

INTRODUCTION
The aim of this tutorial is to review next‐generation sequencing (NGS) techniques and the emerging role of this technology in the diagnosis and treatment of cancer. We provide a basic overview of the different types of NGS and the strengths and weaknesses of each approach in the context of cancer as a heterogeneous disease driven by DNA‐altering mutations. Finally, we discuss how this information can inform personalized clinical development of targeted therapies.
HISTORY AND OVERVIEW OF NGS TECHNOLOGY
Background and basic methodology
Since the first whole‐genome sequence of a free‐living organism—the bacterium Haemophilus influenzae—was reported in 1995,1 DNA sequencing has been a mainstay of life science research and development (Figure 1). During the subsequent decade, using the predominant method of Sanger sequencing,2 large research teams and consortia elucidated the genomes of multiple model organisms, microbes, and ultimately the first draft human genome sequence in 2001.3 Sanger sequencing produces highly accurate sequencing reads up to 1,000 base pairs (bp) long, and is still considered the gold‐standard sequencing method for clinical applications today. The workhorse instruments of this era were capillary electrophoresis instruments that employed fluorescent dye terminators to read out the sequence of up to 96 cloned DNA templates in parallel. Because each instrument run generated fewer than 100 kb of sequence data, sequencing of a human genome was only feasible in a dedicated genome center with a factory‐like floor filled with sequencing and associated instruments.
Figure 1.
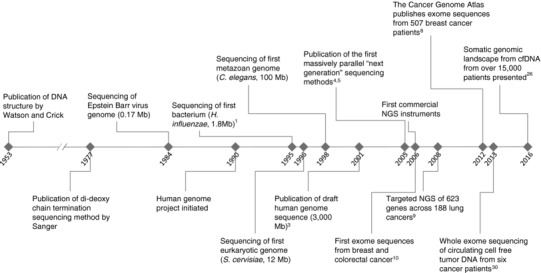
Timeline of key events in development of sequencing technologies and applications to oncology.
The advent of commercially available next‐generation sequencing (NGS) instruments in 2006 made DNA sequencing dramatically simpler and faster by employing microscopic, spatially separated DNA templates to massively parallelize the capture of data.4, 5 Early NGS instruments generated about 1 gigabase (Gb) of sequence, and with the rapid pace of development in this field, the current highest throughput commercial instruments today can generate nearly 1 terabase (Tb) per run (1012 bp). The first instruments could only generate short sequencing reads of fewer than 100 bp, and exhibited higher raw error rates than Sanger sequencing, but the sheer volume of reads allowed reasonably accurate consensus sequences to be inferred by aligning reads to a previously sequenced reference genome. With data generation becoming relatively easy, the bottleneck for sequencing experiments now lies in the data analysis steps, which often require sophisticated algorithms, powerful computing infrastructure, and skilled bioinformaticians. In the decade since the release of the first NGS instruments, short read platforms have continued to improve in data quality and read length, and have been deployed in many clinical laboratories. Additionally, a number of novel technology platforms have emerged, although their utility for clinical tests has not yet been broadly established. Single‐molecule sequencing instruments can generate long sequencing reads over 10 kb in length, albeit with higher error rates. Nanopore‐based instruments represent the latest frontier of sequencing, promising to deliver extremely long reads of over 100 kb, but error rates of 10% or higher in current products often necessitate augmentation with higher‐quality short read data. For more comprehensive information about NGS platforms, please see the excellent reviews by Mardis6 and Goodwin et al.7
DNA sequencing NGS methods
Whole‐genome sequencing (WGS) is the most straightforward application of NGS (see Table 1 for definition). Genomic DNA is isolated, fragmented, and made into a library, using platform‐specific reagents, which can be analyzed by the sequencing instrument. No enrichment step is employed, so the entire genome, including noncoding regions, is sequenced. This method can provide high‐resolution detection of many variant types including single nucleotide variants (SNV), insertions or deletions (indels), copy number aberrations (CNA; also referred to as copy number variations (CNV)), and rearrangements. However, the comprehensive nature of WGS requires massive amounts of sequencing to achieve adequate coverage to detect variants. NGS experiments are typically run to achieve a predetermined coverage, which is the average depth of sequencing over all targeted genome positions. Coverage at any genome coordinate can be calculated, after mapping sequencing reads to the reference genome, by counting the number of independent reads that overlap that position. Due to random variation and sequencing bias caused by nucleotide composition (e.g., GC content), many positions in the genome will have coverage lower than the average, which requires deeper sequencing to ensure most positions are adequately covered for detection of variants. The typical 30x coverage WGS experiment to confidently identify most homozygous and heterozygous germline variants requires about 100 Gb for a human genome, and achieving sufficient depth of coverage to detect rare somatic mutations in tumors may require 10‐fold more sequencing, making WGS expensive and time‐consuming for routine cancer genomics.
Table 1.
Glossary of terms
| Term | Definition |
|---|---|
| Next‐generation Sequencing (NGS) | Any of several high‐throughput approaches to DNA sequencing using the concept of massively parallel processing rather than sequencing individual DNA strands |
| Whole genome Sequencing (WGS) | Sequencing to determine the complete DNA sequence of an organism's genome at a single time, typically the somatic cancer genome in oncology studies |
| Whole exome sequencing (WES) | A method to determine the DNA sequence of all of the exons within protein‐coding genes in a genome |
| RNA sequencing (RNA‐Seq) | Using NGS to determine the sequence of the transcriptome, or complete set of RNA transcripts in a sample |
| Oncogene addiction | The phenomenon whereby the survival of cancer cells depends on the continued activity of a mutated oncogene |
| Circulating tumor cell (CTC) | Tumor cells shed from the tumor mass into peripheral circulation |
| Circulating tumor DNA (ctDNA) | Cell free DNA derived from apoptotic or necrotic tumor cells that is shed into circulation and can be detected in plasma |
| Companion diagnostic (CDx) | A diagnostic test approved by the FDA that is required for the safe and effective administration of a therapeutic |
| Somatic mutation | Genetic alteration acquired by a cell that can be passed to the progeny of the mutated cell but which was not present in germline DNA (sperm or egg) |
| Mutational burden | The total number of mutations in a cancer genome |
| Intratumoral heterogeneity (ITH) | Genetic and phenotypic variation within a tumor, or between individual tumor lesions in the same patient |
| Driver vs. passenger mutation | Driver mutations contribute to the growth and survival of a tumor and therefore confer a selective advantage. Passenger mutations arise during the development of a tumor as a result of increased mutation rates, but do not contribute to tumor growth. |
To make more efficient use of sequencing capacity, many cancer genomics studies employ whole‐exome sequencing (WES) in which library construction is followed by an enrichment step that targets only the exons of protein‐coding genes. By restricting sequencing to less than 2% of the genome, it is possible to sequence more samples per instrument run for lower cost, while still achieving higher coverage for detection of low‐frequency somatic variants. The tradeoff for using WES vs. WGS is loss of the ability to detect noncoding variants, including some rearrangements, which could have important implications on gene regulation. CNA detection may also be impeded relative to WGS‐based methods.
When an investigator seeks to interrogate a specific list of genes or other loci, targeted sequencing panels can be employed. These are prepared in much the same way as WES libraries, but the enrichment reagent—typically a pool of complementary hybridization probes or set of PCR primer pairs—targets a smaller number of loci. These can be manufactured to custom specifications, but many manufacturers offer off‐the‐shelf panels targeting specific pathways or disease areas (e.g., cancer hotspot panels). The small size of such panels allows either high multiplexing of samples with lower cost, or ultra‐deep sequencing of specific loci of interest to detect rare events.
When searching for somatic variants in tumors, many studies sequence a matched normal sample (often blood or tumor‐adjacent normal tissue) from the same patient in order to unambiguously identify germline variants. Genomic aberrations that appear in the tumor but not the normal sample are assumed to be somatically derived. For targeted panels, it is possible to differentiate somatic from germline variants in a single tumor sample, without a matched normal, using a set of heuristics. For example, variants that have been detected in healthy population screens (e.g., 1000 Genomes Project, Exome Aggregation Consortium (ExAC)) are generally assumed to represent germline variants, while those found to be verified somatic mutations in the Catalogue of Somatic Mutations in Cancer (COSMIC) database are often assumed to be of somatic origin. Additional criteria based on allele frequency distributions may also be applied. In the decade since the development of massively parallel NGS platforms, sequencing of cancer genomes has escalated rapidly from targeted panels to WES cohorts of cancer patients across different indications (Figure 1).8, 9, 10
RNA sequencing NGS methods
Sequencing of RNA (RNA‐Seq) is an extension of the methods developed for DNA sequencing that generally requires synthesis of cDNA from an RNA template (Table 1). RNA‐Seq protocols differ in their methods for removing ribosomal RNA (rRNA), which is a critical step given that over 90% of a typical total RNA sample is made up of rRNA molecules. After depleting rRNA or enriching coding transcripts, libraries are constructed and sequenced, and reads are mapped to a reference genome or transcriptome. Quantitative gene expression measurements can be obtained by counting the number of reads mapping to each transcript or noncoding RNA; these expression measurements can be used as biomarkers, either singly or in combinations called gene expression signatures, which are often reflective of relevant biology in a sample (e.g., prostate cancer subtypes11). Fusion transcripts can be detected by searching for “chimeric” reads, and in some cases, genomic variants can be identified. This method also facilitates detection and quantitation of alternative splice variants, which are often differentially expressed in tumor vs. normal cells (reviewed in Feng et al.12). RNA‐Seq is generally performed as a nontargeted assay in order to achieve comprehensive transcriptional profiling, but target enrichment methods described above have been adapted to facilitate assays that interrogate a selected set of transcripts.
By combining genomic (DNA) and transcriptomic (RNA) data from the same sample, it is possible to obtain a deeper understanding of the biology underlying that tumor. The discipline of integrative genomics provides statistical methods and tools that facilitate the merging and analysis of multiple high‐dimensional data sets. Example uses of this approach include identification of gene expression deregulation caused by CNAs,13 characterization of molecular tumor subtypes by combined expression and genomics signatures,14 enhanced mutation detection in low purity tumor samples,15 and identification of oncogenic transcript splicing isoforms.16 For a more detailed review of the utility of integrative genomics in cancer, please see the excellent review from Kristensen et al.17
Recent developments: NGS analysis of cell‐free tumor DNA
Deep sequencing of tumor‐derived DNA offers a powerful methodology to identify potential driver mutations in cancer and match patients with appropriate targeted therapies. While tumor tissue remains the gold standard for molecular diagnostic analysis, collection of fresh biopsies poses risks and significant discomfort to patients.18 Furthermore, a biopsy represents only a single tumor site, and may not reflect the substantial heterogeneity that exists within a single primary tumor19 or between metastatic sites,20 and the fact that many years and intervening therapies may separate collection of archival primary tumor from the need to make a treatment decision (Figure 2).
Figure 2.
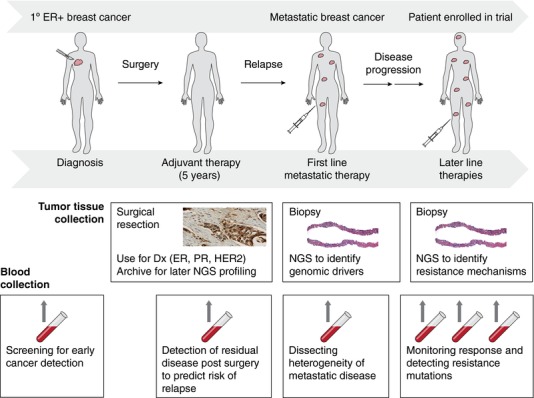
Example of the potential utility of NGS applications in the clinical management of breast cancer.
These limitations have prompted efforts to develop methods to sequence other sources of neoplastic cells, including circulating tumor cells (CTCs) and circulating cell‐free tumor DNA (ctDNA). Recent studies have suggested that rare cells shed from the tumor mass (CTCs), or DNA released into circulation from apoptotic or necrotic cancer cells (ctDNA) may have utility as a source of material for NGS analysis that is representative of tumor tissue and that can be repeatedly sampled by noninvasive blood draws, allowing monitoring of tumor evolution over time and treatment, and identification of resistance mechanisms that may suggest subsequent lines of therapy. A recent study from Lohr et al. compared WES from single CTCs with that from matched tumor tissue and found that 70% of the mutations in the tumor sample could be detected in CTCs, suggesting that CTCs can provide potentially meaningful mutation data.21 However, many patients do not have detectable numbers of CTCs, or have too few CTCs to make meaningful assessments of tumor heterogeneity. Indeed, a study from Bettagowda et al. suggested that ctDNA was often present in patients without detectable circulating tumor cells, suggesting that these two sources of biomarker material may be distinct and have different utility.22
Identifying and accurately measuring the mutant allele fraction of cancer‐specific mutations in ctDNA is complicated by the presence of background cell‐free DNA derived from normal cells, as well as low concentration of ctDNA in many patients.23 Approaches to enhance detection and improve quantitation of ctDNA in these challenging circumstances include sequencing at very high coverage and utilization of molecular barcodes to dramatically lower error rates by redundant sequencing of individual ctDNA molecules (e.g., Kinde et al.24). Methodologies for ctDNA sequencing using NGS are not as sensitive as some other molecular methods (e.g., digital polymerase chain reaction (PCR)25), but NGS has the advantage of interrogating a larger number of genomic loci. In addition, recent technological advances, including molecular barcoding to overcome intrinsic polymerase error rates,24 have increased assay sensitivity. A recent report described the targeted sequencing of a panel of 70 genes from the ctDNA of over 15,000 cancer patients, and suggested that the mutation spectrum observed was largely similar to that from tumor tissue specimens, with the notable exception that known resistance mutations are enriched in ctDNA compared with primary tumors.26 Finally, continued improvements in technology have made targeted sequencing and WES affordable, opening the door to real‐time longitudinal assessment of cancer mutations in circulation.27, 28, 29, 30, 31 Overall, these results suggest significant promise for NGS analysis of ctDNA in the identification of clinically actionable mutations and selection of appropriate therapy. Specific clinical applications of this novel approach will be considered in more detail below.
APPLICATIONS OF NGS IN CLINICAL TRIALS AND CLINICAL PRACTICE
Detection of driver alterations
Detection of somatic driver alterations that occur early in the development of a cancer and result in oncogene addiction is currently the primary application of next‐generation sequencing in oncology.32 Identification of driver alterations can guide the way to treatment with matched targeted therapies. Several recurrent driver mutations have been or are currently being clinically validated (i.e., associated with benefit to specific targeted agents), including BRAF V600E, KIT, EGFR, ERBB2, FGFR3, PIK3CA, AKT1, TSC1, and ROS1 mutations, ERBB2 amplifications, and ALK translocations.33
Unlike traditional molecular assays, which focus on a relatively small panel of commonly mutated sites, NGS offers the capacity to measure somatic allele frequencies from the complete coding sequences of many genes in the same assay. There is general agreement that NGS should be the standard method when several genes must be tested in the same patient. As an illustration, a recent meeting of medical oncologists concluded that patients with non‐small cell lung cancers (NSCLC) should be tested for mutations in EGFR, BRAF, ERBB2, ROS1, and ALK using NGS methods (under specific conditions for ALK).33 Similarly, estrogen receptor‐positive breast cancer patients should be tested for mutations in PIK3CA, ESR1, AKT1, ERBB2, and again, it seems likely that NGS will become the standard method for diagnosis of genomic alterations in breast cancer.33 While NGS has shown analytical validity34 and CLIA‐certified assay vendors can provide NGS results to patients, many approved companion diagnostic tools are based on Sanger sequencing or PCR‐based methods. Nevertheless, health authorities are contemplating regulations for the clinical use of NGS,35 and it seems very likely that companion diagnostic tests for targeted therapies could soon be NGS‐based (discussed in more detail below).
One of the major questions related to the use of NGS in daily practice concerns the number of genes that should be tested (Table 2). We and others have reported previously that the number of recurrent and potentially actionable gene alterations in diseases like lung or breast cancer range from 5 to 10,8, 9, 36 so one could ask why several hundred genes should be tested on an NGS panel. Sequencing studies have shown that, in addition to the high‐prevalence, recurrent drivers, each tumor histology is characterized by a “long tail” of gene alterations that occur each in less than 1% of patients,37 and some of these rare genomic alterations may be associated with drug sensitivity. For example, rare, activating MTOR mutations have been associated with response to everolimus in a patient with thyroid cancer38 and low‐prevalence ERBB3 mutations have been associated with objective response following Her2 inhibition.39
Table 2.
Key open questions for clinical NGS testing in oncology
| Application | Examples | NGS method | Clinical research questions |
|---|---|---|---|
| Oncogenic driver identification | BRAF V600E, KIT, EGFR, ERBB2, FGFR3, PIK3CA, AKT1, TSC1, ROS1 mutations, ERBB2 amplifications, ALK translocations | WES, or targeted sequencing of tumor DNA or ctDNA | Is it useful to sequence large panels of genes vs actionable drivers? |
| Characterization of resistance | EGFR T790M, ESR1 mutations | WES, or targeted sequencing of tumor DNA or ctDNA | Is it useful to detect genetic mechanisms of resistance earlier, using ctDNA? |
| Identification of patients sensitive to immunotherapy | Mutational burden; neoantigens; Gene expression profiles | WES, or targeted (large panel) sequencing of tumor DNA; RNA‐Seq of tumor | Can identification of mutational process and clonality improve prediction based on mutational burden? Can personalized cancer vaccines boost responses to immune checkpoint inhibitors? Can gene expression signatures identify immune‐responsive tumors? |
| Germline mutations | BRCA1, BRCA2, PTEN, TSC1, CDH1 | WGS or WES of normal DNA | Should the number of patients screened for germline mutations be expanded? Is it useful to interrogate a broad panel of genes ? |
| Quantification of intratumor heterogeneity | none | Whole exome sequencing of multiple tumor sites and metastatic lesions, or plasma | Is ITH associated with resistance to therapy and worse outcome? Is it targetable? |
| Quantification of pathway activation | ER, AR, mTOR, and CDK4 signatures | RNA‐Seq of tumors | Can gene expression signatures identify responsive patients for targeted therapies? |
| Cancer screening | none | Ultradeep sequencing of plasma | Can ultradeep sequencing on ctDNA identify some cancers early? Is it useful? |
| Regulatory approval | none | Any | How can clinical validity of NGS tests be established for previously approved biomarkers? What level of evidence is required for biomarkers that are rare in an indication? How will approval be maintained as the field advances? |
Because these genomic alterations are very rare, it is impractical to recruit subjects for clinical trials targeting these alterations. Based on this rationale, several teams have started clinical trials that evaluate the clinical utility of testing large panels of genes and treating based on theoretically actionable alterations. There is no evidence available yet that such an approach actually improves patient outcomes.33 As an illustration, the SAFIR0136 and SHIVA40 trials failed to report outcome improvement when comparative genomic hybridization (CGH) arrays or large NGS panels were used to select patients for targeted therapies. Nevertheless, these trials did not use the latest generation of targeted therapies, and it is hoped that new trials testing large panel of genes and integrating the newest generation of targeted therapies may be more definitive in demonstrating the utility of this approach.41
Detecting resistance
While therapies matched to known driver mutations have clearly improved patient outcomes, the vast majority of patients treated with targeted therapies will develop secondary resistance. Next‐generation sequencing can elucidate the mechanisms of resistance and guide subsequent treatment,42 for example, by identifying new mutations in biopsies at disease relapse. As an illustration, resistance mechanisms to EGFR inhibitors are multigenic and can include T790M EGFR mutations; ERBB2, PIK3CA, and BRAF mutations; and MET amplifications.43 NGS on ctDNA has the potential to detect mutations as early as possible during the disease course,44 but further clinical trials are needed to determine whether using multigene panels for early detection of resistant clones can improve clinical outcomes (Table 2).
Sensitivity to immunotherapeutic agents
Immune checkpoint inhibitors such as anti‐CTLA4, and more recently anti‐PD‐1 and anti‐PD‐L1, have been shown to improve overall survival in certain immunogenic cancers such as melanoma, lung cancer, and bladder cancer. A number of clinical studies have shown that expression of PD‐L1 on tumor or immune cells enriches drug response,45, 46, 47 and several immunohistochemistry (IHC) assays have been approved as companion or complementary diagnostic tests in NSCLC and bladder cancer.48, 49, 50 However, many patients lacking PD‐L1 expression respond to checkpoint blockade and not all patients with PD‐L1 expression respond, suggesting a need for more sensitive and specific diagnostic tests. Several studies have suggested that high tumor mutational burden, determined by NGS, could be associated with increased sensitivity to immune checkpoint inhibitors.47, 51, 52 For example, Rizvi et al. have reported that patients with high mutational burden NSCLC treated with anti‐PD‐1 presented a lower likelihood of progressive disease.51 Further analyses have suggested that taking into account mutational process and intratumoral heterogeneity (ITH, see section below)46 could improve the prediction. Rizvi et al. indeed reported that a specific pattern of base substitutions (also referred to as a mutational signature) associated with prolonged exposure to tobacco smoke is associated with a higher likelihood of benefit within the group of highly mutated NSCLC,51 and McGranahan, et al. reported that among highly mutated cancers, those with high number of clonal neoantigens (see below) and less heterogeneity are more likely to be sensitive to immunotherapeutics.53 These data suggest that mutational burden, fine‐tuned with analysis of mutational patterns and ITH, could predict which patients will derive benefit from single‐agent immune checkpoint blockers. WES is the most comprehensive method to quantify mutational burden and characterize mutational patterns, and this method has recently been shown to be feasible in the context of a clinical trial,54 but recent results have suggested that targeted NGS panels may also have utility in inferring overall mutational burden at a lower cost.47
Another promising and highly personalized application of WES is the idea of personalized cancer vaccines based on the unique spectrum of protein‐altering mutations in a given patient's tumor.55, 56 This is a complicated approach that requires WES to identify mutations that encode candidate neoantigens, application of sophisticated algorithms to determine which neoantigens are immunogenic, and then formulation of a corresponding peptide‐ or RNA‐based vaccine that can be administered to the patient and hopefully elicit a tumor‐specific immune response. Early results have provided some evidence that CD8 T‐cell responses to tumor neoantigens can be enhanced through vaccination in melanoma patients,57 but many challenges await before this approach is clinically validated.58
Assessing intratumoral heterogeneity
Within a single primary tumor and its metastatic lesions, genomic alterations are heterogeneously distributed. During the evolution of a tumor, alterations that occur very early and are required for neoplastic growth are distributed throughout the tumor and are usually named trunk alterations. Variants that arise later during cancer development (branch alterations) are not homogeneously distributed, and may be private to very restricted tumor regions or single metastases.19 ITH is difficult to accurately assess in most patients because collection of extensive biopsies of multiple tumor lesions is impractical and cannot be implemented in routine clinical practice for the reasons discussed above.
One potential method to overcome the limitations with biopsies is to perform WES on ctDNA. This approach accurately quantifies heterogeneity by detecting subclonal and site‐specific alterations.59 Quantifying ITH could have several clinical implications. As already shown in chronic lymphocytic leukemia,60 high ITH could be associated with worse outcome. ITH could also predict the response to immunotherapeutics (see above). Finally, ITH may also predict treatment failure for targeted therapies by identifying low‐frequency, preexisting, subclonal mutations that confer resistance, a phenomenon previously documented in the case of MET amplification associated with EGFR‐inhibitor sensitivity.61 Prospective trials are currently collecting plasma samples in order to further assess the clinical utility of ITH as characterized by ctDNA sequencing.
Guiding development of combination therapies
A potentially important application of NGS is in guiding patient identification for combination therapies, given that the approach yields parallel information on a large number of genes as opposed to other technologies such as PCR that provide sequence information on a small number of recurrent mutations. There are numerous preclinical examples that suggest potential utility in simultaneously targeting multiple drivers. For instance, a number of studies have shown that combined targeting of MAP kinase and PI3K pathways can be synergistic when both pathways are activated.62, 63 In addition, recent studies have provided evidence that the combination of immune checkpoint inhibition with MEK inhibition can synergize to result in greater antitumor immune responses,64 suggesting that NGS assays combining overall mutational load with mutations in the MAPK pathway might be effective in treating patients with this combination. In addition to concomitant alterations in baseline primary tumor samples, as discussed above, a variety of reports have identified resistance mechanisms based on the acquisition of mutations or gene amplification events. For instance, preclinical work has shown that treatment of NSCLC cell lines with the EGFR inhibitor erlotinib results in selection for resistant clones harboring MET amplification, suggesting that NGS on a sample collected at relapse could be useful to detect such events and select patients for treatment for combination therapy with EGFR and MET inhibitors.61 One approach to validating NGS as a means of selecting patients for combination studies would be umbrella trials (described in more detail below) involving different arms that match patient with candidate therapies based on particular genomic alterations. In this scenario, patients could be enrolled in the trial and receive a candidate targeted therapy, not just based on single alterations but in some cases based on a multiple alterations where a strong preclinical rationale exists that patients might benefit from a combination of therapeutics. While conceptually appealing, a challenge to this approach will be the need to conduct phase Ib studies in advance to make sure the investigational agents can be safely combined at exposures sufficient to result in efficacy prior to inclusion in an umbrella study, since in some cases (e.g., MEK and PI3K) the clinical combination has been shown to been challenging in terms of tolerability.65
Clinical applications of ctDNA sequencing
As mentioned in previous sections, ctDNA is detectable in plasma and amenable to NGS. Here we consider the current, practical clinical applications of NGS on ctDNA. One application is detection of driver or resistance mutations that are not present in primary tumors but do occur in relapsed or metastatic samples. Examples of this include ESR1 mutations in ER+ breast cancer, which are very rare in primary tumor tissue, but occur after prolonged aromatase inhibitor therapy and can readily be detected in metastatic samples66, 67 or in ctDNA.68, 69 NGS analysis of ctDNA thus may be useful to identify patients with ESR1 mutations and select them for treatment with novel endocrine agents that may have efficacy against tumors harboring these mutations. Similarly, EGFR T790M mutations and MET amplifications that arise during gefitinib or erlotinib therapy can readily be detected in ctDNA, and these alterations could be used to select patients for treatment with next‐generation inhibitors that target these resistance mutations.70 In another example, Murtaza et al.30 detected PIK3CA and RB1 mutations associated with resistance to chemotherapy, suggesting the possibility of testing candidate targeted agents against these drivers in such patients. In addition, ctDNA NGS assays may broaden the population of patients available for clinical testing in indications such as NSCLC in which a substantial fraction of patients may not have tissue available for testing.
One mid‐term application will be to use ctDNA detection to predict relapse in patients with localized cancer. Garcia‐Murillas et al.71 have shown that after first‐line therapy, rising concentration of ctDNA in the plasma, as assessed by NGS‐based mutation detection, is associated with a metastatic relapse in the following months, suggesting that the technology could be used to identify patients at high risk for clinical relapse with the aim of providing more aggressive or targeted adjuvant therapy. From a long‐term perspective, the capacity to perform ultra‐deep sequencing while minimizing errors should open the avenue of cancer screening and early detection using NGS in plasma. Current reports suggest that sequencing circulating DNA can detect around 50% of early‐stage cancers22 and these numbers will increase with new technologies. Intensive efforts are underway in academic laboratories and biotech start‐up companies to develop more robust assays and clinically validate them in large cohorts of patients.44
Assessing target expression and pathway activation using RNA‐Seq
Some cancers express therapeutic targets without underlying alterations at the DNA level. For example, estrogen receptor gene expression is associated with sensitivity to endocrine therapy, but the gene sequence is typically unaltered in endocrine‐naïve patients. Beyond identification of overexpressed oncogenic targets, analyses at the RNA level can also measure oncogenic pathway activation. RNA‐Seq has been shown to be feasible in the context of daily practice and could complement DNA‐based approaches.
In breast cancer, gene expression signatures have been developed to quantify ER, AR, mTOR, and CDK4 activation,72 which could predict efficacy of drugs targeting these pathways. RNA‐Seq may also enable identification of patients most likely to respond to immunotherapy, as a number of reports have suggested that expression signatures indicating the presence of specific immune cell types is associated with differential prognosis73 and may perhaps be associated with benefit or resistance to immunotherapy agents.74, 75 RNA‐Seq is also applicable for molecular subtyping within tumor histologies. For example, subtyping of urothelial carcinoma samples from a phase II study of atezolizumab identified distinct basal and luminal subtypes that differed in their response to the treatment.47
NGS for germline analyses
NGS is already broadly utilized for the detection of germline mutations that cause hereditary illnesses and influence disease risk (e.g., cystic fibrosis).76 In the oncology setting, this is particularly useful when sequencing large genes like BRCA1, BRCA2, and PTEN, or when multiple genes must be tested in the same patient. Several studies indeed suggest that testing multiple genes could be useful for clinical diagnosis. Using a 17‐gene panel, Couch et al.77 recently reported that 3.7% of triple negative breast cancer tumors carry a deleterious germline mutation aside from BRCA1 and BRCA2. In a series of 692 patients with metastatic prostate cancer, 12% were found to have a deleterious germline mutation in genes involved in DNA‐repair.78 Recent evidence‐based guidelines have included the use of multigene testing, comparing its advantages and disadvantages.79 These guidelines state that “only genes with a known (i.e., published and confirmed) relationship between the aberrant genotype and the pathology should be included in the analysis.” Some groups even propose to use WES as a tool for counseling in rare genetic disorders, but this extended approach raises several challenges for clinical interpretation.80
WES performed to identify somatic aberrations often includes simultaneous analysis of germline DNA from a matched normal sample as a control, and as such, could lead to incidental detection of genetic susceptibilities. The American College of Medical Genetics and Genomics (ACMG) has recommended analysis of 56 specific genes associated with hereditary syndromes, including cancer, and reached consensus that reporting some incidental and secondary findings would likely have medical benefit for the patients and families of patients undergoing clinical NGS testing.81 These considerations emphasize the need to train genomics test‐prescribing clinicians in order to correctly inform patients about the possibility and consequences of incidental genetic findings.
New trial designs to validate genomic approaches
Advances in new technologies and knowledge in biology have led to the development of new concepts in clinical trials. One type of study aims to validate that a given genomic alteration is associated with drug sensitivity. One of the key challenges here is patient accrual in clinical trials since many of the genomic alterations are rare. Two trial designs have been developed to facilitate effective molecular screening. One approach, termed a basket trial, enrolls patients across a broad range of indications (e.g., lung cancer, breast, colon, etc.) as long as the patient is positive for the enrollment biomarker.82 Examples of basket trials include the VE‐BASKET study, which showed promising efficacy of vemurafenib in BRAF mutant NSCLC, and other indications where BRAF mutations are rare (e.g., Langerhans cell histiocytosis, colon cancer, and thyroid cancer), demonstrating the clinical validity of this biomarker‐drug pair in these indications.83 So‐called umbrella clinical trials offer a complementary approach to basket clinical trials by focusing on a single indication (e.g., lung cancer), but incorporating multiple investigational biomarkers and treatments within that indication. The Lung Master Protocol (LUNG‐MAP) is an example of such an approach and involves screening squamous cell lung cancer patients with comprehensive NGS‐based testing. If the patient is positive for one of the biomarkers under study (CDK4, PIK3CA, MET, and FGFR) they are enrolled in the appropriate treatment cohort, while in the absence of an alteration they receive the immunotherapy agent nivolumab.41 In addition to these screening approaches, the oncology community is getting more information about rare genomic variants that impact targeted therapy response by applying NGS to exceptional responders. For example, the retrospective sequencing of one exceptional responder to everolimus identified TSC1 mutations as a predictor of exquisite sensitivity to mTOR inhibitors.84
A second type of study aims to prove the clinical utility of new technologies such as NGS by showing that application of the new technology improves a clinical end point in the context of a prospective clinical trial. As mentioned previously, there is no evidence yet that NGS testing of a broad panel of genes is clinically useful in the context of oncology. The prospective SAFIR02_Breast and SAFIR02_Lung trials (ClinicalTrials.gov identifiers NCT02299999 and NCT02117167) aim to establish such clinical utility by randomizing patients who lack genomic alterations detected by conventional tests between the use of large panel NGS testing vs. standard of care. Studies to demonstrate clinical utility of ctDNA sequencing for the early detection of resistance will likely follow in the near future.
Path to companion diagnostics and widespread use of NGS platforms
While it is clear that NGS‐based technologies are already having an impact on patient care, in order for broad, widespread use to occur several factors will need to be addressed in the coming years. These factors include increasing the “actionability” of the results from NGS‐based approaches, evolving the regulatory paradigm to gain approval of NGS‐based tests and securing broad insurance coverage for NGS‐based testing. While the focus of this review is on NGS‐based cancer testing for informing treatment decision making, it should be noted that similar barriers to the widespread use of NGS‐based tests exist in germline sequencing of patients suspected to harbor a genetic disorder, as well as noninvasive prenatal testing (NIPT) that uses NGS to screen for fetal chromosomal abnormalities (e.g., trisomy 21) through a maternal blood draw.
Numerous clinically validated biomarkers in practice guidelines today are detectable via NGS testing (e.g., EGFR mutations and ALK fusions in NSCLC, and BRAF mutations in melanoma). NGS testing for these biomarkers as opposed to singleplex tests (such as Sanger sequencing or PCR) has the advantage of being able to screen a broad set of genes in one comprehensive test, which utilizes the scarce biopsy tissue available in the most efficient manner possible so that the most appropriate treatment decision can be made for an individual patient. As additional biomarkers are clinically validated in a given indication, the clinical utility of converting singleplex testing paradigms to NGS‐based tests will continue to grow. However, traditional clinical trial designs may not always be appropriate for demonstrating this clinical validity, given the therapeutics’ mechanism of action and the large amount of data available from an NGS‐based test. Several biomarker‐driven clinical trials have been implemented in the field and offer efficient and scientifically driven ways to demonstrate clinical validity of new biomarkers and therapeutics.85 As discussed above, there are now numerous examples of basket and umbrella approaches to clinical trial designs either in development or underway, and these offer a promising path to further demonstration of the clinical validity of NGS‐based tests. Appropriate discussions are underway between trial sponsors and health authorities such that positive outcomes in one or more cohorts from these studies should result in the approval of an NGS‐based companion diagnostic (CDx) test.41
NGS‐based cancer tests are driving an evolution of the regulatory aspects of testing as well.86 In the US, the Food and Drug Administration (FDA) requires premarket approval or clearance before the sale of an in vitro diagnostic test. To date, all of the NGS‐based cancer testing performed in the US has been performed via a Laboratory Developed Test (LDT) paradigm, where the clinical laboratory designs, develops, and analytically validates the test in‐house under the guidelines of the Clinical Laboratory Improvement Amendments (CLIA). The FDA has in the past practiced “enforcement discretion” over LDTs and not used their authority in this area of clinical testing. However, the FDA has recently indicated that they will stop practicing enforcement discretion towards LDTs over the next few years and instead require FDA clearance or approval of these tests in a manner similar to IVD manufacturers.87 This change is in part driven by the rapid increase of NGS testing as LDTs in clinical practice including cancer testing. Under CLIA guidelines, clinical labs do not necessarily have to demonstrate the clinical validity of their test; only analytical validity must be shown. However, for FDA approval or clearance clinical validity of the test must be demonstrated. Several challenges need to be addressed in the regulatory paradigm for FDA approval of an NGS‐based cancer test.88 For instance, how does a test developer demonstrate the clinical validity of a previously approved biomarker (e.g., EGFR mutations in NSCLC) when there are no patient specimens remaining from the original, pivotal clinical trial, and given that it would not be feasible or ethical to rerun the trial? For biomarkers that are rare in an indication (e.g., ROS1 fusions in NSCLC), what level of evidence (e.g., number of patients, need for randomized control treatment, etc.) is required given the difficulty in identifying these patients? Given the translational nature of NGS testing in cancer, the pace of new biomarker discoveries and clinical validation, and the large amount of information from an NGS test, how will FDA approval be maintained such that the approved test keeps pace with discoveries and advancements in the field? While these are not small issues and will take time to work through, the FDA and other health authorities have shown a willingness to work with the stakeholders in the field to find appropriate, balanced solutions that advance patient care.
CONCLUSION
In the 15 years since the publication of the draft human genome, sequencing technologies have advanced rapidly (Figure 1) and the cost to sequence a genome has dropped precipitously from several billion dollars down to several thousand dollars. NGS now enables analysis of a large number of DNA bases in a cost‐ and time‐effective manner. In recent years, this promising technology has been transferred to clinical diagnostics in oncology with the aim to better define which therapy should be given in each patient. In this tutorial we discussed multiple applications for NGS in clinical oncology including driver identification, detection of resistance mechanisms, quantification of mutational burden, evaluation of tumor gene expression, and diagnosis of germline mutations. The FDA recently approved the Illumina MiSeqDx platform for diagnosis of cystic fibrosis associated mutations,76 paving the way for approval of NGS‐based tests as CDx assays for oncology indications. In the near term, comprehensive testing of actionable mutations will be accomplished via NGS‐based CDx assays and used to match patients with appropriate targeted therapies. Longer term, the utility of NGS to predict responders to immunotherapy, either through RNA‐Seq based gene expression or mutational burden, could also drive significant clinical uptake of clinical NGS testing. Recent data suggest that NGS may be broadly applicable to ctDNA, which can be safely and repeatedly obtained through a “liquid biopsy” and may actually be more representative of tumor heterogeneity than a single biopsy. Pending further clinical validation, this approach could open entire new avenues, including detection of early‐stage cancers, determination of residual disease, and longitudinal monitoring for resistance mechanisms. Realizing the promise of these various approaches to change patient care will require close collaboration between drug development companies, medical oncologists, and health authorities.
Acknowledgment
The authors thank Allison Bruce for assistance with graphic design of the figures.
Author Contributions
C.A.C., E.P., L.L., F.A. and M.R.L. conceived and wrote the manuscript.
Conflict of Interest
C.A.C., E.P. and M.R.L. are employees of Genentech, Inc.
References
- 1. Fleischmann, R.D. et al Whole‐genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 269, 496–512 (1995). [DOI] [PubMed] [Google Scholar]
- 2. Sanger, F. & Coulson, A.R. , A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 94, 441–448 (1975). [DOI] [PubMed] [Google Scholar]
- 3. Lander, E.S. et al Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 4. Margulies, M. et al Genome sequencing in microfabricated high‐density picolitre reactors. Nature 437, 376–380 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shendure, J. et al Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732 (2005). [DOI] [PubMed] [Google Scholar]
- 6. Mardis, E.R. Next‐generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 6, 287–303 2013. [DOI] [PubMed] [Google Scholar]
- 7. Goodwin, S. , McPherson, J.D. & McCombie, W.R. , Coming of age: ten years of next‐generation sequencing technologies. Nat Rev Genet 17, 333–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Atlas, C.G. , N. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ding, L. et al Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sjoblom, T. et al The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 (2006). [DOI] [PubMed] [Google Scholar]
- 11. Smith, B.A. et al A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc Natl Acad Sci U S A. 112, E6544–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feng, H. , Qin, Z. & Zhang, X. , Opportunities and methods for studying alternative splicing in cancer with RNA‐Seq. Cancer Lett 340, 179–191 (2013). [DOI] [PubMed] [Google Scholar]
- 13. Chin, K. et al Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 10, 529–541 (2006). [DOI] [PubMed] [Google Scholar]
- 14. Kristensen, V.N. et al Integrated molecular profiles of invasive breast tumors and ductal carcinoma in situ (DCIS) reveal differential vascular and interleukin signaling. Proc Natl Acad Sci U S A. 109, 2802–2807 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilkerson, M.D. et al Integrated RNA and DNA sequencing improves mutation detection in low purity tumors. Nucleic Acids Res. 42, e107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu, J. et al Integrated exome and transcriptome sequencing reveals ZAK isoform usage in gastric cancer. Nat Commun. 5, 3830 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kristensen, V.N. et al Principles and methods of integrative genomic analyses in cancer. Nat Rev Cancer. 14, 299–313 (2014). [DOI] [PubMed] [Google Scholar]
- 18. Overman, M.J. et al Use of research biopsies in clinical trials: are risks and benefits adequately discussed? J Clin Oncol. 31, 17–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gerlinger, M. et al Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 366, 883–892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Juric, D. et al Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature 518, 240–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lohr, J.G. et al Whole‐exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 32, 479–484 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bettegowda, C. et al Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med. 6, 224ra24 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Diaz, L.A., Jr. & A. Bardelli, Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 32, 579–586 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kinde, I. et al Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 108, 9530–9535 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jovelet, C. et al Circulating Cell‐Free Tumor DNA Analysis of 50 Genes by Next‐Generation Sequencing in the Prospective MOSCATO Trial. Clin Cancer Res. 22, 2960–2968 (2016). [DOI] [PubMed] [Google Scholar]
- 26. Zill, O.A. , et al., Somatic genomic landscape of over 15,000 patients with advanced‐stage cancer from clinical next‐generation sequencing analysis of circulating tumor DNA. Journal of Clinical Oncology (supplement). 34 (2016) [Google Scholar]
- 27. Dawson, S.J. , Rosenfeld, N. & Caldas, C. , Circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 369, 93–94 (2013). [DOI] [PubMed] [Google Scholar]
- 28. Frenel, J.S. et al Serial Next‐Generation Sequencing of Circulating Cell‐Free DNA Evaluating Tumor Clone Response To Molecularly Targeted Drug Administration. Clin Cancer Res. 21, 4586–4596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murtaza, M. et al Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 6, 8760 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murtaza, M. et al Non‐invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497, 108–112 (2013). [DOI] [PubMed] [Google Scholar]
- 31. Newman, A.M. et al An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 20, 548–554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vogelstein, B. et al Cancer genome landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Swanton, C. et al Consensus on precision medicine for metastatic cancers: a report from the MAP conference. Ann Oncol. 27, 1443–1448 (2016). [DOI] [PubMed] [Google Scholar]
- 34. Frampton, G.M. et al Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 31, 1023–1031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Toward Better Oversight of NGS Tests. Cancer Discov, (2016). [DOI] [PubMed]
- 36. Andre, F. et al Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol. 15, 267–274 (2014). [DOI] [PubMed] [Google Scholar]
- 37. Lawrence, M.S. et al Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wagle, N. et al Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med. 371, 1426–1433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bidard, F.C. et al Response to dual HER2 blockade in a patient with HER3‐mutant metastatic breast cancer. Ann Oncol. 26, 1704–1709 (2015). [DOI] [PubMed] [Google Scholar]
- 40. Le Tourneau, C. et al Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open‐label, proof‐of‐concept, randomised, controlled phase 2 trial. Lancet Oncol. 16, 1324–1334 (2015). [DOI] [PubMed] [Google Scholar]
- 41. Ferrarotto, R. et al Lung‐MAP–framework, overview, and design principles. Chin Clin Oncol. 4, 36 (2015). [DOI] [PubMed] [Google Scholar]
- 42. Lackner, M.R. , Wilson, T.R. & Settleman, J. , Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol. 8, 999–1014 (2012). [DOI] [PubMed] [Google Scholar]
- 43. Piotrowska, Z. & Sequist, L.V. , Treatment of EGFR‐Mutant Lung Cancers After Progression in Patients Receiving First‐Line EGFR Tyrosine Kinase Inhibitors : A Review. JAMA Oncol. 2, 948–954 (2016). [DOI] [PubMed] [Google Scholar]
- 44. Chi, K.R. , The tumour trail left in blood. Nature 532, 269–271 (2016). [DOI] [PubMed] [Google Scholar]
- 45. Fehrenbacher, L. et al Atezolizumab versus docetaxel for patients with previously treated non‐small‐cell lung cancer (POPLAR): a multicentre, open‐label, phase 2 randomised controlled trial. Lancet 387, 1837–1846 (2016). [DOI] [PubMed] [Google Scholar]
- 46. Herbst, R.S. et al Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosenberg, J.E. et al Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet 387, 1909–1920 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. FDA Nods for Atezolizumab and Nivolumab . Cancer Discov, (2016). [DOI] [PubMed]
- 49. Kazandjian, D. et al FDA Approval Summary: Nivolumab for the Treatment of Metastatic Non‐Small Cell Lung Cancer With Progression On or After Platinum‐Based Chemotherapy. Oncologist 21, 634–642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sul, J. et al FDA Approval Summary: Pembrolizumab for the Treatment of Patients With Metastatic Non‐Small Cell Lung Cancer Whose Tumors Express Programmed Death‐Ligand 1. Oncologist 21, 643–650 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rizvi, N.A. et al Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Snyder, A. et al Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med. 371, 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McGranahan, N. et al Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Van Allen, E.M. et al Whole‐exome sequencing and clinical interpretation of formalin‐fixed, paraffin‐embedded tumor samples to guide precision cancer medicine. Nat Med. 20, 682–688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ledford, H. , Researchers push for personalized tumour vaccines. Nature 532, 425 (2016). [DOI] [PubMed] [Google Scholar]
- 56. Yadav, M. et al Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014). [DOI] [PubMed] [Google Scholar]
- 57. Carreno, B.M. et al Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen‐specific T cells. Science 348, 803–808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Delamarre, L. , Mellman, I. & Yadav, M. , Cancer immunotherapy. Neo approaches to cancer vaccines. Science 348, 760–761 (2015). [DOI] [PubMed] [Google Scholar]
- 59. De Mattos‐Arruda, L. et al Capturing intra‐tumor genetic heterogeneity by de novo mutation profiling of circulating cell‐free tumor DNA: a proof‐of‐principle. Ann Oncol. 25, 1729–1735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Landau, D.A. et al Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Turke, A.B. et al Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 17, 77–88 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Engelman, J.A. et al Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 14, 1351–1356 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hoeflich, K.P. et al In vivo antitumor activity of MEK and phosphatidylinositol 3‐kinase inhibitors in basal‐like breast cancer models. Clin Cancer Res. 15, 4649–4664 (2009). [DOI] [PubMed] [Google Scholar]
- 64. Ebert, P.J. et al MAP Kinase Inhibition Promotes T Cell and Anti‐tumor Activity in Combination with PD‐L1 Checkpoint Blockade. Immunity 44, 609–621 (2016). [DOI] [PubMed] [Google Scholar]
- 65. Bedard, P.L. et al A phase Ib dose‐escalation study of the oral pan‐PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 21, 730–738 (2015). [DOI] [PubMed] [Google Scholar]
- 66. Robinson, D.R. et al Activating ESR1 mutations in hormone‐resistant metastatic breast cancer. Nat Genet. 45, 1446–1451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Toy, W. et al ESR1 ligand‐binding domain mutations in hormone‐resistant breast cancer. Nat Genet. 45, 1439–1445 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schiavon, G. et al Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med. 7, 313ra182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Spoerke, J.M. et al Heterogeneity and clinical significance of ESR1 mutations in ER‐positive metastatic breast cancer patients receiving fulvestrant. Nat Commun. 7, 11579 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chabon, J.J. et al Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 7, 11815 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Garcia‐Murillas, I. et al Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 7, 302ra133 (2015). [DOI] [PubMed] [Google Scholar]
- 72. Arnedos, M. et al Precision medicine for metastatic breast cancer–limitations and solutions. Nat Rev Clin Oncol. 12, 693–704 (2015). [DOI] [PubMed] [Google Scholar]
- 73. Foekens, J.A. , Martens, J.W. & Sleijfer, S. , Are immune signatures a worthwhile tool for decision making in early‐stage human epidermal growth factor receptor 2‐positive breast cancer? J Clin Oncol. 33, 673–675 (2015). [DOI] [PubMed] [Google Scholar]
- 74. Chen, P.L. et al Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hugo, W. et al Genomic and Transcriptomic Features of Response to Anti‐PD‐1 Therapy in Metastatic Melanoma. Cell 165, 35–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Grosu, D.S. et al Clinical investigational studies for validation of a next‐generation sequencing in vitro diagnostic device for cystic fibrosis testing. Expert Rev Mol Diagn. 14, 605–622 (2014). [DOI] [PubMed] [Google Scholar]
- 77. Couch, F.J. et al Inherited mutations in 17 breast cancer susceptibility genes among a large triple‐negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 33, 304–311 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pritchard, C.C. et al Inherited DNA‐Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Matthijs, G. et al Guidelines for diagnostic next‐generation sequencing. Eur J Hum Genet. 24, 2–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sun, Y. et al Next‐generation diagnostics: gene panel, exome, or whole genome? Hum Mutat. 36, 648–655 (2015). [DOI] [PubMed] [Google Scholar]
- 81. Green, R.C. et al ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 15, 565–574 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Redig, A.J. & Janne, P.A. , Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol. 33, 975–977 (2015). [DOI] [PubMed] [Google Scholar]
- 83. Hyman, D.M. et al Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 373, 726–736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Iyer, G. et al Genome sequencing identifies a basis for everolimus sensitivity. Science 338, 221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Simon, R. , Genomic Alteration‐Driven Clinical Trial Designs in Oncology. Ann Intern Med. (2016). [DOI] [PubMed] [Google Scholar]
- 86. Pant, S. , Weiner, R. & Marton, M.J. , Navigating the rapids: the development of regulated next‐generation sequencing‐based clinical trial assays and companion diagnostics. Front Oncol. 4, 78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sapsford, K.E. et al Biomarkers to improve the benefit/risk balance for approved therapeutics: a US FDA perspective on personalized medicine. Ther Deliv. 1, 631–641 (2010). [DOI] [PubMed] [Google Scholar]
- 88. Javitt, G.H. & Carner, K.S. , Regulation of next generation sequencing. J Law Med Ethics. 42 (Suppl 1)9–21 (2014). [DOI] [PubMed] [Google Scholar]