

Abstract
The selective alkylation of a single meso-N atom of a corrolazine macrocycle is reported. Alkylation has a dramatic impact on the physicochemical properties of ReV(O)(TBP8Cz). New electron-transfer and hydrogen-atom-transfer reactivity is also seen for this complex, including one-electron reduction, which gives an air-stable 19π-electron aromatic radical complex.
Graphical Abstract
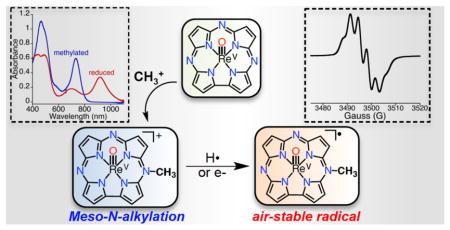
Meso-N-methylation of a corrolazine macrocycle results in changes in both the physicochemical properties and in the reactivity of a ReV(O) corrolazine complex.
The development of novel porphyrinoid compounds is essential for applications in a range of fields that rely on porphyrinoid structures, including bioinorganic models of heme proteins,1 electrochemical energy storage systems,2 new dye-sensitized solar cells (DSSCs),3a,b and molecules for photodynamic therapy (PDT).3c The synthesis of ring-contracted porphyrins, and in particular, ring-contracted tetraazaporphyrins, or corrolazines (Cz),1a,b, 4 has been a focus of our research group. The corrolazine ligands are able to support a range of metal ions in various oxidation states, including high-valent metal-oxo and metal-imido species.5 However, synthetic modification of the corrolazine ring has thus far been limited to the peripheral substituents on the β-carbon positions.4
Porphyrins and corroles can be modified at the meso-carbon atoms, leading to a large array of porphyrinoid compounds with varying structural, electronic, and reactivity properties.6 The meso-N atoms in Cz, and in the related porphyrazines and phthalocyanines, appear to preclude facile alkyl-functionalization at the meso position.7 We envisioned that alkylation of the meso-N positions might be feasible given that protonation of these positions was occurring in the former complexes.8 If a generalized alkylation procedure could be devised, it would open a pathway to meso-N substituted porphyrinoid compounds with a range of new steric and electronic properties.9 Such a synthetic strategy would allow access to meso substitution in corrolazines and porphyrazines similar to what has been seen in porphyrin chemistry.
Herein we describe the synthesis of a cationic rhenium corrolazine [ReV(O)(N-MeTBP8Cz)]+[OTf]− (1), which has been alkylated at one of the meso-N atoms of the Cz ring. To our knowledge, this synthesis is the first report of direct, selective alkylation at a meso-N position in a porphyrinoid compound.9 The methylated meso-N site was confirmed by single crystal X-ray diffraction (XRD). The selective alkylation of the starting ReV(O)(TBP8Cz) complex has a profound effect on the spectroscopic features, redox potentials and reactivity of this metallocorrolazine complex. The unalkylated ReV(O) complex is unreactive toward electron- and hydrogen-atom donors, whereas alkylated 1 undergoes one-electron reduction to give an air-stable 19π-electron radical complex. The stabilization of organic radicals with extended porphyrinoid π systems has been the focus of much effort,9b, 10 and reduced 1 is a rare example of such a species. A PCET mechanism is indicated from kinetic and thermodynamic analyses of the reaction of 1 with H-atom donors.
The synthesis of 1 was accomplished by refluxing ReV(O)(TBP8Cz) with excess MeOTf in toluene (Scheme 1). The solution slowly changes from dark-green to brown upon the formation of complex 1. Purified complex 1 can be isolated as a dark brown solid in 38% yield. The meso-N methylation causes a red-shift and a decrease in the extinction coefficient of the Soret band (464 nm), and a red-shifting of the Q-band (732 nm) by 62 nm compared to ReV(O)(TBP8Cz) (Fig. 1a). The changes in the absorbance spectrum are similar to the changes seen for meso-N protonation of ReV(O)(TBP8Cz), as well as Mn corrolazines.8 The 1H NMR spectrum for 1 shows that the two-fold symmetry of the starting ReV(O) complex has been disrupted (Fig. 2). The NMR spectrum indicates that methylation occurs on one of the meso-N atoms that does not lie on the mirror plane bisecting the pyrrole-pyrrole linkage. The singlet at 5.05 ppm arises from the new meso-CH3 group, which is shifted downfield by the aromatic ring current. Similar chemical shifts are seen for porphyrins methylated at the meso-carbon position.11 Additional support of the assignment of 1 as the monoalkylated product is afforded by LDI-MS, where the parent ion was found at 1572.53 m/z (M+) (calc’d 1572.81 m/z).
Scheme 1.

Synthesis of [ReV(O)(N-MeTBP8Cz)]+[OTf]− (1).
Fig. 1.

UV-vis spectra of (a) neutral ReV(O)(TBP8Cz) (green), meso-N-protonated [ReV(O)(TBP8Cz)(H)]+ (red), and meso-N-methylated 1 (blue) in CH2Cl2. (b) UV-vis spectral changes upon addition of excess HBArF to 1 (blue) in CH2Cl2 to form dicationic 2 (red).
Fig. 2.

Comparison of 1H NMR spectra showing the (a) aromatic and (b) t-Bu region of 1 (green) and ReV(O)(TBP8Cz) (violet) in CD2Cl2 at 25 °C.
Crystallization of complex 1 was not successful, but addition of the proton donor [H(OEt2)2]+[B(C6F5)4]− (HBArF) to 1 in CH2Cl2 led to red shifts in both the Soret and Q-bands (Fig. 1b) indicative of meso-N protonation, and resulted in crystallization of the dicationic derivative [ReV(O)(N-MeTBP8Cz)(H)]2+[BArF]−2 (2). The crystal structure is shown in Fig. 3, and confirms that methylation has occurred at a single meso-N atom. The proton on the opposing meso-N position was successfully located from difference Fourier maps (Fig. S9, ESI†), and two BArF− counterions were found in the crystal lattice, confirming the charge balance. The structure exhibits a short Re–O bond of 1.6643(17) Å, and an out-of-plane (Npyrrole) distance for Re of 0.744 Å, both of which are similar to the neutral ReV(O) complex. The phenyl groups adjacent to the N–CH3 substituent are canted to accommodate the methyl group (Cα–Cβ–Cipso–Cortho) (dihedral angle: 64.2°, 65.7°), as compared to the adjacent phenyl groups (dihedral angle: 44.0°, 29.2°). The angles around the N–CH3 group (C(1)–N(7)–C(97) = 117.9(2)°; C(16)–N(7)–C(97) = 117.7(2)°; C(1)–N7–C16 = 124.3(2)°) are indicative of sp2 hybridization, with the CH3 group lying slightly above the mean plane of the 23-atom macrocyclic core (CH3-plane = 0.43 Å). The 1H NMR spectrum of 2 (Fig. S5, ESI†), generated in situ by addition of HBArF to 1 in CD2Cl2, displays a broad resonance at 12.8 ppm consistent with meso-N protonation. Unfortunately, the dicationic species 2 is extremely moisture-sensitive and converts back slowly to 1 even in anhydrous CH2Cl2, making further reactivity studies difficult.
Fig. 3.

Displacement ellipsoid plots (40% probability level) at 110(2) K of (a) the dication of 2 and (b) side view with peripheral aryl groups omitted. All H-atoms except for the meso-H and disorder were removed for clarity.
The influence of remote meso-N-methylation on the Re–O bond was probed by ATR-IR spectroscopy. The IR spectra for 1 with natural abundance 16O (1-16O) and isotopically enriched 18O (1-18O) (>99%, incorporated from H218O)8c in the terminal oxo position is shown in Fig. S3 (ESI†). Comparison of the spectrum for 1-16O with ReV(O)(TBP8Cz) does not reveal any obvious new peaks. However, the IR spectrum for 1-18O reveals a new peak at 953 cm−1 in comparison with 1-16O, which is consistent with a Re–O stretch (Fig. S3, ESI†). A value of ν(Re–16O) = 1011 cm−1 was calculated by using a simple diatomic oscillator model, which falls under an intense corrolazine vibrational mode centered at 1001 cm−1, also seen in other corrolazine compounds.12 The Re–O stretch in 1-18O is shifted to higher energy by 8 cm−1 as compared to ν(Re–18O) = 945 cm−1 for unmethylated ReV(18O)(TBP8Cz).8c This shift is quite similar to the 11 cm−1 upshift observed for ν(Re–O) upon meso-N protonation of ReV(O)(TBP8Cz).8c Thus both remote meso-N methylation and protonation appear to cause a slight strengthening of the Re–O bond in these complexes (an influence from a change in symmetry on ν(Re–O) cannot be ruled out). To support these assignments, frequency calculations were performed via density functional theory (DFT) computations. Geometry-optimized structures for truncated models of 1 and ReV(O)(TBP8Cz) were obtained at the PBE0/LANL2TZ/6-31G** level of theory, (Fig. S15, ESI†). These structures led to calculated values of ν(Re–O) = 1054 cm−1 for the parent compound and 1063 cm−1 for 1. Although these vibrational frequencies are larger than the experimental data,13 the difference between the calculated ν(Re–O) values for 1 and the parent complex is Δν(Re–O)) = 9 cm−1, in excellent agreement with experiment.
Further insights regarding the electronic differences between 1 and ReV(O)(TBP8Cz) were obtained through cyclic voltammetric measurements (Fig. S4, ESI†). Complex 1 exhibits two reversible waves at −561 mV and −906 mV versus Fc+/Fc in CH2Cl2. This electrochemical profile is different from ReV(O)(TBP8Cz), which shows only one reversible wave at +565 mV, assigned to a Cz ring oxidation.8c The addition of the methyl group makes Cz ring oxidation inaccessible over the solvent window, and the redox couples at −561 mV and −906 mV for 1 can be assigned to consecutive Cz ring reductions. We have shown previously that monoprotonation of ReV(O)(TBP8Cz) to form [ReV(O)(TBP8Cz)(H)]+ led to a similar disappearance of the ring oxidation wave at +565 mV and the appearance of an irreversible Cz ring reduction wave at −645 mV. Attempted reaction of this complex with H-atom, O-atom and electron transfer substrates led either to no reaction or deprotonation. These results contrast those for 1, where a reversible ring reduction is observed. The electrochemical data show that complex 1 is significantly more difficult to oxidize than ReV(O)(TBP8Cz), but should be susceptible to reduction by an appropriate one-electron reducing agent.
Addition of the one-electron reducing agent Cr(C6H6)2 to 1 in CH2Cl2 led to the changes in the UV-vis spectrum seen in Fig. 4a. Isosbestic conversion is observed, giving a new species with λmax = 440, 489 nm, 920 nm. The decrease in the extinction coefficient of the Soret band and the appearance of a long-wavelength band at 920 nm is consistent with a radical delocalized on the corrolazine π-system. The 920 nm band is significantly more red-shifted than the analogous red-shifted peaks in corrolazine π-radical-cations.[8c,14] However, a similarly red-shifted band at 915 nm was recently reported for a PV porphyrazine π-radical complex.[10b] The new 920 nm species can be quantitatively converted back to 1 by titration with the one-electron oxidant Cp2Fe+PF6− (Fig. S11, ESI†). Taken together, the data indicate that cationic 1 undergoes a reversible, one-electron reduction to give a neutral π-radical complex, [ReV(O)(N-MeTBP8Cz)]• (3) (Scheme 2). This complex could be isolated as a red-brown solid by removal of solvent in vacuo, which then slowly decays with a half-life of 23 h under aerobic conditions at 23 °C (Fig. S12, ESI†). Thus 3 is a rare example of an isolable, air-stable porphyrinoid π-radical complex, and can be described as a 19π electron aromatic system. Very few other 19π electron porphyrinoid species are known.10
Fig. 4.

(a) UV-vis spectral titrations of 1 + Cr(C6H6)2 (0 – 1 equiv) in CH2Cl2. (b) EPR spectrum of 1 (0.84 mM) and PhNHNH2 (1.2 M) in CH2Cl2 at 295 K.
Scheme 2.

Electron-transfer and hydrogen-atom-transfer reactions with meso-N-methylated 1 and the 19π-electron radical complex 3.
The neutral complex ReV(O)(TBP8Cz) is unreactive toward H-atom donors. However, given that 1 could be reduced by one electron-transfer agents, we examined this complex for its ability to react with H-atom donors. The reaction of 1 with TEMPOH (BDE(O–H)DMSO = 72 kcal/mol)15 in CH2Cl2 at 23 °C led to conversion to 3 (Fig. S13, ESI†). For TEMPOH, E1/2 = 0.71 V (vs Fc+/Fc) makes outer-sphere ET to 1 prohibitively disfavored (ΔGET = +1.27 eV). A second H-atom donor, phenylhydrazine (PhNHNH2 BDE (N–H)DMSO = 75 kcal/mol) also reacts with 1 to give the same UV-vis spectral changes as TEMPOH (Scheme 2). These reactions indicate that 1 is capable of abstracting H atoms from weak O–H and N–H bonds.
Reaction of 1 with excess PhNHNH2 led to the fluid solution EPR spectrum for 3 shown in Figure 4b. A narrow, six-line signal centered near g ≈ 2 is seen. This spectrum can be assigned to an S = ½ radical delocalized over the Cz π system and split by 14N hyperfine coupling. DFT calculations for the doublet state for 3 (PBE0/LANL2TZ/6-31G** level of theory) yield a spin density plot (Figure S16, ESI†) that shows the unpaired electron is delocalized in a Cz π orbital that includes the three meso-N atoms, consistent with the observed 14N hyperfine coupling. In comparison, the π-cation radical complex [ReV(O)(TBP8Cz]•+ exhibits a sharp singlet at g = 2.00 with no 14N hyperfine splitting,8c and the spin density calculated for this complex (Fig. S16, ESI†) is concentrated on the pyrrole carbon atoms.
Mechanistic insights regarding the HAT reactivity of 1 were obtained from kinetic measurements. Reaction of 1 with excess TEMPOH led to pseudo-first-order decay of 1 and production of 3 as seen by UV-vis, and varying the TEMPOH concentration led to a linear second-order plot with k2 = 0.76(2) M−1 s−1. A kinetic isotope effect = 1.4 was found with TEMPOD. Concerted HAT reactions involving metal-oxo complexes often exhibit larger KIEs,16 although with some exceptions.17 The relatively small KIE seen for 1 may suggest a proton-coupled electron-transfer (PCET) process.18 It is reasonable to speculate that a weakly basic meso-N atom of the Cz ring is the initial proton acceptor, which then is likely rapidly deprotonated by the OTf− counterion.8b However, the exact fate of the proton in the PCET reactions with 1 is not known at this time.
In summary, a facile method for alkylation of meso-N-substituted porphyrinoid compounds has been reported. The post-cyclization treatment of other porphyrazines and phthalocyanines with appropriate alkyl electrophiles should be straightforward, opening the door to a wide range of new porphyrinoid compounds. The attachment of the methyl group to the meso-N atom in 1 has a profound influence on spectral properties and redox potentials, and reduction of 1 leads to a rare, air-stable 19π-electron radical species. Complex 1 is also capable of abstracting hydrogen atoms from weak O–H and N–H bonds, suggesting that porphyrinoid π-radical complexes in synthetic systems or heme proteins may have as yet undiscovered potential for oxidative reactivity toward substrates. The strategy of meso-N alkylation may also allow for the future installation of pendant groups orthogonal to the tetrapyrrolic plane of the Cz ligand, which may help in activating or stabilizing metal-oxygen intermediates.
Supplementary Material
Acknowledgments
The authors acknowledge research support from the NIH (GM101153 to D.P.G). E.E.J. is grateful for The Johns Hopkins Dean’s Science Postdoctoral Teaching Fellowship. J.P.T.Z. is grateful for The Glen E. Meyer ‘39 Fellowship. The Maryland Advanced Research Computing Center (MARCC) is thanked for CPU time to J.P.T.Z.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental, crystallographic, and computational details. CCDC 1510757 for 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/x0xx00000x
Notes and references
- 1.(a) Neu HM, Baglia RA, Goldberg DP. Acc Chem Res. 2015;48:2754. doi: 10.1021/acs.accounts.5b00273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McGown AJ, Badiei YM, Leeladee P, Prokop KA, DeBeer S, Goldberg DP. In: The Handbook of Porphyrin Science. Kadish KM, Smith KM, Guilard R, editors. Vol. 14. World Scientific; NJ: 2011. p. 525. [Google Scholar]; (c) Sahu S, Goldberg DP. J Am Chem Soc. 2016;138:11410. doi: 10.1021/jacs.6b05251. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Meunier B. Chem Rev. 1992;92:1411. [Google Scholar]
- 2.Shin JY, Yamada T, Yoshikawa H, Awaga K, Shinokubo H. Angew Chem Int Ed. 2014;53:3096. doi: 10.1002/anie.201310374. [DOI] [PubMed] [Google Scholar]
- 3.(a) Lee CW, Lu HP, Lan CM, Huang YL, Liang YR, Yen WN, Liu YC, Lin YS, Diau EWG, Yeh CY. Chem Eur J. 2009;15:1403. doi: 10.1002/chem.200801572. [DOI] [PubMed] [Google Scholar]; (b) Cid JJ, Yum JH, Jang SR, Nazeeruddin MK, Martínez-Ferrero E, Palomares E, Ko J, Grätzel M, Torres T. Angew Chem Int Ed. 2007;46:8358. doi: 10.1002/anie.200703106. [DOI] [PubMed] [Google Scholar]; (c) O’Regan BC, López-Duarte I, Martínez-Díaz MV, Forneli A, Albero J, Morandeira A, Palomares E, Torres T, Durrant JR. J Am Chem Soc. 2008;130:2906. doi: 10.1021/ja078045o. [DOI] [PubMed] [Google Scholar]
- 4.Joslin EE, Zaragoza JPT, Baglia RA, Siegler MA, Goldberg DP. Inorg Chem. 2016;55:8646. doi: 10.1021/acs.inorgchem.6b01219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Mandimutsira BS, Ramdhanie B, Todd RC, Wang H, Zareba AA, Czernuszewicz RS, Goldberg DP. J Am Chem Soc. 2002;124:15170. doi: 10.1021/ja028651d. [DOI] [PubMed] [Google Scholar]; (b) Baglia RA, Prokop-Prigge KA, Neu HM, Siegler MA, Goldberg DP. J Am Chem Soc. 2015;137:10874. doi: 10.1021/jacs.5b05142. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cho K, Leeladee P, McGown AJ, DeBeer S, Goldberg DP. J Am Chem Soc. 2012;134:7392. doi: 10.1021/ja3018658. [DOI] [PubMed] [Google Scholar]; (d) Lansky DE, Kosack JR, Sarjeant A, Goldberg DP. Inorg Chem. 2006;45:8477. doi: 10.1021/ic0609251. [DOI] [PubMed] [Google Scholar]; (e) Leeladee P, Jameson GNL, Siegler MA, Kumar D, de Visser SP, Goldberg DP. Inorg Chem. 2013;52:4668. doi: 10.1021/ic400280x. [DOI] [PubMed] [Google Scholar]
- 6.(a) Gryko DT. J Porphyrins Phthalocyanines. 2008;12:906. [Google Scholar]; (b) Pinto SMA, Henriques CA, Tomé VA, Vinagreiro CS, Calvete MJF, Dąbrowski JM, Piñeiro M, Arnaut LG, Pereira MM. J Porphyrins Phthalocyanines. 2016;20:45. [Google Scholar]; (c) Lindsey JS. Acc Chem Res. 2010;43:300. doi: 10.1021/ar900212t. [DOI] [PubMed] [Google Scholar]
- 7.(a) Velazquez CS, Fox GA, Broderick WE, Andersen KA, Anderson OP, Barrett AGM, Hoffman BM. J Am Chem Soc. 1992;114:7416. [Google Scholar]; (b) Liang XL, Ellis DE, Gubanova OV, Hoffman BM, Musselman RL. Int J Quantum Chem. 1994;52:657. [Google Scholar]
- 8.(a) Neu HM, Jung J, Baglia RA, Siegler MA, Ohkubo K, Fukuzumi S, Goldberg DP. J Am Chem Soc. 2015;137:4614. doi: 10.1021/jacs.5b00816. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jung J, Neu HM, Leeladee P, Siegler MA, Ohkubo K, Goldberg DP, Fukuzumi S. Inorg Chem. 2016;55:3218. doi: 10.1021/acs.inorgchem.5b02019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zaragoza JPT, Siegler MA, Goldberg DP. Chem Commun. 2016;52:167. doi: 10.1039/c5cc07956j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.A few meso-N-alkylated porphyrinoid compounds have been prepared from multistep syntheses of specific pyrrolic precursors prior to cyclization: Omori H, Hiroto S, Shinokubo H. Org Lett. 2016;18:2978. doi: 10.1021/acs.orglett.6b01323.Satoh T, Minoura M, Nakano H, Furukawa K, Matano Y. Angew Chem Int Ed. 2016;55:2235. doi: 10.1002/anie.201510734.Johnson AW, Kay IT, Rodrigo R. J Chem Soc. 1963:2336.
- 10.(a) Schweyen P, Brandhorst K, Wicht R, Wolfram B, Bröring M. Angew Chem Int Ed. 2015;54:8213. doi: 10.1002/anie.201503624. [DOI] [PubMed] [Google Scholar]; b) Yoshida T, Zhou W, Furuyama T, Leznoff DB, Kobayashi N. J Am Chem Soc. 2015;137:9258. doi: 10.1021/jacs.5b05781. [DOI] [PubMed] [Google Scholar]
- 11.(a) Shelton AH, Rodger A, McMillin DR. Biochemistry. 2007;46:9143. doi: 10.1021/bi700293g. [DOI] [PubMed] [Google Scholar]; (b) Fletcher SJ, Harper SR, Arnold DP. J Porphyrins Phthalocyanines. 2014;18:200. [Google Scholar]
- 12.Fox JP, Ramdhanie B, Zareba AA, Czernuszewicz RS, Goldberg DP. Inorg Chem. 2004;43:6600. doi: 10.1021/ic049384a. [DOI] [PubMed] [Google Scholar]
- 13.Cundari TR, Raby PD. J Phys Chem A. 1997;101:5783. [Google Scholar]
- 14.(a) Prokop KA, Neu HM, de Visser SP, Goldberg DP. J Am Chem Soc. 2011;133:15874. doi: 10.1021/ja2066237. [DOI] [PubMed] [Google Scholar]; (b) Leeladee P, Baglia RA, Prokop KA, Latifi R, de Visser SP, Goldberg DP. J Am Chem Soc. 2012;134:10397. doi: 10.1021/ja304609n. [DOI] [PubMed] [Google Scholar]; (c) Baglia RA, Krest CM, Yang T, Leeladee P, Goldberg DP. Inorg Chem. 2016;55:10800. doi: 10.1021/acs.inorgchem.6b02109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Nam W. Acc Chem Res. 2007;40:522. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; (b) Lansky DE, Goldberg DP. Inorg Chem. 2006;45:5119. doi: 10.1021/ic060491+. [DOI] [PubMed] [Google Scholar]; (c) Mayer JM. Acc Chem Res. 2011;44:36. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bougher CJ, Liu S, Hicks SD, Abu-Omar MM. J Am Chem Soc. 2015;137:14481. doi: 10.1021/jacs.5b09759. [DOI] [PubMed] [Google Scholar]
- 18.(a) Kundu S, Miceli E, Farquhar ER, Ray K. Dalton Trans. 2014;43:4264. doi: 10.1039/c3dt52644e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weatherly SC, Yang IV, Thorp HH. J Am Chem Soc. 2001;123:1236. doi: 10.1021/ja003788u. [DOI] [PubMed] [Google Scholar]; (c) Osako T, Ohkubo K, Taki M, Tachi Y, Fukuzumi S, Itoh S. J Am Chem Soc. 2003;125:11027. doi: 10.1021/ja029380+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.