signal was significant and specific, however, as it was consistently above background detected with the wild-type extract or the mutant pre-mRNA. Pre-mRNA co-precipitation indicated that RES associates with the spliceosome before step 1; the weaker signal relative to Lea1p, Rse1p and Ysf3p suggests that RES interaction is weaker, more transient and/or that the RES-tagged subunit is less accessible when incorporated into spliceosome.
Figure 5.
SF3b and RES associate with the pre-mRNA. Splicing reaction were assembled with a wild-type pre-mRNA and, as a negative control, a branchpoint deletion pre-mRNA mutant, using extracts prepared from an untagged wild-type strain or isogenic strains harbouring the TAP tag, fused either to Lea1p, Rse1p, Ysf3p, Snu17p, Bud13p or Pml1p. RNA immunoprecipitated with IgG sepharose (pellet) and RNA remaining in the supernant (supernatant) were extracted together with one input fraction, fractionated by denaturing acrylamide gel electrophoresis and detected by phosphorimaging. Structures of the various species are indicated on the left.
Snu17p and Bud13p are required for efficient splicing in vitro
To gain further evidence for the function of the newly discovered proteins in splicing, we analysed in vitro splicing and spliceosome formation, by incubation of radiolabelled pre-mRNA in the extract lacking RES subunits. Interestingly, spliceosome formed in extracts lacking Bud13p or Snu17p migrated faster than complexes formed in wild-type extracts, while commitment complex formation and mobility were unaffected (data not shown). In contrast, removal of Pml1p did not alter spliceosome migration. Analysis of the reaction products revealed that in vitro splicing is significantly inhibited before the first catalytic step in the absence of Bud13p (Figure 6) or Snu17p (data not shown). Again, removal of Pml1p had no significant effect. To control for the specificity of this effect, we added TAP-purified RES complex to the reaction, which partially restored the in vitro splicing deficiency of Δbud13 extracts (Figure 6) and Δsnu17 (data not shown). In contrast, purified RES addition to a wild-type extract did not affect splicing. These results demonstrate a direct role for RES in splicing in vitro and, together with the co-precipitation of pre-mRNA, they indicate that it acts before the first splicing step.
Figure 6.
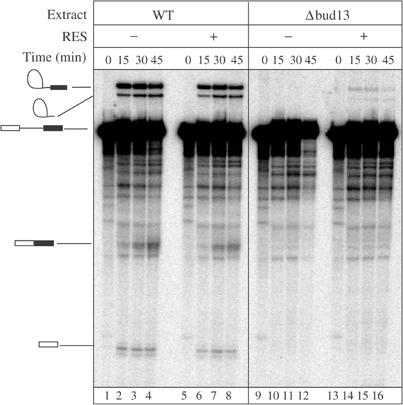
Extracts lacking Bud13p are defective in splicing in vitro and inhibition of splicing can be relieved by addition of purified RES complex. Splicing reactions were performed using extracts prepared from wild-type strain or isogenic Δbud13 strains. Products of 0, 15, 30 and 45 min reactions were fractionated by denaturing gels electrophoresis and detected by phosphorimaging. To reconstitute activity, TAP-purified RES complex was added to the reaction mixtures prior to addition of the pre-mRNA.
Analysis of the new SF3b subunit also provided evidence for their implication in splicing in vitro (data not shown). Strikingly, extracts containing an Rds3p TAP fusion formed commitment complexes, but were unable to assemble pre-spliceosomes (data not shown). This result is consistent with a role of SF3b in the commitment complex–pre-spliceosome transition.
RES is required for efficient splicing in vivo
Taking advantage of the viability and thermosensibility of the Δbud13, Δsnu17 and Δpml1 mutant strains, we tested the effect of RES subunit inactivation on pre-mRNA splicing in vivo. Reporter plasmids containing the RP51A intron or mutants thereof inserted within the lacZ reading frame were introduced in these strains and splicing of these construct assayed by β-galactosidase assays and by primer extension analysis (Jacquier et al, 1985).
Splicing of the wild-type reporter, assayed by the production of β-galactosidase, was essentially normal in the three mutant strains (at most two-fold reduction compared to an isogenic wild-type strain, Figure 7A). We obtained similar results for a branchpoint mutant (data not shown). In contrast, the reporter mRNA carrying a poor 5′ splice site (5′II: GUAUaU) was poorly spliced in strains lacking Snu17p or Bud13p grown at 25°C (100-fold reduction of β-galactosidase levels). This effect was strongly exacerbated at 37°C (500-fold reduction compared to wild type). Inactivation of Pml1p resulted in a milder phenotype as a 15-fold reduction of β-galactosidase could only be detected by combining the 5′ splice site mutation and the nonpermissive temperature.
Figure 7.
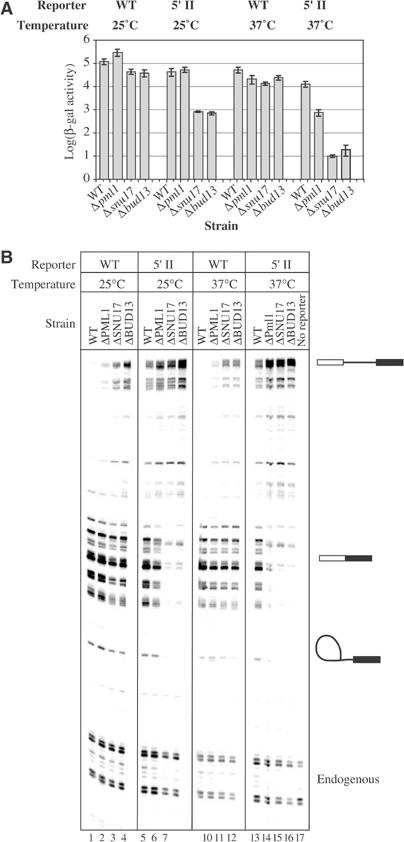
RES subunits are involved in splicing in vivo. The splicing efficiency of reporter constructs was analysed at two different temperatures (25 and 37°C) in a wild-type strain or mutant lacking SNU17, BUD13 or PML1. Splicing reporters contained either the wild-type RP51A intron (WT) or a 5′ splice site mutant derivative (5′II), interrupting the reading frame of the lacZ gene. (A) β-Galactosidase activity of the wild-type and 5′II reporters. Given the wide variation, data are presented on logarithmic scale. In each strain, activities are normalized to the activity of a construct containing no intron in the lacZ gene. (B) Primer extension analysis of RNA present in the various strains. As a control, primer extension using RNA isolated from wild-type strain without reporters is depicted. Owing to multiple transcription initiation sites of the inducible GAL promoter, mRNAs appear as multiple bands. Positions of reporter-derived pre-mRNA, mRNA, lariat intermediate and endogenous RP51A mRNA are indicated.
To ascertain that splicing was affected by RES inactivation and to determine which step was inhibited, we assessed mRNA, pre-mRNA and lariat intermediate levels by a more direct and sensitive primer extension analysis. For the wild-type reporter, we observed an accumulation of pre-mRNA in the Δsnu17 and Δbud13 strains, both at 25 and 37°C (Figure 7B), with no concomitant significant effect on the lariat intermediate or mRNA levels. For the 5′ splice site mutant reporter, mRNA levels were significantly reduced in the Δsnu17 and Δbud13 backgrounds. In the Δpml1 strain, pre-mRNA accumulation was significantly lower, and a combination of high temperature and 5′ splice site mutation was required to detect some mRNA decrease. In all cases, the latter splicing block occurs concomitantly with a reduction of the lariat intermediate level indicative of a first step block. Overall, these data demonstrate that RES is required for efficient splicing. Furthermore, RES appears to act before the first splicing step and to be more critical for introns with weak 5′ splice sites.
RES is required for nuclear pre-mRNA retention in vivo
To check whether, in addition to splicing process itself, RES complex is also involved in pre-mRNA retention in the nucleus, we used a previously described reporter system composed of two related plasmids with either the pre-mRNA (pre-mRNA in-frame) or the mRNA (mRNA in-frame) encoding β-galactosidase (Rain and Legrain, 1997). Henceforth, β-galactosidase activity generated by the pre-mRNA in-frame construct allows for the estimation of pre-mRNA leakage from the nucleus, while enzymatic activity originating from the mRNA in-frame construct reports splicing. As a positive control, we have used isogenic Mlp1 knockout strain, which is known to have pre-mRNA leakage phenotype without associated splicing defect (Galy et al, 2004). For all the three RES knockouts, we observed a significant pre-mRNA leakage at 25°C, evidenced by increased β-galactosidase activity with the pre-mRNA in-frame construct (Figure 8A). This effect was not exacerbated at 37°C (data not shown). Splicing (mRNA in-frame construct) was not more affected in the Δpml1 background than in the Δmlp1 strain in contrast to the significant reduction observed for Δsnu17 and Δbud13. Thus, while mRNA leakage observed in the absence of Snu17 or Bud13 may result from poor splicing, the effect observed in Δpml1 is, like for Δmlp1 (Galy et al, 2004), likely to be direct. This conclusion is further supported by the observation that splicing defects are weak and thermosensitive with this mutant (see above), while pre-mRNA leakage is not. Primer extension analyses confirmed that, for Δpml1 as for Δmlp1, there is no significant increase in pre-mRNA level in comparison to wild type (Figure 8B and C), while splicing of this reporter RNAs was defective for Δbud13 and Δsnu17, with a three-fold increase in pre-mRNA to mRNA ratio. These results indicate that the major function of Pml1 is nuclear pre-mRNA retention. Furthermore, the RES complex, at the edge between splicing and pre-mRNA retention, may connect these processes particularly for introns with weak 5′ splice sites.
Figure 8.
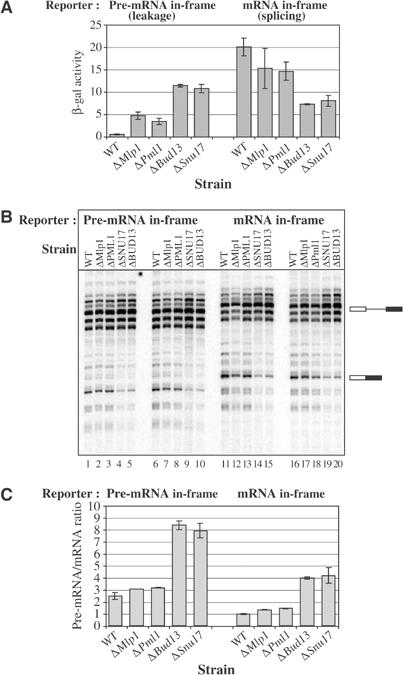
RES subunits are involved in nuclear pre-mRNA retention in vivo. Phenotypes of Δsnu17, Δbud13 or Δpml1 were analysed using reporters containing a synthetic intron reporting pre-mRNA leakage or splicing (‘pre-mRNA in-frame' and ‘mRNA in-frame', respectively). Isogenic wild-type and ΔMlp1 strains were used as controls. (A) β-Galactosidase activity of pre-mRNA retention reporters. Activity is expressed as a % of the activity of a construct containing no intron. (B) Primer extension analysis of the RNA produced by the reporters containing the pre-mRNA and mRNA in-frame. The experiment was made in duplicate using two independent transformants. Owing to multiple transcription initiation sites of the inducible GAL promoter, mRNAs appear as multiple bands. Positions of pre-mRNA and mRNA are indicated. (C) Quantification of the pre-mRNA/mRNA ratio from the primer extension. Error bars are calculated from the analysis of independent transformants.
Discussion
Using a proteomic strategy, we have characterized the yeast SF3a and SF3b splicing factors and identified a new complex, RES, required for efficient intron removal and nuclear pre-mRNA retention. These data reveal interesting similarities and unexpected differences between the yeast and mammalian splicing systems.
Composition, organization and function of SF3 complexes
TAP purification and mass spectrometry analyses were used to determine the composition of the SF3a and SF3b complexes. Our data indicate that yeast SF3a contains only three subunits: Prp9p, Prp11p and Prp21p, with no trace of other partners. Thus, this complex appears to be homologous to its human counterparts (Table I).
Our results revealed the presence of two new subunits in yeast SF3b; it is thus composed of six proteins: Rse1p, Hsh155p, Cus1p, Hsh49p, Rds3p and Ysf3p. All of them are homologous to human SF3b subunits (Table I). However, human SF3b contains an additional protein, p14, which was shown to crosslink to the branchpoint adenosine (Query et al, 1997; Will et al, 2001). It was previously suggested that Snu17p is the yeast orthologue of p14 and a subunit of SF3b (Gottschalk et al, 2001; Wang and Rymond, 2003). Our data strongly argue against this conclusion, for several reasons:
TAP purification of SF3b, using any of the six tagged subunits, failed to reveal a protein migrating at the position expected for Snu17. Furthermore, mass spectrometry analyses failed to identify this factor.
TAP purification of Snu17p reveals that it belongs to a new splicing complex: RES and none of the characteristic SF3b subunits copurified with it. The presence of an unaccessible fraction of Snu17 in SF3b is unlikely because more than 80% of Snu17 TAP can be retained on an IgG column (data not shown). Furthermore, quantitative Western blotting indicates that SF3b and RES subunits are present in roughly similar quantities (Ghaemmaghami et al, 2003).
Tagged Snu17p was unable to precipitate U2 snRNA, a characteristic feature of SF3b subunits.
SNU17 deletion indicates that, like other RES subunits but contrasting with SF3b subunits, it is not an essential gene.
SNU17 inactivation did not lead to commitment complex accumulation, as for SF3b mutants, but to a later-stage defect in the splicing process.
Snu17p is more similar to the human CGI-79 protein than to p14. Furthermore, a true p14 orthologue is not found in S. cerevisiae.
Given this compelling evidence, why was Snu17 suggested to be a yeast SF3b subunit? First, Northern blot analysis of RNAs co-precipitating with Snu17 suggested a weak specific association with U2 snRNA (Gottschalk et al, 2001). However, the large size of U2 snRNA and differences in probe specific activity were not taken into account to correct the strong U2 signal before concluding that U2 snRNA was more specifically precipitated than other snRNAs (see Figure 2 in Gottschalk et al, 2001). It is conceivable that spliceosomes, rather than free snRNPs, were detected. Second, based on this result, Wang and Rymond (2003) detected a protein band corresponding in size to Snu17p in a purified SF3b fraction. However, this protein was not identified by mass spectrometry. It may thus simply correspond to a contaminant or a degradation product.
Taken together, the available data are consistent with the fact that yeast SF3b contains only six subunits. All available evidence, including our results on Rds3p and Ysf3p, indicate that SF3b is required for the commitment complex–pre-spliceosome transition as part of the U2 snRNP. Thus, biochemical characterization of Rds3p revealed that it was required for pre-spliceosome formation as part of the yeast SF3b complex (data not shown). It is unclear whether SF3b remains associated with U2 snRNA in the spliceosome throughout the splicing process. For each of the SF3b subunits that we tested, precipitation of the pre-mRNA was efficient but splicing intermediates were only recovered in small amounts and intron lariat was not detectable. This contrasted with core U2 snRNP protein such as Lea1 (Figure 5). This could suggest that SF3b subunits dissociate from the spliceosome shortly after the first splicing step, during rearrangements leading to the positioning of the 3′ splice site in the catalytic site. However, we cannot formerly rule out that all SF3b subunits are not accessible after the first splicing reaction, even though this seems unlikely. We note that while SF3b subunits were detected in partially purified human spliceosomes blocked after the first catalytic step. However, as this preparation was not homogenous, SF3b may have been associated with the unspliced pre-mRNA still present (Jurica et al, 2002). Crosslinking analyses have shown that, in the spliceosome, SF3b subunits bind to the region surrounding the branchpoint, while p14 has been shown to bind to this sequence directly (Staknis and Reed, 1994; Gozani et al, 1996; Query et al, 1996; McPheeters and Muhlenkamp, 2003). The structural model of human SF3b obtained by cryo-electron microscopy indicates the presence of p14 in the centre of a globular cage-like structure made by the other SF3b subunits (Golas et al, 2003). It is thus tempting to speculate that pre-mRNA threads through SF3b, with the branchpoint contacting its central component. Rearrangement in the association of p14/SF3b with the pre-mRNA, allowing the attack of the 5′ splice site by the branchpoint adenosine, is thus also likely to occur before the first transesterification reaction. The absence of a p14 homologue in yeast SF3b can explain some differences between in vitro spliceosome formation in the yeast and human systems. Thus, yeast pre-mRNA lacking a functional 5′ splice site is unable to assemble pre-spliceosomes (Séraphin and Rosbash, 1989b) although this occurs in human splicing extracts (Query et al, 1997). Thus, due to the presence of p14, mammalian intron recognition may occur through an initial recognition of the branchpoint or the 5′ splice site; however, only the latter pathway would be available to yeast.
A new trimeric complex, RES, is required for efficient splicing and nuclear pre-mRNA retention
Purification of Snu17p-associated proteins identified a new protein complex, RES, containing, in addition to Snu17p, Bud13p and Pml1p proteins. Interaction between Snu17p and Bud13p proteins is also supported by global two-hybrid studies (Uetz et al, 2000; Ito et al, 2001). Furthermore, disruption of the RES subunit genes generates highly related phenotypes including slow growth and thermosensitivity. This supports their association in a complex. Pre-mRNA co-precipitation, together with in vitro and in vivo splicing assays, demonstrates that RES is required for efficient splicing. However, inactivation of PML1 had a weaker effect than SNU17 or BUD13 in all the assays that we performed. In this vein, it is noteworthy that inactivation of either SNU17 or BUD13 also alters the yeast budding pattern (Ni and Snyder, 2001), possibly by reducing the splicing of transcript(s) encoding key factor(s) implicated in this process. All RES subunits have orthologues in higher eukaryotes (Table I). In addition, the Snu17p and Bud13p human orthologues, CGI-79 and MGC13125, respectively, were identified in a purified spliceosome fraction (Rappsilber et al, 2002), supporting their involvement in splicing.
What is the exact function of RES in splicing? Snu17p and RES are clearly not stably associated with the U2 snRNA as proposed previously (Gottschalk et al, 2001). In vivo and in vitro results, as well as the co-precipitation of pre-mRNA, indicate that it acts before the first catalytic step. Interestingly, pre-mRNA precipitation is weak. Furthermore, intermediates and splicing products are not precipitated, suggesting that RES interaction with the spliceosome is transient. As RES is not essential, this interaction is not obligatory either. Thus, RES can be seen as a factor enhancing splicing. This function becomes particularly important for weak introns carrying a poor 5′ splice site, but not for those carrying a weakened branchpoint. Given the similarity between Snu17p and p14, it will be of interest to find out which pre-mRNA and/or snRNA sequence(s) are recognized by the Snu17p RNA-binding domain.
A very interesting characteristic of the RES complex is the pre-mRNA leakage to cytoplasm that occurs in its absence. Moreover, for the Pml1p pre-mRNA leakage, phenotype occurs in the absence of significant splicing defect (at 25°C). By all criteria, Pml1p does not differ from Mlp1p, which has been demonstrated to specifically implicated in nuclear pre-mRNA retention (Galy et al, 2004). We conclude that the main function of Pml1p is also nuclear pre-mRNA retention. Interestingly, disruption of MLP1 similarly to PML1 generates very mild growth defect. Pml1p contains an FHA domain implicated in binding to phosphothreonine residues (Hammet et al, 2000). This raises the interesting possibility that Pml1p could be involved in phophorylation-dependent retention of pre-mRNA in the nucleus. It could thereby play a regulatory role in alternative splicing, particularly for intron carrying a weak 5′ splice site. Further analyses should reveal whether this is indeed the case and whether such feature extends to its mammalian orthologue.
Materials and methods
Yeast strain construction
Gene disruption and tagging on the chromosome were performed using PCR fragments following a published strategy (Puig et al, 1998) SNU17, BUD13 and PML1 genes were disrupted with the S. pombe HIS3 marker from pFA-HIS3MX6 (Wach et al, 1997) in the haploid strain BMA64-1a (MATa, ura3-1, trp1- 2, leu2-3,112, his3-11, ade2-1, can1-101). RDS3 and YSF3 genes were disrupted by integrating K. tactis TRP1 marker from pBS1479 in the diploid strain BSY320 (ade2, arg4, leu2-3 112, trp1-289, ura3-52).
Ysf3 TAP, Snu17 TAP, Bud13 TAP and Pml1 TAP strains were constructed as described previously (Rigaut et al, 1999). The TAP Rse1 fusion strain was described previously (Puig et al, 2001).The Δmlp1 strain was described (Galy et al, 2004).
TAP purification
All TAP purifications were performed as described previously from 2 l of culture (Rigaut et al, 1999). Purified proteins were concentrated by lyophilization, separated by SDS–PAGE and stained with Coomassie Blue or silver.
Mass spectrometry analysis
Proteins were identified following ‘in gel' digestion (Pandey and Mann, 2000; Godovac-Zimmermann and Brown, 2001; Rappsilber and Mann, 2002). Coomassie-stained gels were treated directly, while silver-stained bands were rapidly destained using the Silver Quest decoloration kit (Sigma Aldrich). Digestion was carried out overnight with the addition of 50 ng of trypsin. A MALDI-Tof mass spectrometer (Voyager DE STR, Applied Biosystem) fitted with a pulsed nitrogen laser (337 nm) was used. Mass spectra were acquired in the reflectron mode. The total acceleration voltage was 20 kV, with a grid voltage of 68% and a delay extraction of 240 ns. Close external calibration was realized using a standard peptide mix solution ranging from 573 to 3496 Da (LaserBio Labs, SophiaAntipolis). Samples were prepared in the CHCA matrix at a final concentration of 10 mg/ml in acetonitrile/trifluoroacetic acid (70/0.1%) solution. In all, 1 μl of this sample solution corresponding to a dilution of 1:2 was then deposited on the MALDI target and dried.
Database scans were performed by using MS Fit and Profound search engines. Protein identifications were obtained with a sequence coverage of 55–86% in average, and mass accuracies of about 15–60 ppm.
In vitro splicing analysis
Splicing reactions were as described before, except that the incubation was performed for 30 min at 25°C (Séraphin et al, 1988). Pre-mRNA was generated by in vitro transcription of plasmid pBS195 (wild type) or pBS199 (ΔUACUAAC) digested with DdeI. Reactions were stopped by addition of 200 μl of PK buffer (0.1 M Tris–HCl (pH 7.5), 12.5 mM EDTA (pH 8.0), 150 mM NaCl, 1% SDS) containing 80 μg of proteinase K (Sigma) and 10 μg of Escherichia coli tRNA. After incubation for 20 min at 37°C, RNA was extracted and analysed in a 15% polyacrylamide–7 M urea gel.
For complementation of in vitro splicing reaction with purified RES complex, it was TAP purified and dialysed against buffer D (20 mM HEPES-KOH (pH 7.9), 150 mM KCl, 8% glycerol, 0.5 mM phenylmethylsulphonyl fluoride and 0.5 mM dithiothreitol. A volume of 1 μl of purified material (or as control buffer D) was added to 10 μl splicing reactions.
Immunoprecipitation and primer extension
Immunoprecipitation and primer extension were as described previously (Séraphin, 1995). Immunoprecipitation of pre-mRNA was performed similarly: Briefly, 50 μl splicing reactions were preformed. In all, 10 μl of the reaction was extracted and kept as an input, while the remaining 40 μl was diluted in 500 μl of IPP150 buffer (10 mM Tris (pH 8), 150 mM NaCl, 0.1% NP40) and used for immunoprecipitation.
In vivo splicing assays
The Δsnu17, Δbud13, Δpml1 strains and an isogenic control were transformed with reporters: RP51A wild-type intron (HZ18), 5′II (HZ12) (Jacquier et al, 1985), pre-mRNA in-frame (pLG-Nde°Acc°), mRNA in-frame or no intron (pLG-SD5) (Jacquier et al, 1985; Rain and Legrain, 1997). Reporters were assayed for β-galactosidase activity at two temperatures, 25 and 37°C. Strains were grown overnight at 25°C in a synthetic medium without uracil containing 2% lactate (pH 5.5), 2% glycerol and 0.05% glucose to an OD600 of 0.5–0.8. Cultures were maintained at 25°C or shifted to 37°C for 1 h before a 2 h induction of β-galactosidase. β-Galactosidase activity was tested as described previously (Rutz and Séraphin, 2000). All the experiments were performed in duplicate using two independent transformants. Error bars present standard deviation. Primer extensions to analyse splicing and retention reporters were performed as described previously (Jacquier et al, 1985).
Biocomputing
BLAST was used for database searches. Multiple sequence alignments were done with ClustalW. The neighbour-joining tree was computed, excluding positions with gaps and correcting for multiple substitutions using Clustal W (Thompson et al, 1994).
Acknowledgments
We thank the members of our groups for useful discussions, A Shevchenko and M Wilm for help during a preliminary stage of this work, and M Fromont-Racine for strains and plasmids. AD was supported by fellowships from the Foundation for Polish Science and HFSP. This work was supported by La Ligue contre le Cancer (Equipe Labelisée 2001), the CNRS (particularly the Programme PGP 2003) and the ministry of Research to BS.
References
- Brosi R, Hauri HP, Krämer A (1993) Separation of splicing factor SF3 into two components and purification of SF3a activity. J Biol Chem 268: 17640–17646 [PubMed] [Google Scholar]
- Brow DA (2002) Allosteric cascade of spliceosome activation. Annu Rev Genet 36: 333–360 [DOI] [PubMed] [Google Scholar]
- Caspary F, Séraphin B (1998) The yeast U2A′/U2B complex is required for pre-spliceosome formation. EMBO J 17: 6348–6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary F, Shevchenko A, Wilm M, Séraphin B (1999) Partial purification of the yeast U2 snRNP reveals a novel yeast pre-mRNA splicing factor required for pre-spliceosome assembly. EMBO J 18: 3463–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das BK, Xia L, Palandjian L, Gozani O, Chyung Y, Reed R (1999) Characterization of a protein complex containing spliceosomal proteins SAPs 49, 130, 145, and 155. Mol Cell Biol 19: 6796–6802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy V, Gadal O, Fromont-Racine M, Romano A, Jacquier A, Nehrbass U (2004) Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell 116: 63–73 [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS (2003) Global analysis of protein expression in yeast. Nature 425: 737–741 [DOI] [PubMed] [Google Scholar]
- Godovac-Zimmermann J, Brown LR (2001) Perspectives for mass spectrometry and functional proteomics. Mass Spectrom Rev 20: 1–57 [DOI] [PubMed] [Google Scholar]
- Golas MM, Sander B, Will CL, Lührmann R, Stark H (2003) Molecular architecture of the multiprotein splicing factor SF3b. Science 300: 980–984 [DOI] [PubMed] [Google Scholar]
- Gottschalk A, Bartels C, Neubauer G, Lührmann R, Fabrizio P (2001) A novel yeast U2 snRNP protein, Snu17p, is required for the first catalytic step of splicing and for progression of spliceosome assembly. Mol Cell Biol 21: 3037–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozani O, Feld R, Reed R (1996) Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Genes Dev 10: 233–243 [DOI] [PubMed] [Google Scholar]
- Hammet A, Pike BL, Mitchelhill KI, Teh T, Kobe B, House CM, Kemp BE, Heierhorst J (2000) FHA domain boundaries of the dun1p and rad53p cell cycle checkpoint kinases. FEBS Lett 471: 141–146 [DOI] [PubMed] [Google Scholar]
- Igel H, Wells S, Perriman R, Ares M Jr (1998) Conservation of structure and subunit interactions in yeast homologues of splicing factor 3b (SF3b) subunits. RNA 4: 1–10 [PMC free article] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y (2001) A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA 98: 4569–4574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquier A, Rodriguez JR, Rosbash M (1985) A quantitative analysis of the effects of 5′ junction and TACTAAC box mutants and mutant combinations on yeast mRNA splicing. Cell 43: 423–430 [DOI] [PubMed] [Google Scholar]
- Jurica MS, Licklider LJ, Gygi SR, Grigorieff N, Moore MJ (2002) Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 8: 426–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandels-Lewis S, Séraphin B (1993) Involvement of U6 snRNA in 5′ splice site selection. Science 262: 2035–2039 [DOI] [PubMed] [Google Scholar]
- Konarska MM, Sharp PA (1987) Interactions between small nuclear ribonucleoprotein particles in formation of spliceosomes. Cell 49: 763–774 [DOI] [PubMed] [Google Scholar]
- Krämer A (1996) The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem 65: 367–409 [DOI] [PubMed] [Google Scholar]
- Legrain P, Rosbash M (1989) Some cis- and trans-acting mutants for splicing target pre-mRNA to the cytoplasm. Cell 57: 573–583 [DOI] [PubMed] [Google Scholar]
- Lesser CF, Guthrie C (1993) Mutations in U6 snRNA that alter splice site specificity: implications for the active site. Science 262: 1982–1988 [DOI] [PubMed] [Google Scholar]
- Lührmann R, Kastner B, Bach M (1990) Structure of spliceosomal snRNPs and their role in pre-mRNA splicing. Biochim Biophys Acta 1087: 265–292 [DOI] [PubMed] [Google Scholar]
- McPheeters DS, Muhlenkamp P (2003) Spatial organization of protein–RNA interactions in the branch site-3′ splice site region during pre-mRNA splicing in yeast. Mol Cell Biol 23: 4174–4186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud S, Reed R (1991) An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes Dev 5: 2534–2546 [DOI] [PubMed] [Google Scholar]
- Newman AJ, Norman C (1992) U5 snRNA interacts with exon sequences at 5′ and 3′ splice sites. Cell 68: 743–754 [DOI] [PubMed] [Google Scholar]
- Ni L, Snyder M (2001) A genomic study of the bipolar bud site selection pattern in Saccharomyces cerevisiae. Mol Biol Cell 12: 2147–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A, Mann M (2000) Proteomics to study genes and genomes. Nature 405: 837–846 [DOI] [PubMed] [Google Scholar]
- Parker R, Siliciano PG, Guthrie C (1987) Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell 49: 229–239 [DOI] [PubMed] [Google Scholar]
- Pikielny CW, Rymond BC, Rosbash M (1986) Electrophoresis of ribonucleoproteins reveals an ordered assembly pathway of yeast splicing complexes. Nature 324: 341–345 [DOI] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Séraphin B (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24: 218–229 [DOI] [PubMed] [Google Scholar]
- Puig O, Rutz B, Luukkonen BG, Kandels-Lewis S, Bragado-Nilsson E, Séraphin B (1998) New constructs and strategies for efficient PCR-based gene manipulations in yeast. Yeast 14: 1139–1146 [DOI] [PubMed] [Google Scholar]
- Query CC, McCaw PS, Sharp PA (1997) A minimal spliceosomal complex A recognizes the branch site and polypyrimidine tract. Mol Cell Biol 17: 2944–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Query CC, Strobel SA, Sharp PA (1996) Three recognition events at the branch-site adenine. EMBO J 15: 1392–1402 [PMC free article] [PubMed] [Google Scholar]
- Rain JC, Legrain P (1997) In vivo commitment to splicing in yeast involves the nucleotide upstream from the branch site conserved sequence and the Mud2 protein. EMBO J 16: 1759–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M (2002) What does it mean to identify a protein in proteomics? Trends Biochem Sci 27: 74–78 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ryder U, Lamond AI, Mann M (2002) Large-scale proteomic analysis of the human spliceosome. Genome Res 12: 1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Rutz B, Séraphin B (2000) A dual role for BBP/ScSF1 in nuclear pre-mRNA retention and splicing. EMBO J 19: 1873–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séraphin B (1995) Sm and Sm-like proteins belong to a large family: identification of proteins of the U6 as well as the U1, U2, U4 and U5 snRNPs. EMBO J 14: 2089–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séraphin B, Kretzner L, Rosbash M (1988) A U1 snRNA:pre-mRNA base pairing interaction is required early in yeast spliceosome assembly but does not uniquely define the 5′ cleavage site. EMBO J 7: 2533–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séraphin B, Rosbash M (1989a) Identification of functional U1 snRNA–pre-mRNA complexes committed to spliceosome assembly and splicing. Cell 59: 349–358 [DOI] [PubMed] [Google Scholar]
- Séraphin B, Rosbash M (1989b) Mutational analysis of the interactions between U1 small nuclear RNA and pre-mRNA of yeast. Gene 82: 145–151 [DOI] [PubMed] [Google Scholar]
- Séraphin B, Rosbash M (1991) The yeast branchpoint sequence is not required for the formation of a stable U1 snRNA–pre-mRNA complex and is recognized in the absence of U2 snRNA. EMBO J 10: 1209–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staknis D, Reed R (1994) Direct interactions between pre-mRNA and six U2 small nuclear ribonucleoproteins during spliceosome assembly. Mol Cell Biol 14: 2994–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SW, Ryan DE, Ge HY, Moore RE, Young MK, Lee TD, Abelson J (2002) Composition and functional characterization of the yeast spliceosomal penta-snRNP. Mol Cell 9: 31–44 [DOI] [PubMed] [Google Scholar]
- Tang J, Abovich N, Rosbash M (1996) Identification and characterization of a yeast gene encoding the U2 small nuclear ribonucleoprotein particle B″ protein. Mol Cell Biol 16: 2787–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, Li Y, Godwin B, Conover D, Kalbfleisch T, Vijayadamodar G, Yang M, Johnston M, Fields S, Rothberg JM (2000) A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 403: 623–627 [DOI] [PubMed] [Google Scholar]
- Valadkhan S, Manley JL (2001) Splicing-related catalysis by protein-free snRNAs. Nature 413: 701–707 [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Alberti-Segui C, Rebischung C, Philippsen P (1997) Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast 13: 1065–1075 [DOI] [PubMed] [Google Scholar]
- Wang Q, Rymond BC (2003) Rds3p is required for stable U2 snRNP recruitment to the splicing apparatus. Mol Cell Biol 23: 7339–7349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SE, Neville M, Haynes M, Wang J, Igel H, Ares M Jr (1996) CUS1, a suppressor of cold-sensitive U2 snRNA mutations, is a novel yeast splicing factor homologous to human SAP 145. Genes Dev 10: 220–232 [DOI] [PubMed] [Google Scholar]
- Will CL, Schneider C, MacMillan AM, Katopodis NF, Neubauer G, Wilm M, Lührmann R, Query CC (2001) A novel U2 and U11/U12 snRNP protein that associates with the pre-mRNA branch site. EMBO J 20: 4536–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Schneider C, Reed R, Lührmann R (1999) Identification of both shared and distinct proteins in the major and minor spliceosomes. Science 284: 2003–2005 [DOI] [PubMed] [Google Scholar]
- Will CL, Urlaub H, Achsel T, Gentzel M, Wilm M, Lührmann R (2002) Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J 21: 4978–4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt JR, Sontheimer EJ, Steitz JA (1992) Site-specific cross-linking of mammalian U5 snRNP to the 5′ splice site before the first step of pre-mRNA splicing. Genes Dev 6: 2542–2553 [DOI] [PubMed] [Google Scholar]