

Abstract
Obeticholic acid (OCA), a semisynthetic bile acid, is a selective and potent farnesoid X receptor (FXR) agonist in development for the treatment of chronic nonviral liver diseases. Physiologic pharmacokinetic models have been previously used to describe the absorption, distribution, metabolism, and excretion (ADME) of bile acids. OCA plasma levels were measured in healthy volunteers and cirrhotic subjects. A physiologic pharmacokinetic model was developed to quantitatively describe the ADME of OCA in patients with and without hepatic impairment. There was good agreement between predicted and observed increases in systemic OCA exposure in subjects with mild, moderate, and severe hepatic impairment, which were 1.4‐, 8‐, and 13‐fold relative to healthy volunteers. Predicted liver exposure for subjects with mild, moderate, and severe hepatic impairment were increased only 1.1‐, 1.5‐, and 1.7‐fold. In subjects with cirrhosis, OCA exposure in the liver, the primary site of pharmacological activity along with the intestine, is increased marginally (∼2‐fold).
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓The enterohepatic circulation of endogenous bile acids has been described previously using pharmacokinetic models.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓This study assessed the effects of hepatic impairment on the pharmacokinetic properties of the semisynthetic bile acid, obeticholic acid (OCA), and the subsequent changes in tissue distribution.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
✓This is the first report of a pharmacokinetic model for OCA. This model includes physiologic compartments relevant to drug distribution and enterohepatic recirculation and used a population pharmacokinetic approach to estimate drug exposure variability. This analysis determined that moderate and severe hepatic impairment substantially increased the systemic exposure of OCA, but only marginally increased predicted concentrations of OCA in the liver, the primary site of pharmacological activity and safety.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
✓This physiologic pharmacokinetic model permits plasma OCA concentrations to act as a surrogate marker for hepatic exposure despite disproportionate changes in liver and systemic OCA distribution with hepatic impairment.
Bile acids are the natural endogenous ligands for the farnesoid X receptor (FXR), which is a nuclear receptor with high expression levels in the liver and intestine. Nuclear receptors constitute a family of ligand‐activated transcription factors that can either activate or repress target genes, including those involved in bile acid homeostasis. Obeticholic acid (OCA) is a potent and selective FXR agonist indicated for the treatment of primary biliary cholangitis (PBC), and in development of nonalcoholic steatohepatitis (NASH) and other chronic nonviral liver diseases.1, 2, 3, 4 OCA is a modification of chenodeoxycholic acid (CDCA), the endogenous agonist of FXR, and differs from CDCA by the addition of an α‐ethyl group at the 6 carbon resulting in a ∼100 greater potency compared with CDCA.5
Chronic injury to the liver can lead to the formation of regenerative nodules and fibrous bands in the liver that form after an extended process of fibrosis.6 Cirrhosis causes the interface between the sinusoids and the hepatocytes to fill with fibrotic tissue, leading to increased resistance to hepatic blood flow causing portal hypertension and associated complications.6 Previous reports have shown that patients with cirrhosis have systemic bile acid exposure that is ∼18‐fold higher than in healthy subjects, while hepatic bile acid exposure was only ∼2‐fold higher.7
There are four primary mechanisms of hepatic impairment that are important to the pharmacokinetics of endogenous bile acids and OCA: reduced hepatic uptake (caused in part by capillarization of the sinusoids), portal systemic shunting (both hepatic and extrahepatic), decreased functional liver volume, and increased taurine conjugation. Mild liver disease may be associated with little or no change in the hepatic uptake of bile acids but severe cirrhosis and/or jaundice causes large decreases in the extraction ratio that affects all bile acids similarly.8 Healthy control subjects have hepatic uptakes of bile acids of at least 70%. In contrast, values in cirrhotic patients were reduced, with some values less than 10%.8
Portosystemic shunting occurs secondary to the development of portal hypertension. Ongoing liver injury, resultant fibrogenesis, and the occurrence of nodular regeneration increases intrahepatic resistance. When coupled with increased splanchnic blood flow into the liver due to splanchnic vasodilation in cirrhotics, portal pressures become elevated and blood is shunted away from the liver through collateral veins.8, 9, 10 Johnson et al. summarized the changes in portal and hepatic arterial blood flow with respect to portosystemic shunting for healthy subjects and cirrhotics.11 Relative to normal controls, portal blood flow was 91%, 64%, and 55%, for Child‐Pugh scores A, B, and C, respectively. In response to decreased portal blood flow to the liver, hepatic arterial blood flow increased by 41%, 63%, and 92% for Child‐Pugh A, B, and C, relative to normal controls. The increase in hepatic arterial blood flow in response to decreased portal blood flow is termed hepatic arterial buffer response and allows for relatively constant blood flow to the liver regardless of portosystemic shunting.12
In assessing factors affecting drug disposition in cirrhotic subjects, Johnson et al. found that cirrhotic subjects had decreases of 89%, 71%, and 61% in functional liver size relative to healthy subjects in subjects with Child‐Pugh scores A, B, and C, respectively.11
Bile acids are synthesized in the hepatocyte from cholesterol. After synthesis is completed bile acids are conjugated (N‐acylamidated) to the amino group of glycine or taurine. In healthy adults, the ratio of glycine to taurine bile acid conjugation (“G/T ratio”) is 3:1 with a range of 1:1 to 5:1.13 It has been reported that the G/T ratio is decreased in patients with cirrhosis, possibly because bile acid synthesis decreases and there is preferential conjugation with taurine.10, 14, 15, 16, 17
Molino et al. developed a physiologic pharmacokinetic model describing the metabolism and enterohepatic circulation of CDCA.18 The CDCA model included nine spaces based on anatomical and physiological considerations (systemic circulation, portal circulation, sinusoidal circulation, liver, bile duct, gallbladder, duodenum‐jejunum, ileum, and colon), with each space possessing a compartment of either CDCA, glycine‐conjugated CDCA (glyco‐CDCA), or taurine‐conjugated CDCA (tauro‐CDCA). The model included transfer coefficients describing fluid flow, biotransformation of chemical entities, and transport across membranes. This group's work on CDCA built upon their published model for cholic acid metabolism19 and led to an additional model describing deoxycholic acid metabolism.20
The model developed by Molino et al. was used as a foundation to develop a physiologic pharmacokinetic model for OCA. It was hypothesized that due to the similar structure of CDCA, OCA would have comparable pharmacokinetic properties. The objective of this study was to develop a physiologic pharmacokinetic model in order to quantitatively describe the absorption, distribution, metabolism, and excretion (ADME) of OCA. The model was then used to define the effects of hepatic impairment to predict the systemic and liver exposure to OCA in subjects with varying degrees of hepatic impairment relative to healthy volunteers. The model was developed based on three studies conducted in healthy volunteers and two studies in subjects with varying degrees of hepatic impairment (N = 399).
METHODS
Source data
Participants in each study were ≥18 years of age and provided informed consent for willing participation. These studies were conducted in accordance with the Declaration of Helsinki (Seoul 2008 Revision), adhered to guidelines for Good Clinical Practices, and were approved by all relevant ethics committees.
Data from two phase I clinical studies were used to develop the model. First the model was developed based on healthy volunteers (Study 1, n = 160), which was then adjusted for hepatic impairment (Study 2, n = 32) (Supplementary Methods). After development, the model was validated with data sourced from two phase I clinical studies in healthy volunteers (Studies 3 and 4, n = 24 and n = 160) and one phase II clinical study (Study 5, n = 23) in cirrhotic subjects. Individual study design, dosing, and sampling information are described in the Supplementary Methods. Demographic and baseline characteristics of all studies can be found in Supplementary Table S1.
The data used in model development included subject identifiers, time of dosing and sample collection, plasma drug concentrations (OCA, glyco‐OCA, and tauro‐OCA), dose amount, disease status (e.g., Child‐Pugh score), and meal consumption information. Study participants received standardized meals at specified times during inpatient observation. Gallbladder contraction was assumed to last 90 min after the start of a meal. Drug concentrations below the limit of quantification (BLQ) were imputed to half of the lower limit of quantification (LLOQ). Other methods for handling BLQ values were attempted (including the likelihood‐based M3 method21), but provided inferior overall model performance. Observed and BLQ concentrations of OCA, glyco‐OCA, and tauro‐OCA associated with samples drawn prior to the first dose were excluded from the analyses. As the glyco‐ and tauro‐conjugates are nearly equipotent relative to OCA on FXR, total OCA concentrations were calculated as the sum of OCA, glyco‐OCA, and tauro‐OCA.
Bioanalytical
Concentrations of OCA (420.6 g/mol), glyco‐OCA (477.7 g/mol), and tauro‐OCA (527.8 g/mol) were measured from plasma samples using high‐performance liquid chromatography tandem mass spectrometry (LC‐MS/MS) (Shimadzu 10AVP HPLC System, Kyoto, Japan; AB MDX Sciex API‐4000 LC‐MS/MS System, Framingham, MA). The LLOQ for OCA, glyco‐OCA, and tauro‐OCA were 0.594 nM, 0.523 nM, and 0.474 nM, respectively.
Physiologic pharmacokinetic model development
Base model
The physiologic pharmacokinetic model previously developed for CDCA used 27 compartments consisting of nine spaces with anatomical and physiological considerations, each requiring three compartments to accommodate CDCA, glyco‐CDCA, and tauro‐CDCA. Division of each space into three compartments was necessary because while flow rates (e.g., blood flow) were independent of chemical structure, biotransformation rates (e.g., conjugation and deconjugation) and transport rates (e.g., hepatic uptake) differed based on chemical structure.18 Values for the physiologic compartment volumes and transfer rates used in the model were obtained from the literature.18 To construct the model for OCA, the physiologic pharmacokinetic model for CDCA was structurally modified to accurately reflect the interaction of human physiology and OCA.
A number of structural modifications were made to the base CDCA model in order to adapt it for use with OCA. For simplification, a single space was used to represent the gut (small and large intestines). Since OCA is an exogenous molecule, it was assumed that there was no endogenous synthesis. The physiologic pharmacokinetic model used a dosing compartment and first‐order rate constant (Ka) to represent the cumulative processes from oral intake of OCA through entry into the gut. Based on steric hindrance by the 6α‐ethyl group of OCA, no bacterial 7α‐dehydroxylation biotransformation activity (i.e., CDCA to lithocholic acid) was included in the model. Compartment volumes in the model were fixed to physiological values from the base CDCA model.18 The OCA model used the physiological flow values for blood, bile and gastrointestinal transit from the original physiologic pharmacokinetic model for CDCA,18 with the exception of flows from bile duct to gallbladder and from bile duct to gut. The physiological values from the base model led to poor predictions and may not be applicable due to the simplification of the enteral system into a unified gut compartment. The biotransformation and transport rates in the model were estimated by fitting the model to the plasma concentration–time profiles of OCA, glyco‐OCA, and tauro‐OCA. Figure 1 shows a diagram of the OCA physiologic pharmacokinetic model.
Figure 1.
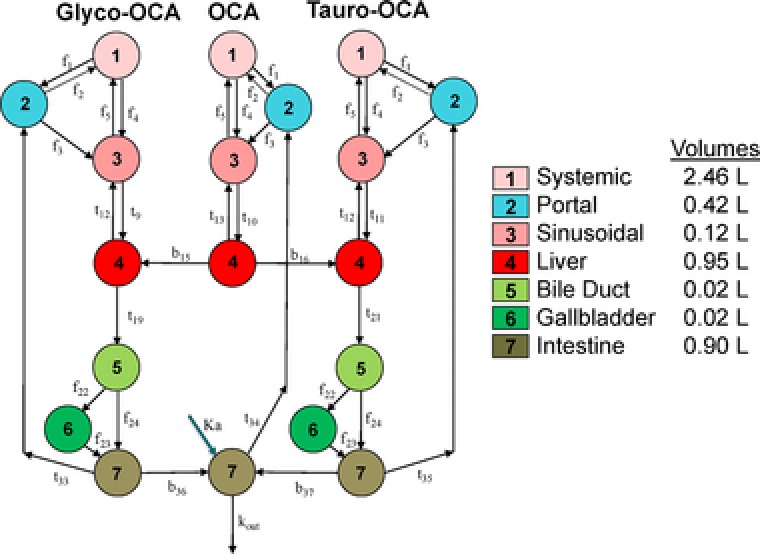
Physiologic Pharmacokinetic Model Diagram. Similar to other bile acids, OCA is conjugated to glycine and taurine. OCA and its conjugates undergo enterohepatic recirculation. Accounting for differences in hepatic impairment is done by modifying the different flow rates “f,” transport rates “t,” and biotransformation rates “b” at different points throughout the model.
The physiologic pharmacokinetic model was first developed using the OCA, glyco‐OCA, and tauro‐OCA plasma concentration–time profiles from healthy volunteers with normal hepatic function. Model parameter estimates related to healthy physiology were then held constant while the model was further developed using OCA, glyco‐OCA, and tauro‐OCA plasma concentration–time profiles from subjects with hepatic impairment (Study 2, Child‐Pugh scores A, B, and C). Only parameters specific to hepatic impairment were estimated during this process.
Hepatic impairment
Four mechanisms of hepatic impairment were incorporated into the OCA physiologic pharmacokinetic model (reduction of hepatic uptake, portal systemic shunting, decreased functional volume, and preferential conjugation to taurine). These mechanisms allowed for parameter estimates in subjects with mild, moderate, and severe hepatic impairment to progressively deviate relative to the parameter values estimated in healthy volunteers. Deviations for the portal systemic shunting and decreased functional liver volume mechanisms used physiological values from the Simcyp library for cirrhotic subjects.11 The deviations for the reduced hepatic uptake and change in OCA conjugation mechanisms were estimated based on plasma drug concentration–time profiles from subjects with hepatic impairment. Supplementary Table S2 lists the model parameters and coding modifications associated with the four mechanisms of hepatic impairment.
For the portal systemic shunting mechanism, the coefficient for portal to sinusoidal flow was progressively decreased as hepatic impairment worsened and was matched by a progressive increase in flow from the portal to systemic circulation of equal magnitude. The latter flow does not occur in healthy individuals. To compensate for reduced blood flow to the liver, the coefficient for hepatic arterial flow from the systemic circulation to the sinusoids was progressively increased with worsening hepatic impairment (i.e., hepatic arterial buffer response).
The biotransformation coefficient for conjugation of taurine to OCA was allowed to change for subjects with mild, moderate, and severe hepatic impairment relative to the coefficient in healthy volunteers, while the coefficient for conjugation to glycine was fixed at the value for healthy volunteers.
Population analysis (between‐subject and residual variability)
The OCA physiologic pharmacokinetic model used a population pharmacokinetic approach and consisted of a description of the relationships between plasma drug concentrations and time as well as components for between‐subject and residual variability. Between‐subject variability (BSV) was modeled assuming a log‐normal distribution as follows:
Where θTVn is the population typical value for the nth pharmacokinetic parameter (e.g., clearance) and ηin (ETA) is the random BSV on the nth parameter for subject i that jointly follow a multivariate normal distribution (MVN) with a mean of zero and variance of Ω. The BSV model assumes that pharmacokinetic parameters are log‐normally distributed. Due to the high level of model complexity, BSV was only incorporated on Ka and the flow rate from bile duct to gallbladder; both parameters are shown to have substantial impact on the plasma concentration–time profiles. The Ka parameter impacted the dosing peaks observed for unconjugated OCA, while the bile duct to gallbladder parameter impacted the meal‐related effects observed primarily for glyco‐OCA and tauro‐OCA.
Residual variability was assumed to have additive and proportional components:
Where and represent the j th observed and predicted plasma drug concentration for the i th subject and is the random residual variability. Each is normally distributed with a mean of zero and a variance of σ2. Distinct residual variability components were estimated for OCA, glyco‐OCA, and tauro‐OCA.
Model assessment
Model development was guided by feedback from various diagnostic plots, including: observed OCA, glyco‐OCA, and tauro‐OCA vs. population prediction (PRED) or individual prediction (IPRED) with a line of unity and trend line, conditional weighted residuals (CWRES) of OCA, glyco‐OCA, and tauro‐OCA vs. PRED or time, and visual predictive checks (VPC; 200 iterations).
A simulation with 200 replicates was performed based on subjects in Study 2 using the OCA physiologic pharmacokinetic model. Dosing and meal consumption history were used to simulate 0–216‐h concentration–time profiles of OCA, glyco‐OCA, and tauro‐OCA. Total OCA was calculated for each observation by summing the molar‐based concentrations for OCA, glyco‐OCA, and tauro‐OCA. Total OCA concentrations from systemic circulation and the liver were subjected to noncompartmental analysis to calculate Cmax and AUC(0‐216h).
Modeling software
Phoenix NLME v. 1.3 (Certara, Princeton, NJ) was used for the physiologic population pharmacokinetic analysis and simulations with Lindstrom‐Bates First‐order Conditional Estimation (FOCE‐LB). Analysis data sets, visualizations, and exploratory analyses were created using R software v. 3.1 (R Foundation, Vienna, Austria) and SAS v. 9.4 (SAS Institute, Cary, NC). GraphPad Prism v. 6.07 (Graphpad Software, La Jolla, CA) was used for the generation of some graphical analyses.
RESULTS
Healthy volunteer model development
The OCA physiologic pharmacokinetic model was initially developed using 8,248 plasma sample concentrations of OCA, glyco‐OCA, and tauro‐OCA from 160 healthy volunteers administered 10 mg OCA in a crossover design (Study 1). Each volunteer contributed pharmacokinetic samples from both study periods. The healthy volunteer pool had normal hepatic function, was primarily male (59%), mean (standard deviation, SD) age was 37.0 (9.8) years, and mean (SD) weight was 76.4 (11.8) kg (Supplementary Table S1). Volunteers were 65.6% white, 32.5% black, 0.6% Asian, and 1.3% other. The percentage of BLQ samples with imputed concentrations (i.e., ½ LLOQ) was 38.2%, 9.4%, and 24.4% for OCA, glyco‐OCA, and tauro‐OCA, respectively.
The physiologic pharmacokinetic model for healthy volunteers contained a total of 22 parameters: seven flow parameters, four biotransformation parameters, and 11 transport parameters (Supplementary Table S3). Many of these were expected to have values specific to exogenous OCA (e.g., hepatic uptake) or did not exist in the base model (e.g., Ka or Kout) and thus required estimation. Most of the flow parameters were fixed to physiological values from the literature with the exception of bile duct to gallbladder and from bile duct to gut.18 All structural parameters (Supplementary Table S3) were well estimated (≤5.5% coefficient of variation, CV). The BSV for flow from bile duct to gallbladder was 78.1% (19.3% CV) and 195% (2.21% CV) for the oral absorption of OCA. The additive portion of residual variability was ≤1 nM (15–50% CV) and the proportional portion ranged from 72% to 88% (30–70% CV) for OCA and its conjugates. The VPC results shown in Figure 2 and the goodness of fit plots shown in Supplementary Figure S1A,B indicate acceptable model performance.
Figure 2.
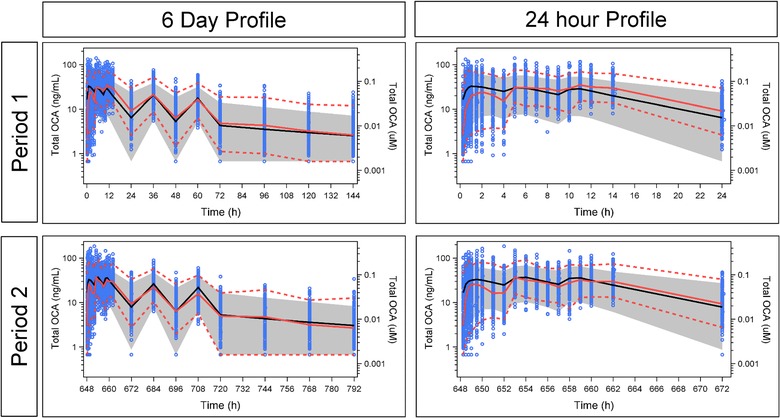
Visual Predictive Check of the Physiologic Pharmacokinetic Model in Healthy Volunteers With Normal Hepatic Function. Blue circles represent observed data points. Solid red line represents median of observed data. Dashed red lines represent the 5th and 95th percentiles of observed data. Solid black line represents predicted median. Gray band represents the 90% prediction interval.
Hepatic impairment model development
Development of the physiologic pharmacokinetic model for the physiological changes associated with hepatic impairment used 928 plasma sample concentrations of OCA, glyco‐OCA, and tauro‐OCA from 32 subjects: eight subjects each with mild, moderate, and severe hepatic impairment and eight subjects with normal hepatic function. Subjects were administered a single 10 mg dose of OCA. The study subjects were primarily male (72%), had a mean (SD) age of 55.0 (5.6) years, and mean (SD) body weight was 81.7 (16.9) kg. Participants were 90.6% white, 3.1% black, 3.1% Asian, and 3.1% other. Mean (SD) Child‐Pugh Score was 8.0 (2.0) (Child‐Pugh Score: Class A/Mild 5–6 points, Class B/Moderate 7–9 points, Class C/Severe 10–15 points) (Supplementary Table S1). The percentage of BLQ samples with imputed concentrations was 16.8%, 5.4%, and 9.6% for OCA, glyco‐OCA, and tauro‐OCA, respectively.
The 22 parameters from the physiologic pharmacokinetic model for healthy volunteers were held fixed during the development of the hepatic impairment model. Portal systemic shunting and reduced functional liver volume parameters were fixed to physiological values from the literature, while parameters associated with reduced hepatic uptake and changes in OCA conjugation were estimated.8, 10, 11, 17 Parameter estimates for hepatic uptake and taurine conjugation obtained from the physiologic pharmacokinetic model for hepatic impairment are presented in Supplementary Table S4. The structural parameters were well estimated (<25% CV) with the exception of the change in hepatic uptake in OCA and its conjugates in moderate hepatic impairment (44% CV) and the change in OCA conjugation for severe hepatic impairment (109% CV). The BSV for flow from bile duct to gallbladder was 168% (299% CV) and for the oral absorption of OCA was 246% (9.87% CV). The additive portion of residual variability was ≤1 nM (>75% CV) and the proportional portion ranged from 112% to 123% for OCA and its conjugates (>75% CV). This magnitude of variability is consistent with a bile acid analog that undergoes extensive enterohepatic recirculation.22, 23 The VPC results shown in Figure 3 and the goodness of fit plots shown in Supplementary Figure S2A,B indicate acceptable model performance.
Figure 3.
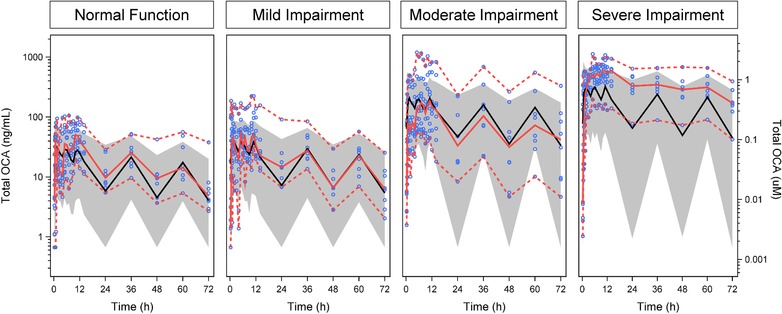
Visual Predictive Check of the Physiologic Pharmacokinetic Model in Subjects With Normal and Impaired Hepatic Function. Blue circles represent observed data points. Solid red line represents median of observed data. Dashed red lines represent the 5th and 95th percentiles of observed data. Solid black line represents predicted median. Gray band represents the 90% prediction interval.
External model validation
The final OCA physiologic pharmacokinetic model, developed for both normal and impaired hepatic function, was validated with external pharmacokinetic data in healthy volunteers (single doses of 10 mg and multiple‐dosing to steady‐state with doses of 5 mg, 10 mg, or 25 mg) (Studies 3 and 4) and subjects with hepatic impairment (cirrhosis and portal hypertension) (Study 5). Figure 4 shows VPC‐based assessments of the models ability to accurately predict the plasma total OCA concentration–time profiles under these varied conditions. The model predicted the profiles well for healthy volunteers with normal hepatic function (Figure 4 a, Supplementary Figure S3) and for subjects with moderate or severe hepatic impairment (Figure 4 b). For mild hepatic impairment, the model tended to underestimate the total OCA concentrations.
Figure 4.
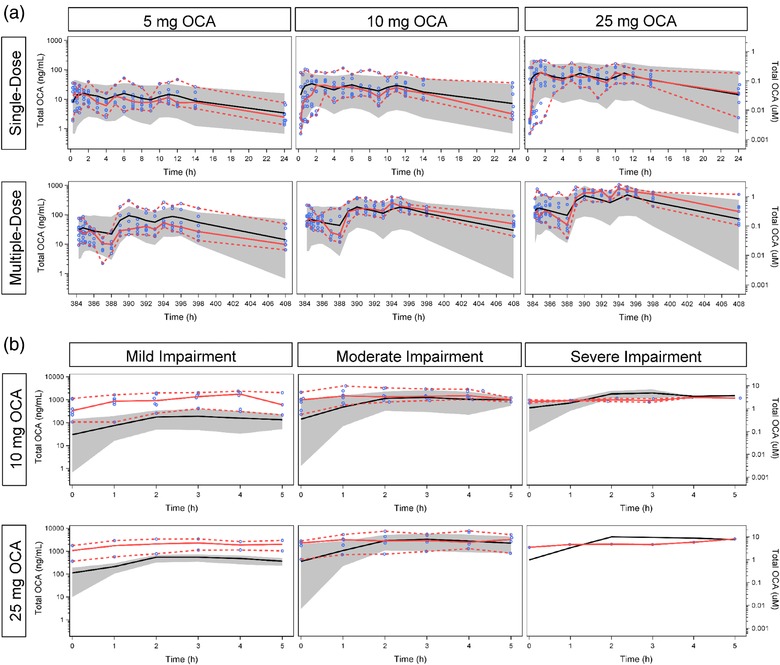
External Validation of the Physiologic Pharmacokinetic Model. (a) Healthy volunteers with normal hepatic function. (b) Subjects with impaired hepatic function. Blue circles represent observed data points. Solid red line represents median of observed data. Dashed red lines represent the 5th and 95th percentiles of observed data. Solid black line represents predicted median. Gray band represents the 90% prediction interval.
Simulation of systemic and liver OCA exposures with hepatic impairment
Simulations of total OCA in the systemic circulation and in the liver after a single 10 mg dose were compared with observed total OCA plasma concentrations from the hepatic impairment clinical study in Figure 5. The total OCA exposures are summarized as either the Cmax or the AUC over the 216‐h sampling period. For both exposure measures, and across all levels of hepatic function, there appeared to be agreement between the exposures observed in the clinical study and the exposures predicted by the physiologic pharmacokinetic model. Simulation data in Table 1 reveal that the systemic AUC of total OCA in subjects with mild, moderate, and severe hepatic impairment increased, respectively 1.4‐, 8.0‐, and 13‐fold relative to healthy volunteers with normal hepatic function. However, in the liver the predicted AUC of total OCA in subjects with mild, moderate, and severe hepatic impairment increased only respectively 1.1‐, 1.5‐, and 1.7‐fold relative to healthy volunteers with normal hepatic function.
Figure 5.
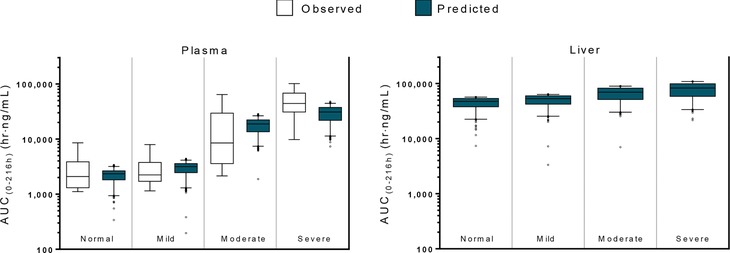
Plasma OCA Concentrations are a Poor Surrogate for Liver OCA Concentrations. Systemic exposure of OCA was predicted to be 1.4‐, 8.0‐, and 13‐fold greater in subjects with mild, moderate, and severe hepatic impairment, respectively, based on Child‐Pugh score, than in healthy volunteers, which is consistent with the observed results. Liver exposure of OCA was predicted to be 1.1‐, 1.5‐, and 1.7‐fold greater in subjects with mild, moderate, and severe hepatic impairment, respectively, than in healthy volunteers. n = 8 for observed values. Predicted values were estimated using a 200 replicate simulation from the physiologic pharmacokinetic model of OCA (n = 1,600). Boxplot whiskers represent 1st to 99th percentile.
Table 1.
Systemic and liver AUC of total OCA in patients with mild, moderate, and severe hepatic impairment
| Liver impairment | Ratio (liver impairment/normal) | |||||||
|---|---|---|---|---|---|---|---|---|
| Exposure | Metric | Normal | Mild | Moderate | Severe | Mild/Normal | Moderate/Normal | Severe/Normal |
| Systemic | AUC (ng×h/mL) | 2,339 | 3,156 | 18,785 | 30,986 | 1.35 | 8.03 | 13.2 |
| Cavg (ng/mL) | 10.8 | 14.6 | 87 | 143 | 1.35 | 8.03 | 13.2 | |
| Cmax (ng/mL) | 99.5 | 131 | 634 | 961 | 1.31 | 6.38 | 9.66 | |
| Liver | AUC (ng×h/mL) | 47,427 | 53,032 | 69,540 | 82,521 | 1.12 | 1.47 | 1.74 |
| Cavg (ng/mL) | 220 | 246 | 322 | 382 | 1.12 | 1.47 | 1.74 | |
| Cmax (ng/mL) | 2,395 | 2,665 | 2,701 | 2,651 | 1.11 | 1.13 | 1.11 | |
| Mean Simulated OCA Distribution (% Nanomoles Total OCA) | ||||||||
| Systemic Circulation | 1.03 | 10.36 | ||||||
| Portal Circulation | 0.77 | 2.33 | ||||||
| Sinusoidal Circulation | 0.05 | 0.45 | ||||||
| Liver | 7.88 | 10.44 | ||||||
| Bile Ducts | 0.14 | 0.13 | ||||||
| Gallbladder | 39.94 | 37.74 | ||||||
| Gut | 50.20 | 38.56 | ||||||
AUC, area under the concentration‐time curve; Cavg, average concentration over 24 h; Cmax, maximum concentration; OCA, obeticholic acid.
DISCUSSION
This OCA physiologic pharmacokinetic model, derived from prior bile acid models, provides valuable insights into the ADME of the drug. Orally administered OCA is absorbed from the intestines, conjugated with glycine or taurine in hepatocytes, and then circulates enterohepatically. Conjugation also results in most of the OCA conjugates being poorly absorbed in the proximal small intestine and reaching the terminal ileum, where bile acids are actively conserved. The predominant pharmacodynamic activity of OCA is thus mediated in the hepatocyte and distal small intestine by its taurine and glycine amidates.
Simulations for healthy volunteers using the physiologic pharmacokinetic model showed that at steady‐state, ∼90% of total OCA mass is distributed to the gut and the gallbladder. Approximately 8% of the total OCA mass resides in the liver, while ∼1% can be found in the plasma of the systemic circulation. In contrast, the simulated total OCA mass increases to ∼10% in the systemic circulation in subjects with Child‐Pugh C hepatic impairment with the liver distribution being approximately equal to healthy volunteers (∼10%). The simulated gut and gallbladder distribution was 79% in patients with Child‐Pugh C hepatic impairment. The low systemic levels of total OCA would in part explain the high variability observed in plasma. Simulations from the physiologic pharmacokinetic model showed that the half‐life for OCA is ∼4 d, indicating a time to steady‐state or a posttreatment washout period of about 2 weeks. The estimated OCA half‐life is consistent with the half‐life values for CDCA reported in the literature.24, 25
Simulations using the physiologic pharmacokinetic model predicted changes in systemic and liver OCA concentrations associated with changes in the severity of hepatic impairment. Model simulations predicted that for mild, moderate, and severe hepatic impairment total OCA concentrations in the plasma increase 1.4‐, 8.0‐, and 13‐fold relative to healthy volunteers while total OCA concentrations in the liver increase 1.1‐, 1.5‐, and 1.7‐fold, respectively. The predicted changes in plasma OCA concentrations were similar to the observed concentration of total endogenous bile acids in subjects with alcoholic cirrhosis (Study 5). The concentration of total endogenous bile acids measured in the plasma were increased 1.6‐, 6.4‐, and 13‐fold for mild, moderate, and severe hepatic impairment relative to normal hepatic function, respectively. Previously, soluble and tissue‐bound hepatic bile acids were measured from the livers of endstage liver disease (ESLD) patients and compared with measurements made from healthy livers (resected for tumors but functionally and histologically normal).7 It was shown that endogenous bile acid levels in the serum of patients with ESLD prior to liver transplantation were 18‐fold greater than those in healthy volunteers. However, in the livers of patients with ESLD bile acid levels were only ∼2‐fold higher relative to healthy livers. The observed twofold increase in the liver exposure of endogenous bile acids is in good agreement with the predicted increase in total OCA exposure in subjects with severe hepatic impairment (1.7‐fold; Child‐Pugh C). These results suggest that the pharmacokinetic characteristics of OCA are similar to endogenous bile acids.
The physiologic pharmacokinetic model underpredicted the exposure of total OCA in subjects with mild hepatic impairment in Study 5. Portal hypertension, which leads to portal systemic shunting and an increase in systemic exposures of OCA and bile acids, may or may not be present in subjects with mild hepatic impairment based on the Child‐Pugh score. Subjects in Study 5 had portal hypertension based on study inclusion criteria, which would explain the higher systemic exposures in subjects with mild hepatic impairment. In contrast, Study 2, which was used to develop the model, did not require portal hypertension for study participation. While the Child‐Pugh score is important for assessing the degree of cirrhosis, it is important to take into account that portal hypertension may occur at different points in disease progression based on the etiology of liver disease.26, 27
In conclusion, the OCA physiologic pharmacokinetic model predicted liver to plasma ratios for OCA concentrations that were similar to the ratios previously observed for endogenous bile acids.7 These results indicate that the liver‐to‐plasma ratio is not consistent between healthy volunteers and subjects with hepatic impairment. The liver is the primary site of action for the safety and efficacy of OCA; therefore, it is important to account for the differences in systemic and liver exposure when assessing an effective dose. The hepatically impaired subjects treated with OCA in Study 5 experienced plasma OCA exposures >10‐fold higher than those experienced by healthy volunteers, yet there was no apparent impact on the overall safety profile experienced by these subjects. The safety results from Study 5 are consistent with only a modest increase in liver exposure of OCA associated with hepatic impairment. Collectively, the results from these analyses and those from the literature for bile acids would suggest that the dose of OCA administered to hepatically impaired subjects should be modestly lower than those for subjects with normal hepatic function to achieve similar hepatic exposure.
Author Contributions
J.E.E., C.L., and A.F.H. wrote the article; J.E.E., A.F.H., and D.S. designed the research; C.L., T.P., N.H.G., and J.F.M. performed the research; C.L., T.P., N.H.G., and J.F.M. analyzed the data.
Conflicts of Interest
J.E.E., C.L., and D.S. are all employees of Intercept Pharmaceuticals, Inc.
Supporting information
Supplementary Methods: Study and Sampling Description
Supplementary Table 1. Demographic Summary for Model Development
Supplementary Table 2. Model Parameters and Coding Modification Associated with Mechanism of Hepatic Impairment
Supplementary Table 3. Model Parameters in Normal Hepatic Function
Supplementary Table 4. Modified Model Parameters in Impaired Hepatic Function
Supplementary Figure 1. (A) Goodness of Fit: Observed Concentrations vs Predicted Concentrations of OCA (Study 1). The black circles represent individual data points. The solid blue line is the line of unity. The solid red line is a loess fit of the relationship between predicted and observed. (B) Goodness of Fit: Residuals (Study 1). The black circles represent individual conditional weighted residuals. The solid blue line is a loess fit of the conditional weighted residuals. The solid red lines are loess fits of the absolute values of the conditional weighted residuals.
Supplementary Figure 2. (A) Goodness of Fit: Observed Concentrations vs Predicted Concentrations of OCA (Study 2). The black circles represent individual data points. The solid blue line is the line of unity. The solid red line is a loess fit of the relationship between predicted and observed. (B) Goodness of Fit: Residuals (Study 2). The black circles represent individual conditional weighted residuals. The solid blue line is a loess fit of the conditional weighted residuals. The solid red lines are loess fits of the absolute values of the conditional weighted residuals.
Supplementary Figure 3. External Validation of Model in Subjects with Normal Hepatic Function (Study 4). The blue circles represent observed data points. The solid red line represents median of observed data. The dashed red lines represent the 5th and 95th percentiles of observed data. The solid black line represents predicted median. The gray band represents the 90% prediction interval.
Acknowledgments
These studies and the work presented in this article were supported by Intercept Pharmaceuticals, Inc. Alexander Liberman, provided medical writing support and was an employee of Intercept Pharmaceuticals, Inc. during the authoring of this article. Study 747‐103 ClinicalTrials.gov Identifier: NCT01904539https://clinicaltrials.gov/ct2/show/NCT01904539 Study 757‐105 ClinicalTrials.gov Identifier: NCT01933503 https://clinicaltrials.gov/ct2/show/NCT01933503 Study 747‐204 EudraCT Number: 2010‐023241‐29 https://www.clinicaltrialsregister.eu/ctr-search/trial/2010-023241-29/GB
References
- 1. Hirschfield, G.M. et al Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 148, 751–761 (2015). English. [DOI] [PubMed] [Google Scholar]
- 2. Walters, J.R. , Johnston, I.M. , Nolan, J.D. , Vassie, C. , Pruzanski, M.E. & Shapiro, D.A. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment. Pharmacol. Ther. 41, 54–64 (2015). PubMed PMID: 25329562. [DOI] [PubMed] [Google Scholar]
- 3. Mudaliar, S. et al Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145, 574–582 e1 (2013). PubMed PMID: 23727264. [DOI] [PubMed] [Google Scholar]
- 4. Neuschwander‐Tetri, B.A. et al Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 385, 956–965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pellicciari, R. et al 6alpha‐ethyl‐chenodeoxycholic acid (6‐ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 45, 3569–3572 (2002). PubMed PMID: 12166927. Epub 2002/08/09. eng. [DOI] [PubMed] [Google Scholar]
- 6. Schuppan, D. , Schattenberg, J.M. Non‐alcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. J. Gastroenterol. Hepatol. 28 (Suppl 1), 68–76 (2013). PubMed PMID: 23855299. [DOI] [PubMed] [Google Scholar]
- 7. Fischer, S. , Beuers, U. , Spengler, U. , Zwiebel, F.M. , Koebe, H.G. Hepatic levels of bile acids in end‐stage chronic cholestatic liver disease. Clin. Chim. Acta 251, 173–186 (1996). PubMed PMID: 8862472. [DOI] [PubMed] [Google Scholar]
- 8. Ostrow, J. Metabolism of bile salts in cholestasis in humans In Hepatic transport and bile secretion. (eds. Tavoloni N., Berk P. 673–712 (Raven Press, New York, 1993). [Google Scholar]
- 9. Martell, M. , Coll, M. , Ezkurdia, N. , Raurell, I. , Genesca, J. Physiopathology of splanchnic vasodilation in portal hypertension. World J. Hepatol. 2, 208–220 (2010). PubMed PMID: 21160999. Pubmed Central PMCID: 2999290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ohkubo, H. , Okuda, K. , Iida, S. , Ohnishi, K. , Ikawa, S. , Makino, I. Role of portal and splenic vein shunts and impaired hepatic extraction in the elevated serum bile acids in liver cirrhosis. Gastroenterology 86, 514–520 (1984). [PubMed] [Google Scholar]
- 11. Johnson, T.N. , Boussery, K. , Rowland‐Yeo, K. , Tucker, G.T. , Rostami‐Hodjegan, A. A semi‐mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin. Pharmacokinet. 49, 189–206 (2010). PubMed PMID: 20170207. [DOI] [PubMed] [Google Scholar]
- 12. Eipel, C. , Abshagen, K. , Vollmar, B. Regulation of hepatic blood flow: the hepatic arterial buffer response revisited. World J. Gastroenterol. WJG 16, 6046–6057 (2010). PubMed PMID: 21182219. Pubmed Central PMCID: 3012579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garbutt, J.T. , Lack, L. , Tyor, M.P. Physiological basis of alterations in the relative conjugation of bile acids with glycine and taurine. Am. J. Clin. Nutr. 24, 218–228 (1971). PubMed PMID: 5545849. [DOI] [PubMed] [Google Scholar]
- 14. Harada, T. Studies on biliary bile acid metabolism in hepato‐biliary diseases. Gastroenterol. Jap. 10, 174–178 (1975). PubMed PMID: 1234096. [DOI] [PubMed] [Google Scholar]
- 15. Turnberg, L.A. , Grahame, G. Bile salt secretion in cirrhosis of the liver. Gut 11, 126–133 (1970). PubMed PMID: 5441881. Pubmed Central PMCID: 1411355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murphy, G.M. , Ross, A. , Billing, B.H. Serum bile acids in primary biliary cirrhosis. Gut 13, 201–206 (1972). PubMed PMID: 5024725. Pubmed Central PMCID: 1412131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Neale, G. , Lewis, B. , Weaver, V. , Panveliwalla, D. Serum bile acids in liver disease. Gut 12, 145–152 (1971). PubMed PMID: 5548561. Pubmed Central PMCID: 1411536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Molino, G. , Hofmann, A.F. , Cravetto, C. , Belforte, G. , Bona, B. Simulation of the metabolism and enterohepatic circulation of endogenous chenodeoxycholic acid in man using a physiological pharmacokinetic model. Eur. J. Clin. Investig. 16, 397–414 (1986). PubMed PMID: 3100308. Epub 1986/10/01. eng. [DOI] [PubMed] [Google Scholar]
- 19. Hofmann, A.F. , Molino, G. , Milanese, M. , Belforte, G. Description and simulation of a physiological pharmacokinetic model for the metabolism and enterohepatic circulation of bile acids in man. Cholic acid in Healthy Man. J. Clin. Investig. 71, 1003–1022 (1983). PubMed PMID: 6682120. Pubmed Central PMCID: 436958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hofmann, A.F. , Cravetto, C. , Molino, G. , Belforte, G. , Bona, B. Simulation of the metabolism and enterohepatic circulation of endogenous deoxycholic acid in humans using a physiologic pharmacokinetic model for bile acid metabolism. Gastroenterology 93, 693–709 (1987). PubMed PMID: 3623017. [DOI] [PubMed] [Google Scholar]
- 21. Beal, S.L. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodynam. 28, 481–504 (2001). PubMed PMID: 11768292. [DOI] [PubMed] [Google Scholar]
- 22. Steiner, C. et al Bile acid metabolites in serum: intraindividual variation and associations with coronary heart disease, metabolic syndrome and diabetes mellitus. PLoS One 6, e25006 (2011). PubMed PMID: 22110577. Pubmed Central PMCID: 3215718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bathena, S.P. et al Urinary bile acids as biomarkers for liver diseases I. Stability of the baseline profile in healthy subjects. Toxicol. Sci. 143, 296–307 (2015). PubMed PMID: 25344562. [DOI] [PubMed] [Google Scholar]
- 24. Danielsson, H. , Eneroth, P. , Hellström, K. , Lindstedt, S. , Sjövall, J. On the turnover and excretory products of cholic and chenodeoxycholic acid in man. J. Biol. Chem. 238, 2299–2304 (1963). PubMed PMID: 14024949. [PubMed] [Google Scholar]
- 25. Pedersen, L. , Arnfred, T. Kinetics and pool size of chenodeoxycholic acid in cholesterol gallstone patients. Scand. J. Gastroenterol. 10, 557–560 (1975). PubMed PMID: 1153952. [PubMed] [Google Scholar]
- 26. Kew, M.C. , Varma, R.R. , Santos, H.A.D. , Scheuer, P.J. Portal hypertension in primary biliary cirrhosis. Gut 12, 830–834 (1971). PubMed PMID: PMC1411882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duché, M. , Fabre, M. , Kretzschmar, B. , Serinet, M.‐O. , Gauthier, F. , Chardot, C. Prognostic Value of Portal Pressure at the Time of Kasai Operation in Patients With Biliary Atresia. J. Pediatr. Gastroenterol. Nutr. 43, (2006):640‐5. PubMed PMID: 00005176‐200611000‐00017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods: Study and Sampling Description
Supplementary Table 1. Demographic Summary for Model Development
Supplementary Table 2. Model Parameters and Coding Modification Associated with Mechanism of Hepatic Impairment
Supplementary Table 3. Model Parameters in Normal Hepatic Function
Supplementary Table 4. Modified Model Parameters in Impaired Hepatic Function
Supplementary Figure 1. (A) Goodness of Fit: Observed Concentrations vs Predicted Concentrations of OCA (Study 1). The black circles represent individual data points. The solid blue line is the line of unity. The solid red line is a loess fit of the relationship between predicted and observed. (B) Goodness of Fit: Residuals (Study 1). The black circles represent individual conditional weighted residuals. The solid blue line is a loess fit of the conditional weighted residuals. The solid red lines are loess fits of the absolute values of the conditional weighted residuals.
Supplementary Figure 2. (A) Goodness of Fit: Observed Concentrations vs Predicted Concentrations of OCA (Study 2). The black circles represent individual data points. The solid blue line is the line of unity. The solid red line is a loess fit of the relationship between predicted and observed. (B) Goodness of Fit: Residuals (Study 2). The black circles represent individual conditional weighted residuals. The solid blue line is a loess fit of the conditional weighted residuals. The solid red lines are loess fits of the absolute values of the conditional weighted residuals.
Supplementary Figure 3. External Validation of Model in Subjects with Normal Hepatic Function (Study 4). The blue circles represent observed data points. The solid red line represents median of observed data. The dashed red lines represent the 5th and 95th percentiles of observed data. The solid black line represents predicted median. The gray band represents the 90% prediction interval.