

Abstract
Myotonic dystrophy type 1 (DM1) is caused by an expanded trinucleotide (CTG)n tract in the 3′ untranslated region (UTR) of the dystrophia myotonica protein kinase (DMPK) gene. This results in the aggregation of an expanded mRNA forming toxic intranuclear foci which sequester splicing factors. We believe down‐regulation of DMPK mRNA represents a potential, and as yet unexplored, DM1 therapeutic avenue. Consequently, a computational screen for agents which down‐regulate DMPK mRNA was undertaken, unexpectedly identifying the sodium channel blockers mexiletine, prilocaine, procainamide, and sparteine as effective suppressors of DMPK mRNA. Analysis of DMPK mRNA in C2C12 myoblasts following treatment with these agents revealed a reduction in the mRNA levels. In vivo analysis of CD1 mice also showed DMPK mRNA and protein down‐regulation. The role of DMPK mRNA suppression in the documented efficacy of this class of compounds in DM1 is worthy of further investigation.
Keywords: muscle, molecular genetics, molecular biology, drug therapy, drugs
Introduction
Autosomal dominant myotonic dystrophy type 1 (DM1) is among the most common forms of muscular dystrophy with an estimated incidence of 1 in 8000.1 DM1 is caused by a tract of pathologically expanded trinucleotide CTG repeats in the 3′ untranslated region of the dystrophia myotonica protein kinase (DMPK) gene.2, 3 While the exact functions of DMPK remain to be elucidated, the mechanisms by which the pathogenic DMPK trinucleotide repeat interferes with normal cell function have been well characterized; it has become clear that mutated DMPK mRNA is the chief DM1 pathogenic mediator. The expanded DMPK mRNA forms a hairpin structure which accumulates in intranuclear foci leading to the sequestration and misregulation of several proteins involved in pre‐mRNA splicing. The resulting missplicing gives rise to the array of symptoms experienced and physical signs seen in DM1. These include myotonia, insulin resistance, cardiac conduction effects and muscle wasting. The greater the number of trinucleotide repeats the greater the disease severity.4, 5 Importantly, the ablation of the murine DMPK gene results only in a mild phenotype.6, 7 Given the centrality of DMPK mRNA to DM1 pathology and the apparent innocuous impact of its absence, we believe down‐regulation of this transcript represents a credible DM1 therapeutic avenue worthy of exploration.
In this regard, drugs have more than the primary effect for which they are utilized, affecting the transcription of genes and the activation or inhibition of diverse signaling pathways. The spectrum of effects that the modern pharmacopeia may have on gene expression is currently largely unknown and difficult to predict. Over the past decade, the Broad Institute compiled a connectivity map capturing the global genomic response of mammalian cell lines to drugs and drug‐like compounds. The database was brought together as a resource to identify connections between drugs of similar mechanisms, chemicals and physiological processes, and diseases and drugs.8 We identified those agents showing down‐regulation of DMPK mRNA, identifying a class effect for compounds already shown to have some therapeutic efficiency for DM1; sodium channel blockers.
Currently, the main treatment for DM1 is symptomatic including pacemaker insertion for cardiac conduction abnormalities and noninvasive ventilation for central or obstructive sleep apnea. Some small studies have suggested that mexiletine, imipramine, clomipramine, and taurine may be useful in the treatment of myotonia.9, 10 The altered splicing of the muscle specific chloride channel 1 (ClC‐1) has been shown to cause the myotonic phenotype of DM1 and is reversible in mouse models using morphilino antisense to modify splicing of ClC‐1 mRNA.11 Recently a DMPK antisense RNA approach has been shown to hold considerable potential in knocking down DMPK mRNA and improving murine DM1.12 In this study we have explored the effect of sodium channel blockers on DMPK expression that can be utilized to develop novel therapeutics for DM1.
Materials and Methods
Connectivity Map Data Mining
Data mined from the Broad Institute's Connectivity Map project served as the starting point for this project. The Broad Institute's Connectivity Map is constituted of AffymetrixGeneChip U133‐A microarray data generated with cDNA isolated from cell lines incubated with approximately 1,300 drugs individually at a concentration of 10 μM for 6 hours. The two different DMPK cDNA tags 37996_s_at, 217066_s_at on the GeneChip enabled the generation of two different lists (builds) of candidate compounds mitigating the variability inherent in microarray analyses. To compare across builds we took an average of the relative expression of both builds and ranked compounds according to their average effect. Connectivity Map drugs are tested from 1 to 100 times. In a further attempt to reduce false positive drug identification, we restricted most of our analyses to those compounds which had a minimum of four trials per build, leaving us with an aggregate minimum of eight trials. This number was chosen as it reflects the highest number of trials that would allow a reasonable number of candidate drugs to be returned. This resulted in our candidate drug list shrinking to approximately 400 compounds that we included for further study. A small number of compounds were included that did not meet the four trial cut‐off but whose suppression/induction capabilities were high. A very small number of compounds were removed after this stage due to the lack of availability. The majority of these were discontinued compounds, making them less than ideal candidate drugs.
Cell culture and drug treatment conditions
C2C12 myoblasts were cultured under standard conditions on 15 cm plates (Sigma‐Aldrich, St. Louis, MO, USA; Greiner Bio‐One, Monroe, NC, USA) and kept at 37°C in a water‐saturated environment which contained 5% CO2. Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal calf serum and 100 units/mL of penicillin–streptomycin was used as growth media. To prevent any differentiation of the myoblasts into myotubes, all growth plates were carefully monitored and were split when cells reached 70% confluence.
For trials where RNA was to be extracted for qPCR or protein for Western blotting, cells were split from 25 cm growth plates into 12‐well (BD Biosciences, San Jose, CA, USA; Falcon cell culture, Tewksbury, MA, USA) plates (BD Biosciences, Falcon cell culture). Depending on trial length (4, 16, 24 hours), cells were seeded at densities so as to never surpass 70–80% confluence. Cells were monitored before treatment and before lysis to ensure equal cell number between trials.
The mid‐throughput screens conducted on DMPK suppressors candidate drugs (purchased from Sigma‐Aldrich) were screened at 2 concentrations (1 μM and 25 μM). All drugs were diluted as per manufacturers' recommendation in either sterile H2O or dimethyl sulfoxide. All compounds were diluted so that no vehicle surpassed 0.1% in cell media.
For transcriptional inhibitor treatment, C2C12 cells were seeded in 12‐well plates and treated 24 hours later with Amanitin (2.5 μg/mL) for up to 24 hours.
Animal studies
Six‐week‐old CD1 mice were purchased from Charles River Laboratories (Boston, MA, USA). They were cared for in approval with the University of Ottawa Animal Care and Use Committee, which is compliant with the Guidelines of the Canadian Council on Animal Care and the Animals for Research Act. Animals were assessed daily by an animal care technician and were monitored before, during, and after treatment to ensure no negative effects of treatment. All treatments were given to mice through intraperitoneal injection (I.P.), once daily. To reduce stress on animals, small gauge needles (30 ½) were used. All tissues were extracted from mice within 8 hours of the last dose.
qPCR analysis
Total RNA was isolated according to the protocol provided by the manufacturer using the RNAeasy gDNA eliminator kit (Qiagen, San Jose, CA, USA). This extraction included the gDNA elution step. For qPCR, cDNA was reverse transcribed from isolated RNA employing the provided primer mix (oligodT primers/random primers) provided with the Quantitect Reverse Transcription kit (Qiagen). cDNA synthesis was conducted following manufacturer's instructions. The optional gDNA wipeout treatment was included during cDNA synthesis. The synthesized cDNA template was used for qPCR with PerfeCTa© Sybr green (Quanta Biosciences, Gaithersburg, MD, USA) and analyzed on the EppendorfMastercyclerrealplex using Eppendorf 96‐well qPCR plates (white plastic bottom). β‐2‐microglobulin was chosen as the housekeeping gene and commercially available primers were obtained from Origene (Rockville, MD, USA). Following qPCR, the delta delta Ct method of analysis was used to determine relative expression of our transcript of interest. Standard curves were generated for each primer pair to ensure our amplification and detection were occurring efficiently and within the linear range.
The following primer sequences were used to detect DMPK and β‐2‐microglobulin
DMPK (5′ → 3′)
Forward: CTGCTCGACCTTCTCCTGG
Reverse: CACGCCCGATCACCTTCAA
Amplicon length: 166
Location: Spanning exon 1 (219)‐exon 2 (384) of DMPK transcript
β‐2‐microglobulin (5′→3′)
Forward: ACAGTTCCACCCGCCTCACATT
Reverse: TAGAAAGACCAGTCCTTGCTGAAG
Product length: 105
Location: Exon 2 (195–299)
Mice were individually euthanized by exposure to CO2 and tissue (gastrocnemius and heart) were obtained, washed in sterile PBS, and frozen in liquid nitrogen. Dissection instruments were cleaned with RNAse Away (Molecular Bioproducts, Thermo Fisher Scientific Waltham, MA, USA) and RNAase free H2O water after each dissection to prevent contamination between samples. Samples were then stored at −80 °C. For RNA isolation, samples were lysed using the Qiagen Tissue Lyser II in RNAse free tubes utilizing steel balls. Total RNA was extracted using the RNeasy fibrous tissue midi kit (Qiagen), with the optional gDNA elimination step included, as per the manufactures instructions. cDNA synthesis and qPCR were performed on mouse samples in the same manner as described above.
Western blot analysis
Cells were washed twice with 1 × PBS and lysed with either 75 μL (12 well plate) or 300 μL (10 cm plate) of RIPA buffer containing 10 mg/mL each of aprotinin, phenylmethylsulfonyl fluoride (PMSF), and leupeptin for 30 min at 4°C. Following lysis, the samples were centrifuged at 13,000 × g for 20 minutes at 4°C and supernatants were collected and frozen at −20°C. Protein concentrations were determined by Bradford protein assay using a Bio‐Rad protein assay kit (Richmond, CA, USA).
Before analysis, samples were boiled for 5 minutes at 95°C and equal amount of protein extracts were separated by 11% SDS‐Page (80 V for 30 minutes, 120 V for 1 hour). Proteins were subsequently transferred (300 mA, 3 hours) to nitrocellulose membrane and the membrane was incubated in blocking solution (1× PBS, 5% milk, 0.2% Tween‐20) for 1 hour at room temperature followed by overnight incubation with the primary antibody. Anti‐DMPK polyclonal provided by Dr. Chris Storbeck, Luke Sabourin (Ottawa)13 was used at a dilution of 1:1,000. A commercially available Tubulin antibody (Abcam, Toronto, ON, Canada) was used at a concentration of 1:10,000. Membranes were washed with PBS‐T (1× PBS, 0.2% Tween‐20) three times for 10 minutes followed by incubation with secondary antibody (antirabbit [DMPK], antimouse [Tubulin], cell signaling) for 1 hour at room temperature. Antibody complexes were visualized by autoradiography using ECL Western blotting system (GE Healthcare, Mississauga, ON, Canada) and X‐Ray film (Kodak, Washington, NJ, USA). Quantification was performed by scanning the autoradiographs and signal intensities were determined by densitometry using the ImageJ software.
Mice were individually euthanized by exposure to CO2 and tissue (gastrocnemius and heart) were obtained, washed in sterile PBS, and frozen in liquid nitrogen. Samples were then stored at −80°C. Samples were prepared by lysing in either 500 μL (Heart tissue), or 300 μL (gastrocnemius tissue) of RIPA buffer containing 10 mg/mL each of aprotinin, PMSF, and leupeptin in the Qiagen Tissue Lyser II until a smooth consistency was obtained. Following lysis, the samples were centrifuged at 13,000 × g for 30 minutes at 4°C and supernatants were collected and frozen at −80°C. Protein concentrations were determined by Bradford protein assay using a Bio‐Rad protein assay kit (Richmond). Samples were analyzed in the same manner as was described above.
Statistical analysis
For statistical analysis of transcript/protein increase or decrease, the Student's t‐test (two‐tailed, two‐sample unequal variance test) was used for analysis. All graph error bars represent the standard error of the mean (SEM).
Results
Assessment of connectivity map to identify compounds that decrease DMPK mRNA: Initial screen for DMPK suppressing compounds
We initially turned to the compilation of system‐wide cell line transcriptional profiles elicited by 1,300 small molecules (approximately 75% FDA‐approved drugs) contained in the Connectivity Map.17 Our analysis of the Connectivity Map data resulted in a list of compounds that decrease DMPK mRNA in a number of cell lines. We noted that of the top 11 DMPK mRNA suppressing agents (Table 1), four were sodium channel blockers. The degree of DMPK reduction was comparatively modest (25–30%) but the class effect was such that the agents were further investigated.
Table 1.
Table summarizing the connectivity map data for DMPK suppressors
| Drug | Drug Class | DMPK mRNA level | |
|---|---|---|---|
| Connectivity | |||
| Map Data | |||
| 10 μM: 6 hours | |||
| (fold expression) | |||
| 1 | Tomelukast | Leukotriene antagonist | 0.56 |
| 2 | Sulindac sulfide | XSAID | 0∼64 |
| 3 | 8‐Azaguanine | Purine analog | 0.71 |
| 4 | Santonin | Anthelminthic | 0.72 |
| 5 | Prilocaine | Na channel blocker | 0.75 |
| 6 | Procainamide | Na channel blocker | 0.75 |
| 7 | Splitomicin | Sirtuin inhibitor | 0.76 |
| 8 | Apigenin | Flavone (yellow) | 0.80 |
| 9 | Liothyronine | Thyroid hormone | 0.80 |
| 10 | Mexiletine | Na Channel Blocker | 0.80 |
| 11 | Sparteine | Na Channel Blocker | 0.80 |
Connectivity map data indicates the original fold expression values provided by the Broad Institute used initial ranking of the compounds. Sodium channel inhibitors are indicated by bold type.
Secondary validation of sodium channel blockers as DMPK suppressors in vitro
The recapitulation of DMPK mRNA suppression observed with the Connectivity Map cell lines (HL60, PC3, MCF7) in the more disease relevant C2C12 mouse myoblast cell line was next assessed. The effect of sodium channel blockers mexiletine, prilocaine, procainamide (50 nM, 1 μM, and 25 μM) in DMPK mRNA suppression in C2C12 cells after 4 hours was next assessed. Statistically significant decrease in DMPK mRNA levels was observed with all doses of mexiletine and prilocaine, (Figure 1). No statistically significant decrease in DMPK mRNA was observed following 4 hours of treatment with procainamide (Figure 1 C).
Figure 1.
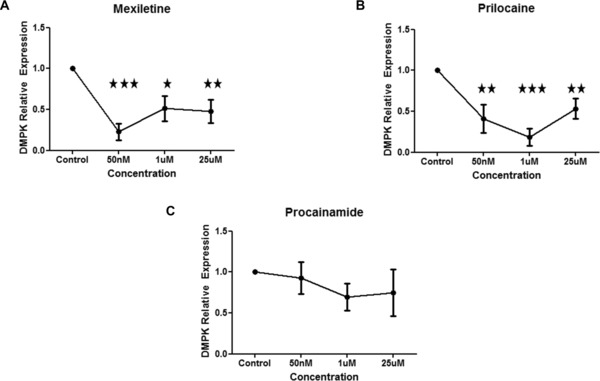
Sodium channel blockers mexiletine and prilocaine decrease DMPK mRNA in vitro. C2C12 cells were treated with range of doses of sodium channel blockers (0 [n = 6], 50 nM [n = 3], 1 μM [n = 6], and 25 μM [n = 6])} for 4 hours and then harvested for RT‐PCR. (A) Quantification of DMPK mRNA relative to β‐2‐microglobulin (the ratio of control group is set as 1) upon treatment with mexiletine. (B) Quantification of DMPK mRNA relative to β‐2‐microglobulin (the ratio of control group is set as 1) upon treatment with prilocaine. (A) Quantification of DMPK mRNA relative to β‐2‐microglobulin (the ratio of control group is set as 1) upon treatment with procainamide. Mean ± SEM (bars) of three independent experiments are shown. *p < 0.05; **p < 0.01; ***p < 0.001, t‐test.
Figure 2.
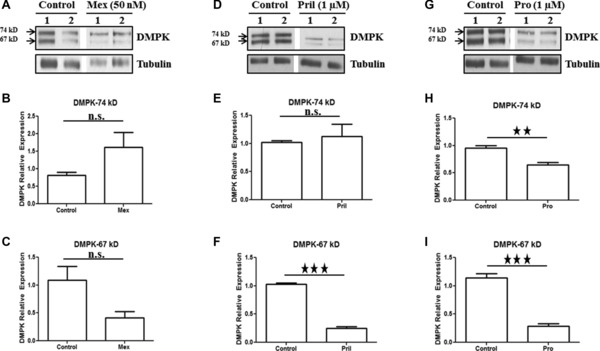
Sodium channel blockers prilocaine and procainamide decrease DMPK protein (67 KDa isoform) in vitro. C2C12 cells were treated with sodium channel blockers for 4 hours and then harvested for Western blot analysis. Representative Western blot (A) and densitometric quantification of DMPK protein (B and C) relative to tubulin (the ratio of control group is set as 1) upon treatment with mexiletine (50 nM). Representative Western blot (D) and densitometric quantification of DMPK protein (E and F) relative to tubulin (the ratio of control group is set as 1) upon treatment with prilocaine (1 μM). Representative Western blot (G) and densitometric quantification of DMPK protein (H and I) relative to tubulin (the ratio of control group is set as 1) upon treatment with procainamide (1 μM). Mean ± SEM (bars) of three independent experiments are shown. All lanes were run on the same gel but were noncontiguous. *p < 0.05; **p < 0.01; ***p < 0.001, t‐test.
We also evaluated the impact of sodium channel blockers on DMPK protein levels. Although we are aware it is the mRNA which is the pathogenic agent in DM1, we utilized protein levels as a biomarker which might help validate any observed DMPK mRNA decrease. No statistically significant decrease in the 67 and 74 kDa DMPK isoforms was observed in response to mexiletine. Prilocaine treatment significantly reduced the 67 kDa DMPK protein isoform but had no effect on 74 kDa isoform. Procainamide treatment significantly reduced both 67 and 74 kDa DMPK protein isoforms.
In vivo confirmation of DMPK suppression
To establish whether sodium channel blockers recapitulate their effect of DMPK suppression in vivo, wild type CD‐1 mice were treated with the sodium channel blockers. Dose selection was guided both by typical human clinical dosing, as well as attempting to approximate murine serum levels with the optimal DMPK suppressing concentrations observed in cell culture. CD‐1 mice were therefore treated with daily IP injections of all three sodium channel blockers over a range of doses (6.25 mg/kg, 12.5 mg/kg, 25 mg/kg, or 50 mg/kg of mexiletine; 1.25 mg/kg, 2.5 mg/kg, and 5 mg/kg of prilocaine and 6.25 mg/kg, 12.5 mg/kg, or 25 mg/kg of procainamide). No statistically significant DMPK mRNA decrease was observed in gastrocnemius or heart samples following a 5‐day treatment with mexiletine (Figures 3 A and S1A), although a nonsignificant reduction in DMPK mRNA levels in response to 25 mg/kg dose of mexiletine was noted in gastrocnemius (Figure 3 A). Similarly, a statistically significant decrease in DMPK RNA levels in response to prilocaine was observed in gastrocnemius tissue but not in heart (Figures 3 B and S1B). Although procainamide was not effective at decreasing DMPK in cell culture, it was effective in decreasing DMPK mRNA levels in gastrocnemius muscle in CD‐1 mice (Figure 3 C).
Figure 3.
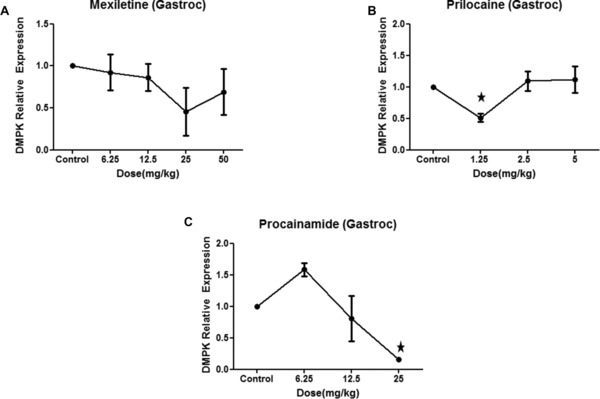
Sodium channel blockers mexiletine and prilocaine decrease DMPK mRNA in vivo. CD‐1 wild type mice were treated with daily IP injections of all three sodium channel blockers over a range of doses (6.25 mg/kg, 12.5 mg/kg, 25 mg/kg, or 50 mg/kg of mexiletine; 1.25 mg/kg, 2.5 mg/kg, and 5 mg/kg of prilocaine and 6.25 mg/kg, 12.5 mg/kg, or 25 mg/kg of procainamide) for 5 days. Control mice were treated with equal volume of vehicle. Gastrocnemius muscle was harvested for RT‐PCR. (A) Quantification of DMPK mRNA relative to β‐2‐microglobulin (the ratio of control group is set as 1) upon treatment with mexiletine. (B) Quantification of DMPK mRNA relative to β‐2‐microglobulin (the ratio of control group is set as 1) upon treatment with prilocaine. (C) Quantification of DMPK mRNA relative to β‐2‐microglobulin (the ratio of control group is set as 1) upon treatment with procainamide. Three mice per treatment group (control and treatment) were used. Mean ± SEM (bars) is shown. *p < 0.05, t‐test.
We next looked at DMPK protein in gastrocnemius in response to mexiletine (25 mg/kg), prilocaine (1.25 mg/kg), and procainamide (25 mg/kg) treatment in CD‐1 mice. Treatment with prilocaine conferred a significant reduction of DMPK protein in gastrocnemius (Figures 4 D‐F). A statistically nonsignificant decrease in 74 kDa and 67 kDa DMPK protein was observed in gastrocnemius in response to mexiletine and procainamide (Figures 4 A‐C and G–I)
Figure 4.
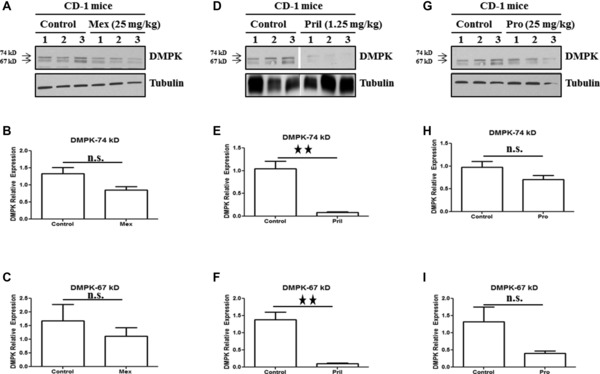
Sodium channel blocker prilocaine decrease DMPK protein in vivo. CD‐1 wild‐type mice were treated with daily IP injections of all three sodium channel blockers for 5 days. Control mice were treated with equal volume of vehicle. Gastrocnemius muscle was harvested for Western blot analysis. Representative Western blot (A) and densitometric quantification of DMPK protein (B and C) relative to tubulin upon treatment with mexiletine (25 mg/kg). Representative Western blot (D) and densitometric quantification of DMPK protein (E and F) relative to tubulin upon treatment with prilocaine (1.25 mg/kg). Representative Western blot (G) and densitometric quantification of DMPK protein (H and I) relative to tubulin upon treatment with procainamide (25 mg/kg). Three mice per treatment group (control and treatment) were used. Mean ± SEM (bars) is shown. All lanes were run on the same gel but were noncontiguous. *p < 0.05; **p < 0.01; t‐test.
Transcriptional inhibition of DMPK mRNA by prilocaine
Prilocaine was the most effective of our candidate compounds in decreasing DMPK mRNA both in vitro and in vivo. To determine whether the decrease in steady‐state levels of DMPK mRNA and protein observed following prilocaine treatment is conferred through mRNA destabilization or by transcriptional inhibition, C2C12 cells were treated with prilocaine in the presence or absence of RNA pol II (and thus transcriptional) inhibitor amanitin. Total RNA was harvested at different time points (0, 4, 8, 12, and 24 hours) for RT–PCR amplification. Prilocaine‐mediated DMPK mRNA reduction was unaffected by the presence of transcriptional inhibitor amanitin suggesting that prilocaine treatment does not destabilize DMPK mRNA but rather blocks its transcription (Figures 5 A‐C). In contrast, the level of TNFα mRNA known to be inherently unstable used as a positive control for transcriptional inhibition, decreased more rapidly upon treatment with amanitin (Figure 5 D).
Figure 5.
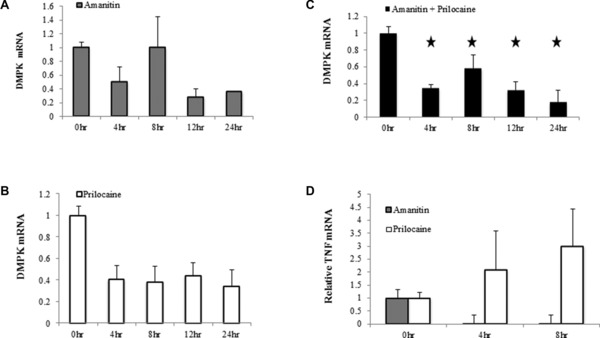
Transcriptional inhibition of DMPK mRNA in vitro by prilocaine. C2C12 cells were treated with prilocaine (1 μM) in the presence or absence of RNA pol II (and thus transcriptional) inhibitor amanitin (2.5 μg/mL). Total RNA was harvested at different time points (0, 4, 8, 12, and 24 hours) for RT–PCR. (A) Quantification of DMPK mRNA relative to β‐2‐microglobulin (the ratio of control group is set as 1) upon treatment with amanitin alone. (B) Quantification of DMPK mRNA relative to β‐2‐microglobulin (the ratio of control group is set as 1) upon treatment with prilocaine alone. (C) Quantification of DMPK mRNA relative to β‐2‐microglobulin (the ratio of control group is set as 1) upon treatment with amanitin + prilocaine. (D) Quantification of TNF mRNA relative to β‐2‐microglobulin (the ratio of control group is set as 1) upon treatment with amanitin and prilocaine. Mean ± SEM (bars) of three independent experiments are shown. *p < 0.05, t‐test.
Discussion
Given that small molecules can affect a substantial proportion of the human transcriptome and the cost and time required for new therapeutic compounds, the prospect of finding new uses for approved drugs by studying drug gene interaction is appealing. In the case of DM1, this might occur by down‐regulation of the pathogenic DMPK mRNA especially given that the ablation of the DMPK gene leads to either no or minimal pathology in the mouse. We therefore elected to screen for compounds which can down‐regulate DMPK mRNA levels.
Lamb et al. have used DNA microarray technology to measure system‐wide mRNA profiles in mammalian cell lines (MCF7, HL60, and PC3) in response to drugs. This high‐throughput drug‐based screening interrogation of the transcriptome8 was used to identify the p38 kinase pathway in activating the spinal muscular atrophy modulating SMN2 gene.14, 15, 16 The purpose of the current study was to determine whether databases such as the Connectivity Map could yield similarly successful results for DM1. Interestingly a number of the sodium channel blockers we identified in this screen have already been used as DM1 treatments. Procainamide was identified as an effective antimyotonia treatment in 1955.17 While the sodium channel blocking properties of the compound may account for the amelioration of myotonia, procainamide appears to treat other symptoms of DM1 as well, dyspnea (shortness of breath), for example.18 Mexiletine has also recently been recognized as an effective treatment for myotonia in DM1 patients.9 In keeping with the broader effects seen with procainamide, the impact of mexiletine on a number of DM1 signs and symptoms beyond myotonia has also been observed, triggering an effort to have the drug relabeled as a broader therapeutic for myotonic dystrophy.
Prilocaine was the most consistent DMPK suppressor in our study. Although the effect of procainamide and mexiletine on DM1 has been the subject of published reports, no analogous studies have been conducted utilizing prilocaine. Although the impact on DMPK mRNA of prilocaine in combination with the transcriptional inhibitor amanitin did not differ from amanitin alone, suggesting that prilocaine inhibits DMPK transcription, the mechanism by which this occurs is not known. The optimal in vitro DMPK mRNA suppressing concentrations approximate serum levels reported in patients receiving prilocaine infusions (approximately 390 nM)19 but are much lower than that required for complete block of neuronal sodium channels (approximately 17 μM)20 suggesting that the suppression of DMPK mRNA is occurring by a mechanism other than that of the sodium channel blockade.
Interestingly DMPK itself appears to modulate sodium channel expression; DMPK knockout mice have a reduced concentration of sodium channels present in skeletal muscle.21 Furthermore, a direct interaction between the DMPK protein and sodium channels has been shown to occur whereby the kinase phosphorylates sodium channels, thereby decreasing their activity.22, 23 One might therefore envision that with the reduction in sodium channel activity, an attempt is made by the cell to offset this by blocking the inhibitory DMPK phosphorylation of sodium channel by rapidly reducing DMPK mRNA with the attendant drop in DMPK protein that we have shown; a reduction in channel inhibition results from a reduction in channel activity. Clearly the precise mechanism by which this may occur remains to be elucidated, although it is noteworthy that a link between sodium channel blockade and modulation of sodium channel alpha subunit mRNA has been previously observed.24
The tissue specific response we observed in our treatment with prilocaine, mexiletine, and procainamide may represent another clue to the relationship between DMPK levels and sodium channel activity. Prilocaine worked well as DMPK suppressor in skeletal muscle tissue (gastrocnemius) but failed to have any effect on DMPK levels in heart tissue. It has been observed that although DMPK appears to modulate skeletal muscle expression of sodium channels (Nav 1.4), it has no effect on the sodium channel isoform (Nav 1.5) present in heart tissue.22 There may exist an element of reciprocity; only in tissues in which DMPK modulates sodium channel activity will blocking the sodium channel have an effect on DMPK mRNA and protein levels.
We have shown here a consistent decrease in DMPK mRNA levels in response to our candidate compound prilocaine at both in vitro and in vivo. The next phase of research shall be to characterize these compounds effects on the pathogenic DMPK RNA. The documentation of a decrease in pathogenic DMPK mRNA would represent a novel therapeutic target for this drug class, it may be that a dosing scheme different than that used to block channels elicits a more robust DMPK down‐regulation. Whether it be sodium channel blockers and/or another drug class that is shown to be effective, the next step shall be exploration in an adult DM1 population. We hope that our success in compound identification may lead to potential therapeutics for this disease and others, as well as highlighting the usefulness of in silico screens as a tool for drug repurposing.
Conflict of Interest
The authors declare that they have no conflict of interest.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Figure S1: Sodium Channel blockers treatment have no effect on heart DMPK mRNA in vivo.
Acknowledgments
We want to thank Dr. Chris Storbeck and Dr. Luke Sabourin for providing anti‐DMPK polyclonal antibody. We would also like to thank Fahad Shamim and the Animal Care and Veterinary Service staff at the University of Ottawa for all their help. This work was supported by operating grants from Tori's Buddies, CML Healthcare, FightSMA, the SMA Foundation, the Canadian Gene Cure Foundation, Physicians Services Incorporated, Ilsa Mae SMA Research Fund and the Canadian Institutes of Health Research (to A. MacKenzie) and the Canadian Gene Cure Foundation and CIHR Institute of Genetics (to F. Farooq).
References
- 1. Vignaud A, Ferry A, Huguet A, Baraibar M, Trollet C, Hyzewicz J, Butler‐Browne G, Puymirat J, Gourdon G, Furling D. Progressive skeletal muscle weakness in transgenic mice expressing CTG expansions is associated with the activation of the ubiquitin‐proteasome pathway. Neuromuscul Disord. 2010; 20(5): 319–325. [DOI] [PubMed] [Google Scholar]
- 2. Brouwer JR, Willemsen R, Oostra BA. Microsatellite repeat instability and neurological disease. Bioessays. 2009; 31(1): 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans. 2009; 37(Pt 6): 1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell. 1992; 68(4): 799–808. [DOI] [PubMed] [Google Scholar]
- 5. Hunter A, Tsilfidis C, Mettler G, Jacob P, Mahadevan M, Surh L, Korneluk R. The correlation of age of onset with CTG trinucleotide repeat amplification in myotonic dystrophy. J Med Genet. 1992; 29(11): 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jansen G, Groenen PJ, Bachner D, Jap PH, Coerwinkel M, Oerlemans F, van den Broek W, Gohlsch B, Pette D, Plomp JJ, et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet. 1996; 13(3): 316–324. [DOI] [PubMed] [Google Scholar]
- 7. Reddy S, Smith DB, Rich MM, Leferovich JM, Reilly P, Davis BM, Tran K, Rayburn H, Bronson R, Cros D, et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet. 1996; 13(3): 325–335. [DOI] [PubMed] [Google Scholar]
- 8. Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The Connectivity Map: using gene‐expression signatures to connect small molecules, genes, and disease. Science. 2006; 313(5795): 1929–1935. [DOI] [PubMed] [Google Scholar]
- 9. Logigian EL, Martens WB, Moxley RTt, McDermott MP, Dilek N, Wiegner AW, Pearson AT, Barbieri CA, Annis CL, Thornton CA, et al. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology. 2010; 74(18): 1441–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turner C, Hilton‐Jones D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry. 2010; 81(4): 358–367. [DOI] [PubMed] [Google Scholar]
- 11. Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA. Correction of ClC‐1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007; 117(12): 3952–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, Thornton CA. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012; 488(7409): 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Whiting EJ, Waring JD, Tamai K, Somerville MJ, Hincke M, Staines WA, Ikeda JE, Korneluk RG. Characterization of myotonic dystrophy kinase (DMK) protein in human and rodent muscle and central nervous tissue. Hum Mol Genet. 1995; 4(6): 1063–7102. [DOI] [PubMed] [Google Scholar]
- 14. Farooq F, Abadia‐Molina F, MacKenzie D, Hadwen J, Shamim F, O'Reilly S, Holcik M, MacKenzie A. Celecoxib increases SMN and survival in a severe spinal muscular atrophy mouse model via p38 pathway activation. Hum Mol Genet. 2013; 22(17): 3415–3424. [DOI] [PubMed] [Google Scholar]
- 15. Farooq F, Balabanian S, Liu X, Holcik M, MacKenzie A. p38 mitogen‐activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum Mol Genet. 2009; 18(21): 4035–4045. [DOI] [PubMed] [Google Scholar]
- 16. Hadwen J, MacKenzie D, Shamim F, Mongeon K, Holcik M, MacKenzie A, Farooq F. VPAC2 receptor agonist BAY 55–9837 increases SMN protein levels and moderates disease phenotype in severe spinal muscular atrophy mouse models. Orphanet J Rare Dis. 2014. 9(1): 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geschwind N, Simpson JA. Procaine amide in the treatment of myotonia. Brain. 1955; 78(1): 81–91. [DOI] [PubMed] [Google Scholar]
- 18. Fitting JW, Leuenberger P. Procainamide for dyspnea in myotonic dystrophy. Am Rev Respir Dis. 1989; 140(5): 1442–1445. [DOI] [PubMed] [Google Scholar]
- 19. Arthur GR, Scott DH, Boyes RN, Scott DB. Pharmacokinetic and clinical pharmacological studies with mepivacaine and prilocaine. Br J Anaesth. 1979; 51(6): 481–485. [DOI] [PubMed] [Google Scholar]
- 20. Simon MA, Gielen MJ, Alberink N, Vree TB, van Egmond J. Intravenous regional anesthesia with 0.5% articaine, 0.5% lidocaine, or 0.5% prilocaine. A double‐blind randomized clinical study. Reg Anesth. 1997; 22(1): 29–34. [DOI] [PubMed] [Google Scholar]
- 21. Mounsey JP, Mistry DJ, Ai CW, Reddy S, Moorman JR. Skeletal muscle sodium channel gating in mice deficient in myotonic dystrophy protein kinase. Hum Mol Genet. 2000; 9(15): 2313–2320. [DOI] [PubMed] [Google Scholar]
- 22. Chahine M, Jr George AL, . Myotonic dystrophy kinase modulates skeletal muscle but not cardiac voltage‐gated sodium channels. FEBS Lett. 1997; 412(3): 621–624. [DOI] [PubMed] [Google Scholar]
- 23. Mounsey JP, Xu P, John JE 3rd, Horne LT, Gilbert J, Roses AD, Moorman JR. Modulation of skeletal muscle sodium channels by human myotonin protein kinase. J Clin Invest. 1995; 95(5): 2379–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duff HJ, Offord J, West J, Catterall WA. Class I and IV antiarrhythmic drugs and cytosolic calcium regulate mRNA encoding the sodium channel alpha subunit in rat cardiac muscle. Mol Pharmacol. 1992; 42(4): 570–574. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Figure S1: Sodium Channel blockers treatment have no effect on heart DMPK mRNA in vivo.