

Abstract
Suppression of the myostatin (GDF‐8) pathway has emerged as an important therapeutic paradigm for muscle‐wasting disorders. In this study, we conducted a translational pharmacokinetic/pharmacodynamic (PK/PD) analysis of MYO‐029, an anti‐myostatin monoclonal antibody, using PK data in mice, rats, monkeys, humans, mouse tissue distribution data with 125I‐labeled MYO‐029, muscle weight increase in SCID mice, and muscle circumference changes in monkeys. This analysis revealed significant in vivo potency shift between mice and monkeys (72 nM vs. 1.3 μM for 50% effect on quadriceps). Estimated central clearance of MYO‐029 (0.38 mL/h/kg) in humans was greater than twofold higher than typical IgG mAbs. Peak and trough steady‐state exposures of MYO‐029 in patients at biweekly intravenous doses of 10 mg/kg MYO‐029 are predicted to achieve only 50% and 10% of the maximum effect seen in monkeys, respectively. These retrospective analyses results suggest that the MYO‐029 exposures in this trial had a low probability of producing robust efficacy.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ MYO‐029 antibody has been dosed in healthy volunteers and patients with muscular dystrophy in multiple phase I/II studies. No improvement was noted in exploratory end points related to muscle strength or function.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ Was the extent of MYO‐029 antibody exposures and resulting target modulation at the clinical doses sufficient to elicit muscle mass increase?
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
✓ Based on the PK/PD relationships in monkeys, steady‐state exposures of MYO‐029 at the clinical doses (≤ 10 mg/kg i.v.) may have been insufficient to produce a robust muscle mass increase in patients with muscle dystrophy. It is not clear from these data that variability in clinical end points and sample size did not also contribute to the observed lack of efficacy.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
✓ Future trials with new therapeutic agents targeting myostatin (GDF8) should be designed to ensure high target coverage, and target/pharmacology biomarkers should be developed to enable rational decision‐making.
Despite substantial progress in understanding the pathobiology of muscular dystrophies (MDs), effective therapies that increase muscle strength are yet to be approved. Current standards of care, corticosteroids, provide only modest effects on a limited set of patients.1 Recently, blockade of myostatin (also known as growth and differentiation factor‐8 [GDF‐8]) has emerged as a new therapeutic strategy for the treatment of muscle‐wasting disorders, such as muscular dystrophy, cachexia, and sarcopenia.2, 3 Myostatin is expressed in developing and adult skeletal muscle, heart, and adipose tissue, and acts as a negative regulator of muscle fiber growth and mass.4 Naturally occurring myostatin mutations in cattle, sheep, mice, dogs, and humans are associated with hypermuscularity and improved muscle performance.5, 6, 7, 8 Transgenic myostatin null/mdx mice not only featured muscle hypertrophy with decreased dystrophic features on histology, but also increased strength compared with the dystrophin‐deficient (mdx) mice.9
Supported by the data from preclinical animal models and human genetic studies on myostatin pathway, a neutralizing antibody against myostatin, MYO‐029, was developed and taken to clinic in phase I/II trials with healthy volunteers and patients with MD.10 MYO‐029 is a recombinant human immunoglobulin G (IgG)1(λ) antibody that binds to myostatin and neutralizes its activity by preventing binding to its endogenous high‐affinity receptor ActRIIB. Clinical development of MYO‐029 included a double‐blind, placebo‐controlled, randomized trial designed to evaluate its safety, tolerability, and biological activity in 116 patients with facioscapulohumeral, Becker, and limb‐girdle muscular dystrophy, following 1, 3, or 10 mg/kg i.v. administration every 2 weeks for 6 months.10 The outcome of this study indicated favorable safety end points with relatively few patients showing evidence of cutaneous hypersensitivity, likely due to repeated protein administration. Approximately 6% of the patients showed more severe side effects, but anaphylactic shock or death was not observed. No significant improvements were noted on the exploratory end points of muscle strength and function, as measured by manual and quantitative resting of muscle force, muscle magnetic resonance imaging, dual‐energy x‐ray absorptiometry for muscle mass, and muscle biopsy. There was a trend for improvement in the latter two parameters in the 10 mg/kg dose group, but the lack of statistical significance of the effect was thought to be due to the limited size of the study. Subsequent to the MYO‐029 development efforts, ACE‐031, a recombinant Fc fusion protein of the soluble form of ActRIIB, was taken to clinic by Acceleron Pharma.11 Similar to MYO‐029 antibody, ACE‐031 binds to and inhibits biological function of myostatin; but unlike MYO‐029, it is able to suppress in vivo function of other TGF‐β ligands, such as BMPs and activins. However, recent trials of ACE‐031 in patients with Duchenne muscular dystrophy were halted pending further analysis of safety findings related to minor nose and gum bleeding and dilation of blood vessels in the skin.12
In light of inconclusive results from the MYO‐029 clinical trials, it is unclear whether suppression of myostatin pathways is likely to produce effective therapies for muscle‐wasting diseases, considering the exposure‐response translation from preclinical species to humans. In this study, we conducted a retrospective analysis of the MYO‐029 program to answer whether the extent and duration of drug exposure achieved in humans was sufficient to achieve adequate target suppression, and could result in robust muscle mass increase efficacy in patients. We conducted an integrated analysis of extensive data on MYO‐029 obtained in a series of pharmacokinetic (PK), pharmacodynamic (PD), and toxicokinetic studies conducted in mice, rats, and Cynomolgus monkeys. As a first step, compartmental modeling of PK data in preclinical species and healthy humans was conducted to explore the PK scaling from preclinical to clinic. Subsequently, PK/PD modeling analysis of muscle efficacy end points in mice and monkeys was conducted to estimate in vivo potencies. Finally, estimated in vivo potencies in preclinical species were used as benchmarks to assess whether antibody exposures observed in phase I/II trials were sufficient to produce robust muscle efficacy response.
MATERIALS AND METHODS
All animals used in the in vivo studies were handled in compliance with the National Research Council Guidelines. The studies were performed in a laboratory approved by the American Association for Accreditation of Laboratory Animal Care, according to protocols that were reviewed and approved by the Institutional Animal Care and Use Committee.
MYO‐029 serum enzyme‐linked immunosorbent assay methods
Enzyme‐linked immunosorbent assay (ELISA) methods were developed and validated for the quantitation of MYO‐029 in the serum of mice, rats, monkeys, and humans. In these assays, MYO‐029 was captured by biotinylated GDF‐8 adsorbed onto a streptavidin‐coated microtiter plate. The captured MYO‐029 was detected with a mouse anti‐human immunoglobulin G (IgG) horseradish peroxidase conjugate. Tetramethylbenzidine substrate was utilized for signal generation and colorimetric readout. The range of quantitation was 10–80 ng/mL in 100% mouse serum, 21.1–240 ng/mL in 100% rat serum, 63.2–720 ng/mL in 100% monkey serum, and 63.2–720 ng/mL in 100% human serum.
Mouse pharmacokinetic studies
MYO‐029 was dosed i.v. at 1, 5, 20, and 100 mg/kg for 4 weeks in C57/SCID mice. Serum samples (1–2 per mouse) were collected at 3, 6, 48, 144, 312, and 648 h.
Mouse tissue distribution studies
Tissue distribution of 125I‐labeled MYO‐029 was investigated in male C57/SCID mice after i.v. and i.p. administration of 1 mg/kg dose. One group of 30 animals received an i.v. injection via the tail vein and the other group of 30 animals received an i.p. injection. At 0.083, 3, 24, 120, and 336 h postdosing, the animals were euthanized, whole‐body perfused, and following tissues were removed, weighed and counted for total radioactivity: spleen, lungs, heart, liver, kidneys, skeletal muscle (quadriceps and triceps), stomach, small intestines, large intestines, skin, and fat. Plasma sampling time points were 0.083, 0.25, 1, 3, 6, 24, 48, 120, 168, and 336 h postdose. Total radioactivity was counted for each tissue with a gamma counter. Precipitable counts per min (cpm) were calculated by subtracting two times the supernatant cpm from the total cpm. Concentrations of MYO‐029 were calculated based on the specific activity of the dosing solution. The areas under the serum concentration‐time curve (AUC) were calculated using the noncompartmental method (WinNonlin, version 4.0).
Mouse efficacy studies
Female SCID mice were distributed into groups of eight with respect to their body weight, and injected i.p. with MYO‐029 at various doses ranging from 1–10 mg/kg/week for 12 weeks. Mice received a double dose at the first injection. Mice receiving a phosphate‐buffered saline (PBS) injection were used as vehicle controls. At the end of the treatment periods (12 weeks), mice were euthanized and the gastrocnemius, quadriceps, and extensor digitorum longus (EDL) muscles were collected and immediately weighed. Morphometric analysis was performed on EDL muscles of mice treated with 10 mg/kg/week MYO‐029 or PBS for 4 weeks.
Rat pharmacokinetics studies
The PKs of MYO‐029 in male Sprague Dawley rats were determined after a single i.v. dose of 2, 10, or 50 mg/kg. Eighteen animals were randomly assigned into three treatment groups with six animals in each group. Each animal received an i.v. infusion (15 min) of test material through the tail vein. Blood samples were collected through the jugular vein before dosing and at 0.25, 1, 4, 24, 48, 72, 168, 336, 504, 672, 840, 1,008, and 1,056 h (44 days) after dosing.
Five‐week monkey study
Male and female monkeys were administered weekly doses of 10, 30, and 100 mg/kg/week of MYO‐029 for 5 weeks (doses given on days 1, 8, 15, 22, and 29) by i.v. injection. Blood samples were collected for PK analysis after dose administration on days 1 and 22 from all animals and before dose administration and at approximately 10 min, 1, 8, 24, 48, 72, and 168 h (before dosing on days 8 and 29, respectively); an additional sample was taken before dose administration on day 15 for trough level. The concentrations of MYO‐029 in the monkey serum samples were determined using a validated ELISA, as described in the Methods section.
Thirty‐nine week monkey study
Male and female cynomolgus monkeys (n = 4/sex/group) were administered once‐weekly doses (0, 10, 30, and 100 mg/kg/week) of MYO‐029 by i.v. injection for 39 weeks. Seven plasma samples on day 169 (at 0.083, 6, 24, 48, 72, 120, and 168 h after dose) were collected over a 1‐week period. Muscle circumference was measured by tape once predose and during weeks 20 and 39–40 of the dosing phase. Specifically, circumference data were collected on six muscles, biceps, quadriceps, and gastrocnemius muscles on both the left and right side.
MYO‐029 clinical trials
As reported previously,10 clinical development of MYO‐029 antibody included three phase I clinical studies in healthy subjects and one phase I/II study10 with MYO‐029 in subjects with Becker muscular dystrophy (BMD), facioscapulohumeral muscular dystrophy, or limb‐girdle muscular dystrophy. The PK data from the phase I study conducted in US healthy volunteers with single ascending doses of MYO‐029 were used for analysis. This study (n = 61) was a randomized, double‐blind, placebo‐controlled, single‐center, sequential‐group, inpatient/outpatient study of MYO‐029. The study was designed to assess the safety, tolerability, PK, and PD of single ascending i.v. doses of MYO‐029 (0.3, 1, 3, 7.5, and 11.25 mg/kg) or placebo administered to healthy subjects aged 18 to 45 years. For PK evaluation, blood samples were collected before test article administration and at 0.5, 1, 4, 12, 24, 48, 72, 168, 312, 480, 648, 816, 984, 1,152, and 1,320 h after the start of the infusion.
PK/PD modeling analysis
For each species, plasma data from all individual animals were fitted simultaneously using the first‐order conditional estimation with interaction option (FOCE INTERACTION) in NONMEM, version 7. During the model‐building procedure, several structural PK models were tested. These included, among others, a two‐compartment model and a three‐compartment model. Furthermore, linear, nonlinear, or a combination of both were tested for the clearance of MYO‐029. Discriminations between hierarchical models were based on the objective function value (OFV) provided by NONMEM at a significance level of 0.001, equal to a decrease of 10.8 in the OFV, graphical analysis of residuals and predictions in model diagnostics using S‐Plus scripts, and visual predictive checks. When appropriate, attempts were made to estimate interindividual variability terms on various model parameters in each species. Different error models were tested in each species model. When modeling a PK/PD relationship, an effect compartment model, and various forms of direct or indirect response models were tested.
RESULTS
MYO‐029 PKs in mice, rats, monkeys, and humans
MYO‐029 exposures in mice, rats, monkeys, and humans were analyzed using a mixed effects modeling approach. PK data were collected from the following in vivo studies post‐i.v. dosing: (1) C57/SCID mice dosed with 1, 5, 20, and 100 mg/kg/week for 4 weeks; (2) Sprague Dawley rats dosed with a single injection of 2, 10, or 50 mg/kg; (3) 5‐week and 39‐week studies in Cynomolgus monkeys dosed with 10, 30, and 100 mg/kg/week; and (4) phase I human healthy volunteer trials with single ascending doses of 0.3, 1, 3, 7.5, and 11.25 mg/kg.
The i.v. PK in C57/SCID mice were best fitted using one‐compartment model with linear and nonlinear clearance component, whereas a two‐compartment model with linear clearance best described the data in rats, monkeys, and humans (Figure 1). As presented in Table 1, central clearance (CL) of MYO‐029 was estimated to be 0.25, 0.54, 0.23, and 0.38 mL/h/kg in mice, rats, monkeys, and humans, respectively. Central volume of distribution (V1) was estimated to be 103, 59, 42, and 65 mL/kg in mice, rats, monkeys, and humans, respectively. Distributive clearance in rats, monkeys, and humans ranged within 0.31–1.80 mL/h/kg, and peripheral volume of distribution (V2) ranged within 33–95 mL/kg in the three species. In mice, maximum clearance rate (Vmax) through the nonlinear route and Michaelis binding constant (Km) were estimated to be 189 ug/h/kg (or 0.11 mg/day) and 2.7 ug/mL, respectively. Supplementary Figure S1 data include an illustration of goodness of fit for human PK data.
Figure 1.
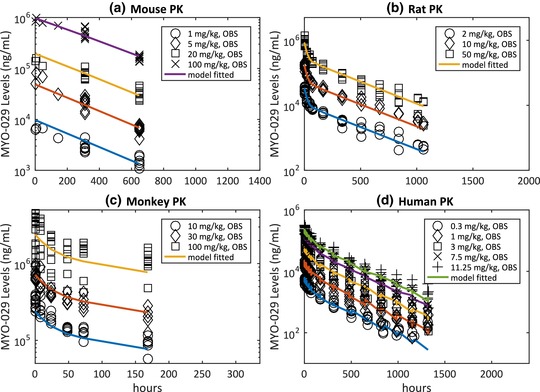
MYO‐029 serum exposures (observed vs. population fit). (a) Mouse pharmacokinetic (PK) at 1, 5, 20, and 100 mg/kg i.v.; (b) rat PK at 2, 10, and 50 mg/kg i.v.; (c) monkey PK at 10, 30, and 100 mg/kg i.v.; and (d) healthy human PK at 0.3, 1, 3, 7.5, and 11.25 mg/kg i.v.
Table 1.
MYO‐029 pharmacokinetic parameters estimated for mice, rats, monkeys, and humans
| Parameter | Units | Definition | Mouse | Rat (%RSE) | Monkey (%RSE) | Human (%RSE) |
|---|---|---|---|---|---|---|
| CL | mL/h/kg | Clearance from central compartment | 0.245 | 0.542 (4%) | 0.228 (4%) | 0.38 (4%) |
| V1 | mL/kg | Volume of central compartment | 103 | 58.9 (4%) | 41.8 (6%) | 65 (3%) |
| Q | mL/h/kg | Distributive clearance | – | 1.79 (9%) | 0.815 (6%) | 0.31 (12%) |
| V2 | mL/kg | Volume of peripheral compartment | – | 95.3 (5%) | 27.8 (11%) | 33 (6%) |
| Vmax | ug/h/kg | Maximum clearance rate from nonlinear route | 189 | – | – | – |
| Km | ug/mL | Apparent binding constant | 2.67 | – | – | – |
| ω CL(IIV) 2 | – | IIV on CL | – | 0.15 (1%) | 0.27 (2%) | 0.04 (27%) |
| ω V1(IIV) 2 | – | IIV on V1 | – | 0.12 (1%) | 0.35 (3%) | 0.03 (25%) |
| σ1 | – | Proportional error | – | – | – | 0.16 (8%) |
RSE, relative standard error.
Mouse pharmacokinetic was fitted with a one‐compartment model with linear and nonlinear elimination whereas the two‐compartment model with only linear elimination produced the best fit for rat, monkey, and human data.
MYO‐029 tissue distribution
Tissue distribution of 125I‐labeled MYO‐029 were investigated in male C57/SCID mice after i.v. and i.p. administration of 1 mg/kg dose. Figure 2 shows serum‐to‐tissue area under the mean radioactive‐equivalent concentration‐time curve of 125I‐MYO‐029 (AUC0–336h) in serum, spleen, lungs, heart, kidneys, skin, liver, fat, skeletal muscle (quadriceps and triceps), stomach, small intestines, and large intestines. The highest concentrations of 125I‐MYO‐029 at most time points evaluated (up to 336 h) were found in serum, as expected. For the quadriceps and triceps, AUC0–336h ratio was approximately 5–6%. Because the muscle interstitial volume is approximately 15% of the total muscle tissue13 and most of the antibody is expected to be confined within that space, the actual concentration of antibody in muscle interstitial space is estimated to be approximately 30–40% of the serum concentration.
Figure 2.
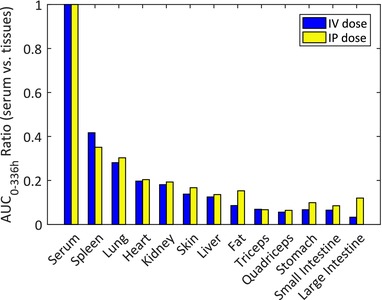
Ratio of tissue area under the serum concentration‐time curve (AUC)0–336h to serum AUC0–336h after i.v. or i.p. administration of 125I‐MYO‐029 at 1 mg/kg to C57/SCID mice.
Muscle growth in mice
The effect of MYO‐029 on skeletal muscle mass was explored in SCID mice dosed at 1, 2.5, 5, and 10 mg/kg/week for 12 weeks via the i.p. route. At the end of the treatment period, mice were euthanized and the gastrocnemius (gastro), quadriceps (quad), and EDL muscles were collected and immediately weighed. As shown in Figure 3 a,b,c, MYO‐029 increases skeletal muscle mass in vivo in a concentration‐dependent manner. To explore exposure‐response relationships, mouse data were fitted using a direct effect maximum effect (Emax) model and vehicle response (E0), maximum % change over vehicle (Emax), weekly steady state AUC50 for half maximal (50%) response, and corresponding average antibody concentration (Cave50) were estimated for each muscle type. As shown in Table 2, Emax values for EDL, gastro, and quad muscles ranged between 23% and 30% and Cave50 range was estimated to be 47–72 nM. In order to determine whether the increase in skeletal muscle mass caused by MYO‐029 treatment is due to hypertrophy or hyperplasia, morphometric analysis (data not shown) was performed on EDL muscles after 4 weeks of treatment in the 10 mg/kg/week dose group. The results indicated that MYO‐029 treatment leads to muscle fiber hypertrophy and not hyperplasia.
Figure 3.

MYO‐029 exposure‐response in mice and monkeys. (a) Extensor digitorum longus (EDL), (b) gastrocnemius (gastro), and (c) quadriceps (quad) muscle mass increase in mice after i.p. administration of MYO‐029 at weekly doses of 1, 2.5, 5, and 10 mg/kg for 12 weeks. (d) Growth in muscle circumference in Cynomolgus monkeys (n = 4/sex/group) following once‐weekly doses (0, 10, 30, and 100 mg/kg/week) of MYO‐029 by i.v. injection for 39 weeks. Muscle circumference was measured by tape once pretest, weeks 20 (day 135) and 39–40 (day 271) of the dosing phase. F, female; R, right; L, left; d, day.
Table 2.
MYO‐029 PD parameters estimates for mouse and monkey
| Species | Parameter | Units | Estimate | CV% | |
|---|---|---|---|---|---|
| Mouse | EDL | Emax | % | 29.9 | 5 |
| E0 | Gram | 16.7 | 1 | ||
| AUC50 | ug*h/mL | 1,152.6 | 21 | ||
| Cave50 a | nM | 47.2 | NA | ||
| Gastro | Emax | % | 23.2 | 13 | |
| E0 | Gram | 0.24 | 2 | ||
| AUC50 | ug*h/mL | 1,560.5 | 50 | ||
| Cave50 a | nM | 63.9 | NA | ||
| Quad | Emax | % | 27.2 | 15 | |
| E0 | Gram | 0.34 | 3 | ||
| AUC50 | ug*h/mL | 1,762.6 | 54 | ||
| Cave50 a | nM | 72.2 | NA | ||
| Monkey | Female quad‐L (135d) | Emax | % | 274.1 | 57 |
| E0 | % | 4.1 | 40 | ||
| AUC50 | ug*h/mL | 32,092 | 98 | ||
| Cave50 a | nM | 1,313.8 | NA | ||
| Female quad‐L (271d) | Emax | % | 200.5 | 2 | |
| E0 | % | 6.1 | 1 | ||
| AUC50 | ug*h/mL | 32428 | 4 | ||
| Cave50 a | nM | 1,327.5 | NA |
CV, coefficient of variation; EDL, extensor digitorum longus; Gastro, gastrocnemius; NA, not applicable; PD, pharmacodynamic; Quad, quadriceps.
Cave50(nM) = AUC50/1,000/168/145.4; MYO‐029 MW: 145.4 kDa.
Direct effect Emax model was used to describe effect of steady‐state exposures (AUC0–168h) on % increase in muscle mass over baseline in mouse, and rate of growth in muscle circumference in monkeys. Parameters are defined as: maximum % change over vehicle (Emax), vehicle baseline (E0), steady‐state area under the curve to achieve 50% effect (AUC50), steady‐state average concentration to achieve 50% effect (Cave50).
Muscle growth in monkeys
MYO‐029 was administered to male and female Cynomolgus monkeys (4/sex/group) at i.v. dosages of 10, 30, and 100 mg/kg/week once weekly for a total of 39 consecutive doses and mean muscle (biceps, gastro, and quad) circumference of the right and left limbs was measured at pretest, week 20, and finally at week 39 or 40. Comprehensive data from this study are presented in Table 3. In general, male monkeys have statistically significant increases (p value < 0.05) in mean muscle circumference that occurred primarily in animals administered 100 mg/kg/week, with the exception of the left biceps, in which an increased circumference was detected for all groups based on change from predose to day 135. In female monkeys, the increase in mean muscle circumference generally occurred at all dosages and was not dosage‐dependent in magnitude, with the exception of the right and left quadriceps measurement, which increased with increasing dosage.
Table 3.
Muscle circumference data from Cynomolgus monkeys post‐MYO‐029 treatment in 39‐week study
| Male Cynomolgus monkeys | Female Cynomolgus monkeys | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Predose | Change (predose to day 135) | Change (predose to day 271) | Predose | Change (predose to day 135) | Change (predose to day 271) | ||||||||
| Type | Group | Mean | (SD) | Mean | (SD) | Mean | (SD) | Mean | (SD) | Mean | (SD) | Mean | (SD) |
| Bicep (right) | Vehicle | 9.2 | (0.9) | 0.7 | (0.3) | 1.1 | (0.4) | 8.4 | (0.3) | 0.6 | (0.2) | 0.8 | (0.3) |
| 10 mg/kg | 8.8 | (0.8) | 0.9 | (0.3) | 1.3 | (0.4) | 8.5 | (0.3) | 0.9 | (0.3) | 1.1 | (0.3) | |
| 30 mg/kg | 9.1 | (0.8) | 1.0 | (0.3) | 1.2 | (0.5) | 8.6 | (0.5) | 1.2 | (0.2) | 1.5a | (0.3) | |
| 100 mg/kg | 9.3 | (1.0) | 1.3b | (0.2) | 1.9 | (0.4) | 8.5 | (0.2) | 1.0 | (0.3) | 0.9 | (0.3) | |
| Bicep (left) | Vehicle | 9.1 | (0.6) | 0.6 | (0.2) | 1.0 | (0.4) | 8.6 | (0.4) | 0.5 | (0.3) | 0.7 | (0.3) |
| 10 mg/kg | 8.8 | (0.8) | 1.2b | (0.3) | 1.6 | (0.4) | 8.5 | (0.1) | 0.8 | (0.3) | 0.9 | (0.2) | |
| 30 mg/kg | 8.9 | (0.7) | 1a | (0.1) | 1.3 | (0.6) | 8.5 | (0.5) | 1b | (0.1) | 1.5b | (0.2) | |
| 100 mg/kg | 9.2 | (0.9) | 1.3b | (0.4) | 1.9 | (0.4) | 8.6 | (0.3) | 0.9a | (0.3) | 1.0 | (0.4) | |
| Gastro (right) | Vehicle | 8.6 | (1.0) | 0.9 | (0.1) | 1.1 | (0.4) | 8.1 | (0.4) | 0.4 | (0.2) | 0.6 | (0.2) |
| 10 mg/kg | 8.2 | (0.5) | 1.0 | (0.1) | 1.0 | (0.3) | 8.0 | (0.2) | 0.6 | (0.2) | 0.7 | (0.2) | |
| 30 mg/kg | 8.4 | (0.6) | 1.0 | (0.3) | 1.0 | (0.3) | 8.0 | (0.3) | 0.5 | (0.4) | 0.8 | (0.4) | |
| 100 mg/kg | 8.8 | (0.8) | 1.2 | (0.2) | 1.4 | (0.3) | 8.0 | (0.3) | 0.8 | (0.3) | 1.0 | (0.5) | |
| Gastro (left) | Vehicle | 8.8 | (0.7) | 0.6 | (0.3) | 1.1 | (0.2) | 8.3 | (0.3) | 0.5 | (0.3) | 0.4 | (0.3) |
| 10 mg/kg | 8.4 | (0.4) | 1.0 | (0.5) | 1.2 | (0.5) | 8.5 | (0.2) | 0.6 | (0.1) | 0.6 | (0.4) | |
| 30 mg/kg | 8.6 | (0.6) | 1.3a | (0.1) | 1.1 | (0.5) | 8.4 | (0.4) | 0.7 | (0.2) | 0.9 | (0.3) | |
| 100 mg/kg | 8.9 | (0.7) | 1.3a | (0.4) | 1.4 | (0.1) | 8.3 | (0.2) | 0.7 | (0.3) | 0.7 | (0.6) | |
| Quad (right) | Vehicle | 10.5 | (1.1) | 0.4 | (0.3) | 0.7 | (0.3) | 9.7 | (0.3) | 0.6 | (0.2) | 0.5 | (0.2) |
| 10 mg/kg | 10.2 | (0.8) | 0.6 | (0.4) | 0.8 | (0.4) | 9.5 | (0.1) | 1.1 | (0.4) | 1.2a | (0.4) | |
| 30 mg/kg | 10.9 | (1.1) | 0.3 | (0.3) | 0.5 | (0.7) | 9.8 | (0.4) | 1.2 | (0.4) | 1.6b | (0.4) | |
| 100 mg/kg | 11.7 | (1.4) | 0.5 | (0.3) | 1.4 | (0.5) | 9.5 | (0.3) | 1.0 | (0.3) | 1a | (0.3) | |
| Quad (left) | Vehicle | 10.7 | (1.1) | 0.1 | (0.8) | 0.3 | (0.7) | 9.9 | (0.5) | 0.4 | (0.4) | 0.7 | (0.3) |
| 10 mg/kg | 10.6 | (0.7) | 0.5 | (0.6) | 0.6 | (0.5) | 9.9 | (0.5) | 1.2a | (0.1) | 1.3a | (0.1) | |
| 30 mg/kg | 10.6 | (0.9) | 0.8 | (0.3) | 0.9 | (0.5) | 10.1 | (0.6) | 1.2a | (0.4) | 1.6b | (0.6) | |
| 100 mg/kg | 11.6 | (1.3) | 0.6 | (0.4) | 1.3 | (0.9) | 10.0 | (0.4) | 1.5b | (0.3) | 1.7b | (0.3) | |
Gastro, gastrocnemius; Quad, quadriceps.
Muscle circumference was measured by tape once pretest, weeks 20 (day 135) and 39–40 (day 271) of the dosing phase
p < 0.05
p < 0.01: significantly different from Vehicle arm.
To explore the exposure‐response relationships, observed data from the following cohorts were used: left biceps in males at day 135 (Bicep‐L, M[135d]), left quad in females at day 135 (quad‐L, F[135d]), and right and left quad in females at day 271 (quad‐R, F[271d] and quad‐L, F[271d]). These were the only groups in which statistically significant response in all dose groups was seen when compared with the vehicle arm. Percentage change in vehicle and treated arms at day 135 or 271 over day 0 predose baseline was fitted with direct effect Emax model to estimate % vehicle response (E0), % maximal change over vehicle (Emax), weekly steady‐state AUC50 for half maximal (50%) response, and corresponding average antibody concentration (Cave50). Results obtained from Emax model fitting are shown in Figure 3 d. As depicted, only two groups (quad‐L, F[135] and quad‐L, F[271]) showed robust exposure‐response. Maximum % change (Emax) in circumference of female quad‐L muscles was estimated to be 274% and 201% over vehicle on day 135 and quad‐L on day 271, respectively, when adjusted for day 0 baseline (Table 2). Estimated AUC50 and Cave50, approximately 32 mg*h/mL and 1.3 μM, respectively, were comparable for day 135 and 271. However, only day 271 data resulted in precise estimation of PD parameters.
Prediction of MYO‐029 PK in multiple dose studies in clinic
Simulations were conducted to predict steady‐state exposures of MYO‐029 after biweekly administration of antibody (1 h infusion). The two‐compartmental PK parameters estimated from healthy volunteer data (Table 1) were used for this purpose. Median value of Cmax was estimated to be 130, 390, and 1,301 nM, respectively, for 1, 3, and 10 mg/kg (Supplementary Table S1). For the same doses, median value of Ctrough was estimated to be 24, 72, and 241 nM, respectively (Supplementary Table S1). Predicted PK profiles for 1 mg/kg, 3 mg/kg, and 10 mg/kg biweekly, respectively, are summarized in Supplementary Figure S1.
DISCUSSION
MYO‐029 was developed by Wyeth Pharma (now Pfizer) and dosed in healthy volunteers and patients with muscular dystrophy in multiple phase I/II studies.10 However, a lack of statistical power in study size did not allow for the observance of robust efficacy beyond a trend in improvements at the higher doses. Results from MYO‐029 clinical trials left several important questions, for example, was the extent of antibody exposure and resulting target modulation at the clinical doses sufficient to elicit the adequate muscle growth? Is the blockage of myostatin pathways still a valid candidate for developing new therapies for patients with myriad forms of muscular dystrophies? How well do the rodent and nonhuman primate models designed to assess the muscle growth translate to the pharmacological effects seen in humans? Addressing these questions is critical to bring forward the next generation of clinical candidates with higher probability of success in muscle‐wasting diseases, for which there are presently no effective treatment options.
As an important step toward addressing these questions, we conducted an integrated PK/PD analysis of preclinical studies designed to explore muscle growth in the form of lean mass increase in mice or muscle fiber circumference in monkeys. In addition, the extent of distribution of MYO‐029 into the muscle tissues was determined using radiolabeled studies in mice. Finally, the phase I/II exposures in healthy volunteers and patients with muscular dystrophy were analyzed in relation to the exposure response observed in preclinical species.
Compartmental modeling analysis of MYO‐029 concentration‐time data indicated that its CL was higher in humans (0.38 mL/h/kg) than in monkeys (0.23 mL/h/kg; Table 1). The human CL value is also >2× higher compared with population mean value (0.26 day/L or 0.15 mL/h/kg) reported for typical IgG1 monoclonal antibodies (mAbs).14 In mice, the presence of nonlinear CL at 1 mg/kg i.v. dose suggested target‐mediated drug disposition (TMDD), however, MYO‐029 exhibited linear PKs in rats at doses higher than 2 mg/kg, in monkeys at doses higher than 10 mg/kg, and in healthy volunteers at doses 0.3 mg/kg or above. The impact of TMDD is expected to be greater in mice compared with monkeys and humans because myostatin levels in mice (approximately 100 ng/mL) are about 10× higher than those in monkeys and humans.15, 16 Thus, lack of nonlinear response in rats, monkeys, and humans, at the doses studied, can potentially be explained by significantly lower target levels compared with those in mice.17
Myostatin and its receptor are expressed in skeletal muscle; therefore, it is important to ask whether sufficient levels of MYO‐029 antibody are achievable in muscle interstitial space. To confirm this, radiolabeled studies with 125I‐MYO‐029 were conducted in mice at 1 mg/kg dose via the i.v. or i.p. route. Data from this study (Figure 2) showed that MYO‐029 levels in the interstitial space of triceps and quadriceps were estimated to be 30–40% of systemic exposures. Based on these data, it can be concluded that MYO‐029 is able to distribute to muscle interstitial space and can be available to bind to its target.
Efficacy studies in mice showed a 23–30% increase in EDL, gastro, and quad muscle mass after 12 weeks of dosing. Modeling of these data indicated that the average MYO‐029 concentration to cause 50% effect on muscle mass (Cave50) in mice was in the range of 47–72 nM. In monkeys, we studied growth in muscle circumference of the right and left biceps, quads, and gastro muscles in a 39‐week study with weekly dosing of MYO‐029. Modeling of these data showed that maximum growth in the left quad muscles for female cohorts was two‐ to threefold (201–274%) higher compared with vehicle. Average MYO‐029 concentration to cause 50% effect on growth rates (Cave50) in monkey muscle circumference was estimated in the range of 1.3 μM. Thus, Cave50 in monkeys was approximately 20× higher than in mouse Cave50 of 47–72 nM, suggesting a significant potency shift between the two species. For mAb therapeutics, potency shift across species is usually due to the target protein sequence difference across species. However, myostatin is known to be 100% homologous across mice, rats, monkeys, and humans.2 Therefore, it is unlikely that the in vivo potency shift from mice to monkeys was due to differences in binding affinities across the species. It is unclear whether the reason for this species difference is due to the physiologically distinct role of myostatin in rodents vs. nonhuman primates (NHPs), or other compensatory mechanism driving higher target coverage requirements in NHPs.
In light of significantly weaker potency in monkeys, it was important to explore whether sufficient levels of antibody exposures were seen in clinical trials. Due to similarity in myostatin expression and muscle physiology between monkeys and humans, MYO‐029 potency in monkeys is an appropriate benchmark for predicting human efficacy. Figure 4 presents an integrated view of efficacious concentrations in mice, monkeys, and observed steady‐state in humans Cmax and Ctrough at 10 mg/kg biweekly dose (1,301 nM and 241 nM, respectively). In this plot, Emax response is normalized over vehicle to afford a clear comparison of potencies. Exposure‐response curves corresponding to monkey muscle effect are right‐shifted when compared with those for the mouse. It is also worth noting that PD effects in both of these species occur at much higher levels compared with its myostatin binding Kd (11 pM by BiAcore) of MYO‐029. When compared with monkey potency, maximum antibody concentration at the highest dose (Cmax: 1.3 uM) in the clinic only provides 50% effect, and the trough concentration at the steady state only maintained approximately 10% effect.
Figure 4.
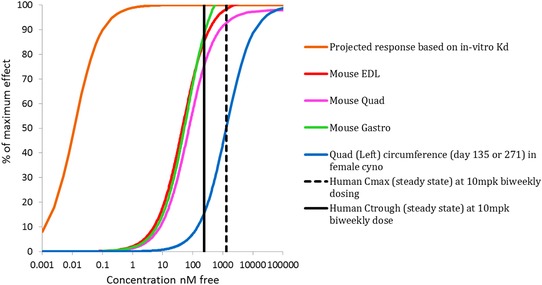
Integrated plot of pharmacodynamic end points to compare exposure‐response of MYO‐029 in mice, monkeys, and humans. Data included projected coverage based on Kd, muscle weight increase post‐MYO‐029 treatment from an efficacy study in SCID mice, muscle circumference from a 39‐week toxicology study in monkeys, and predicted peak/trough levels of MYO‐029 in patients at steady‐state following 10 mg/kg biweekly i.v. doses.
Based on the analysis presented in this report, it is reasonable to conclude that exposures of MYO‐029 in humans did not demonstrate high target coverage and had low probability of success in producing robust muscle mass increase efficacy. Interestingly, Wagner et al.10 pointed out that, although statistical significance was not observed for muscle strength and function, muscle mass, as estimated by lean mass by dual‐energy x‐ray absorptiometry, was found to increase by approximately 2.4% in the 3 mg/kg cohort, which was statistically significant from control subjects in BMD subjects and approached significance in all treated subjects. In addition, a dose‐dependent increase in fiber diameter was noted in the 3 and 10 mg/kg cohorts. Although these results are encouraging, they are exploratory in nature and should be used with caution. A definitive statement on whether the mechanism was tested in the clinical trials is precluded by the fact that robust target engagement and pharmacology biomarkers in animals and humans were not available. It is recommended that future trials with therapeutic agents1 targeting myostatin pathway should ensure high myostatin coverage, and target/pharmacology biomarkers should be developed to enable rational decision‐making.
Current analysis does not deal with the uncertainty in imaging assays themselves, or how much they might have contributed toward the lack of statistical significance in the clinical trial, given the small sample size. Use of translational research in improving diagnostic and patient‐selection methods will be important for future success. Furthermore, innovative and flexible clinical trial designs supported by the statistical methods and novel techniques are critically needed for describing, monitoring, and interpreting disease outcomes and effects of intervention in small and/or enriched populations.18
Author Contributions
P.S., H.R., and I.B. wrote the manuscript. P.S. and H.R. designed the research. I.B. performed the research. P.S., T.G., J.B., and I.B. analyzed the data.
Conflict of Interest
This work was funded by Pfizer. P.S., H.R., and I.B. were Pfizer employees when the study was conducted. T.G. and J.B. were employees of Rosa Pharma when the study was conducted.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Information
Acknowledgments
The authors thank Steven Arkin M.D. for insightful comments on the manuscript draft.
References
- 1. Khurana, T.S. & Davies, K.E. Pharmacological strategies for muscular dystrophy. Nat. Rev. Drug Discov. 2, 379–390 (2003). [DOI] [PubMed] [Google Scholar]
- 2. Tobin, J.F. & Celeste, A.J. Myostatin, a negative regulator of muscle mass: implications for muscle degenerative diseases. Curr. Opin. Pharmacol. 5, 328–332 (2005). [DOI] [PubMed] [Google Scholar]
- 3. Tsuchida, K. Targeting myostatin for therapies against muscle‐wasting disorders. Curr. Opin. Drug Discov. Devel. 11, 487–494 (2008). [PubMed] [Google Scholar]
- 4. Patel, K. & Amthor, H. The function of myostatin and strategies of myostatin blockade‐new hope for therapies aimed at promoting growth of skeletal muscle. Neuromuscul. Disord. 15, 117–126 (2005). [DOI] [PubMed] [Google Scholar]
- 5. Grobet, L. et al A deletion in the bovine myostatin gene causes the double‐muscled phenotype in cattle. Nat. Genet. 17, 71–74 (1997). [DOI] [PubMed] [Google Scholar]
- 6. McPherron, A.C. & Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 94, 12457–12461 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mosher, D.S. et al A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 3, e79 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schuelke, M. et al Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 350, 2682–2688 (2004). [DOI] [PubMed] [Google Scholar]
- 9. Wagner, K.R. , McPherron, A.C. , Winik, N. & Lee, S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 52, 832–836 (2002). [DOI] [PubMed] [Google Scholar]
- 10. Wagner, K.R. et al A phase I/II trial of MYO‐029 in adult subjects with muscular dystrophy. Ann. Neurol. 63, 561–571 (2008). [DOI] [PubMed] [Google Scholar]
- 11. Acceleron Pharma . Acceleron presents preliminary ACE‐031 results from a phase 1 multiple ascending dose study in healthy volunteers. (Acceleron Pharma, Cambridge, Massachusetts, 2010). http://www.acceleronpharma.com/wp-content/uploads/2011/09/October-13-2010-Acceleron-Press-Release.pdf. [Google Scholar]
- 12. Acceleron Pharma Update: ACE‐031 clinical trials in Duchenne muscular dystrophy. (Acceleron Pharma, Cambridge, Massachusetts, 2013). http://quest.mda.org/news/update-ace-031-clinical-trials-duchenne-md. [Google Scholar]
- 13. Shah, D.K. & Betts, A.M. Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. MAbs 5, 297–305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dirks, N.L. & Meibohm, B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 49, 633–659 (2010). [DOI] [PubMed] [Google Scholar]
- 15. Han, D.S. et al Serum myostatin levels and grip strength in normal subjects and patients on maintenance haemodialysis. Clin. Endocrinol. (Oxf.) 75, 857–863 (2011). [DOI] [PubMed] [Google Scholar]
- 16. Lakshman, K.M. et al Measurement of myostatin concentrations in human serum: circulating concentrations in young and older men and effects of testosterone administration. Mol. Cell. Endocrinol. 302, 26–32 (2009). [DOI] [PubMed] [Google Scholar]
- 17. Singh, A.P. et al Quantitative prediction of human pharmacokinetics for mAbs exhibiting target‐mediated disposition. AAPS J. 17, 389–399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pariser, A.R. & Gahl, W.A. Important role of translational science in rare disease innovation, discovery, and drug development. J. Gen. Intern. Med. 29 (Suppl 3), S804–S807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Information