

Abstract
Cell migration potency is essential in cancer metastasis and is often regulated by extracellular stimuli. Oral squamous cell carcinoma cell lines include those that are sensitive, as well as resistant, to the effects of the epidermal growth factor receptor (EGFR) inhibitor cetuximab on cell migration. In the present study, the molecular differences in the EGFR response to cell migration between the SAS cetuximab-sensitive and HSC4 cetuximab-resistant cell lines was examined. Treatment with the EGFR inhibitors AG1478 and cetuximab reduced the migration potency of SAS cells, but not HSC4 cells. The migration of the two cell lines was inhibited under serum-free culture conditions, and the addition of EGF to the serum-free medium promoted the migration of SAS cells, but not HSC4 cells. In addition, SAS cell migration was reduced by the mitogen-activated protein kinase kinase and protein kinase B (Akt) inhibitors PD98059 and MK2206, whereas HSC4 cell migration was only inhibited by MK2206. EGF induced an increase in extracellular signal-regulated kinase phosphorylation levels in HSC4 cells, and stimulated Akt phosphorylation levels in SAS cells. Furthermore, the staining of actin filaments with phalloidin was significantly increased by the inhibition of EGFR in SAS cells, but was not observed as altered in HSC4 cells. Conversely, the addition of EGF to the culture medium decreased the accumulation of actin filaments in SAS cells. The results suggest that the EGF-EGFR signaling pathway has an important role in SAS cell migration via the modulation of actin dynamics, and that HSC4 cell migration is regulated by a serum component other than EGFR.
Keywords: EGF, EGFR, cetuximab, AG1478, MK2206, PD98059
Introduction
Despite recent advances in surgery, radiotherapy and chemotherapy for the treatment of various types of cancer, morbidity remains at a high level (1) and the five-year survival rate for oral cancer has only moderately improved (2,3). Therefore, novel therapeutic strategies are required. As the majority of types of oral cancer are oral squamous cell carcinoma (OSCC), one feature of which is progressive local invasion (4,5), it is necessary to elucidate its underlying invasion mechanisms in order to improve currently available treatments for OSCC. The processes involved in tumor invasion include cell migration, interaction between the tumor and stroma at the invasive front and the involvement of growth factors and external stimuli that affect the invading cells (6–10). It is important to understand the signaling mechanisms underlying the regulation of cell migration and invasive growth, in order to facilitate the identification of novel therapeutic targets (11–13).
Signal transduction via receptor tyrosine kinases (RTKs) is stimulated by the respective extracellular ligands, which regulate critical cellular processes, including cell proliferation and cell migration (14). Therefore, genetic changes and abnormalities in RTKs often lead to a malignant transformation (14). A notable example of extracellular growth factors activating RTKs is the epidermal growth factor (EGF) family, the members of which function via the EGF receptor (EGFR) (15). Although EGFR is expressed in the normal oral epithelium as well as in the majority of OSCC cells (15,16), it is also a therapeutic target for the treatment of oral cancer (12,13). Previous studies using various cell types have demonstrated that the downstream signaling pathways of numerous RTKs are involved in the regulation of cell motility (7,17).
Certain mitogen-activated protein (MAP) kinases, including extracellular-regulated kinase (ERK), Jun kinase and tumor protein (p)38, are able to affect various cell functions, including migration (18). Phosphatidylinositol-3 kinase (PI3K) controls cell motility through the activation of protein kinase B (Akt) and other targets (19,20); however, cell-dependent differences in these regulatory mechanisms exist (18,21–23).
In head and neck cancer, the signaling pathways involved in RTK-mediated migration have yet to be elucidated and understood. EGFR stimulation induces cell migration through the activation of matrix metalloproteinases (MMPs) (24), signal transducer and activator of transcription 3 (STAT3) (25,26) or the MEK/ERK and PI3K signaling pathways (9), and may be associated with an epithelial-mesenchymal transition (EMT)-like phenotype (27). Furthermore, cross-talk between EGFR and G-protein-coupled receptors contributes to cell migration (28,29).
Our previous study reported that cetuximab, an EGFR-specific monoclonal antibody, inhibits migration of the SAS OSCC cell line, but not of the HSC4 OSCC cell line; however, the proliferation of HSC4 cells was observed to be sensitive to cetuximab (30). These results suggested that EGFR signaling may induce cell migration in a cell type-dependent manner. In the present study, the underlying mechanisms of EGFR signal transduction involved in the migration of the SAS and HSC4 OSCC cell lines were investigated and compared.
Materials and methods
Cell culture and reagents
The HSC4 and SAS OSCC cell lines were purchased from RIKEN Bioresource Center (Ibaraki, Japan). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) at 37°C in a humidified atmosphere of 5% CO2. DMEM and FBS were purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The antibodies used consisted of anti-AKT (rabbit monoclonal; cat. no. 4681; dilution, 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-phospho-AKT (rabbit polyclonal; cat. no. 9271; dilution; 1:1,000, Cell Signaling Technology, Inc.), anti-ERK (rabbit polyclonal; cat. no. sc-93; dilution, 1:1,000; Santa Cruz Biotechnology, Inc. Santa Cruz, CA, USA), anti-phospho-ERK (mouse monoclonal; cat. no. sc-7383; dilution, 1:1,000; Santa Cruz, Inc.), anti-profilin-1 (rabbit polyclonal; cat. no. 3237; dilution, 1:1,000; Cell Signaling Technology, Inc.), anti-cofilin (rabbit polyclonal; cat. no. sc-33729; dilution, 1:1,000; Santa Cruz Biotechnology, Inc.), anti-phospho-cofilin (rabbit polyclonal; cat. no. sc-12912; dilution, 1:1,000; Santa Cruz Biotechnology) and anti-α-tubulin (mouse monoclonal; cat. no. T6074; dilution, 1:1,000; Sigma-Aldrich; Merck Millipore, Darmstadt, Germany). The secondary antibodies used consisted of ECL™ anti-mouse IgG, horseradish peroxidase (HRP)-linked (dilution, 1:10,000; cat. no. NA931V; GE Healthcare Japan, Tokyo, Japan) and ECL™ anti-rabbit IgG, HRP linked (dilution 1, 10,000; cat. no. NA934V; GE Healthcare Japan). Cetuximab (Erbitux®) was purchased from Merck Serono (Tokyo, Japan). EGFR inhibitor AG1478, Akt inhibitor MK2206 and MEK inhibitor PD98059 were from Calbiochem (Merck Millipore, Darmstadt, Germany). Acti-stain™ 488 (Cytoskeleton, Inc., Denver, CO, USA) was used for actin filament staining.
Scratch wound healing assay
Cell migration was evaluated using a scratch wound healing assay, as previously described (30), with certain modifications. Briefly, OSCC HSC4 and SAS cells were seeded at 1–2×104 in 12-well plates, cultured for 24 h, and the semiconfluent cells were treated with 10 µg/ml mitomycin C for 4 h at 37°C to block proliferation and were subsequently wounded with a sterile 200 µl pipette tip in order to generate a cell-free gap. The cells were then washed with PBS and an image was captured using inverted phase-contrast microscopy (IX71; Olympus Corporation, Tokyo, Japan) and a digital CCD camera (DP72-SET; Olympus Corporation) to record the wound width at 0 h. One group of HSC4 or SAS cells was then cultured in DMEM supplemented with 10% FBS as a control. The HSC4 and SAS cells were treated with reagents including cetuximab (10 and 20 µg/ml), AG1478 (10 and 20 µg/ml), FBS (10%), EGF (10, 20 and 50 ng/ml), MK2206 (1, 5 and 10 µM) or PD98059 (1, 5 and 10 µM). Following incubations for 12–20 h at 37°C, images of the cells were captured using inverted phase-contrast microscopy (IX71, Olympus, Tokyo, Japan) in order to evaluate the rate of migration.
Western blotting
The OSCC HSC4 and SAS cells were washed with PBS and then lysed with radioimmunoprecipitation assay buffer, consisting of 150 mM NaCl, 10 mM Tris-HCl, pH 8.0, 1% (v/v) Nonidate P-40, 0.5% (w/v) deoxycholic acid, 0.1% (w/v) SDS, 5 mM EDTA, 1X Halt™ protease inhibitor cocktail (Thermo Fisher Scientific, Inc.) and 1X Halt™ protein phosphatase inhibitor (Thermo Fisher Scientific, Inc). The protein concentration of the lysates was determined using a BCA™ Protein Assay kit (Thermo Fisher Scientific, Inc.) and equal amounts of the proteins (10–50 µg) were subjected to SDS-PAGE analysis. The separated proteins were electrophoretically transferred onto polyvinylidene fluoride membranes (Clear Trans SP; Wako Pure Chemical Industries, Ltd., Osaka, Japan). Non-specific binding was blocked by incubation in 5% (w/v) bovine serum albumin (BSA; Sigma-Aldrich; Merck Millipore) in TBS/Tween-20 (TBS-T) for 1 h at room temperature. The membranes were probed with primary antibodies including anti-ERK, anti-phospho-ERK, anti-Akt, anti-phospho-Akt, anti-profilin, anti-cofilin, anti-phospho-cofilin and anti-α-tubulin in TBS-T overnight at 4°C and then incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Antibody-antigen complexes were detected using the Enhanced Chemiluminescence Plus Western Blotting Detection Reagent (GE Healthcare Japan, Tokyo, Japan).
Staining of actin fiber
Cultured HSC4 and SAS cells were fixed in 3.5% (w/v) paraformaldehyde for 10 min at room temperature, permeabilized in 0.2% (v/v) Triton X-100 for 5 min at room temperature, and blocked in 2% (w/v) BSA for 30 min at room temperature. Fixed cells were incubated in 100 nM of Acti-stain™ 488 phalloidin in the dark for 30 min at room temperature. Phalloidin staining was observed under fluorescent microscopy (Olympus, Tokyo, Japan).
Statistical analysis
All data are presented as the mean ± standard error and an unpaired student's t-test was used to determine the significant differences between the groups. P<0.05 was considered to indicate a statistically significant difference.
Results
The EGF-EGFR signaling pathway is associated with the migration of SAS cells, but not the migration of HSC4 cells
Changes in the EGFR signaling pathway induced by cetuximab have an important role in the migration of SAS cells, but not in the migration of HSC4 cells (30). In the present study, the effects of the AG1478 EGFR inhibitor on cell migration were examined and compared with those of cetuximab (Fig. 1A). The migration of SAS cells was significantly inhibited by AG1478 (10 µM P=0.00036, 20 µM P=0.00000055) as well as cetuximab (10 µM P=0.0027, 20 µM P=0.000045) treatment, whereas the migration of HSC4 cells was not affected by these inhibitors (Fig. 1A). As the migration of HSC4 and SAS cells was significantly stimulated (HSC4 P=2.8×10−18; SAS P=0.00012) by the addition of FBS in culture medium (Fig. 1B), the effects of EGF, an EGFR ligand present in serum, on migration were examined. EGF was observed to significantly stimulate the migration of SAS cells (20 ng/ml, P=0.0022, 50 ng/ml, P=0.000023), but not of the HSC4 cells (Fig. 1C). These results suggest that the EGF-EGFR signaling pathway induces the migration of SAS cells, whereas the signaling pathways induced by various other factors present in serum may contribute to the migration of HSC4 cells.
Figure 1.

Treatment with EGF or EGFR inhibitors affected the migratory potency of SAS cells. (A) Cetuximab and AG1478 inhibited the migration of SAS cells, but not of HSC4 cells. Phase-contrast micrographs of scratch wound healing assays performed following treatment with cetuximab or AG1478 for 24 h in HSC4 cells, or 12 h in SAS cells. The graph represents the wound width measurements of treated cells, as compared with non-treated cells (1.0). (B) FCS-induced migration of HSC4 and SAS cells. Phase-contrast micrographs of scratch wound healing assays performed in HSC4 and SAS cells with (+) or without (−) serum. (C) EGF induced migration in SAS cells, but not in HSC4 cells. Phase-contrast micrographs of scratch wound healing assays performed following treatment with EGF for 20 h in HSC4 cells, or 12 h in SAS cells. Error bars, SEM; n=3; **P<0.01; ***P<0.001. EGF, epidermal growth factor; EGFR, EGF receptor; FBS, fetal bovine serum.
The EGFR-Akt signaling pathway is associated with the migration of SAS cells
The ability of distinct RTKs to stimulate the migration of certain cell lines (14) indicates that various signal transduction pathways may be involved in the motility of HSC4 and SAS cells. In order to investigate the downstream signaling factors involved in OSCC cell motility, the effects of specific Akt and MEK inhibitors on the migration of these cells were evaluated. The Akt inhibitor MK2206 significantly suppressed the migration of HSC4 (1 µM, P=0.0097; 5 µM, P=0.000018; 10 µM, P=0.000023) and SAS cells (5 µM, P=0.047; 10 µM, P=0.0056; Fig. 2A). In addition, EGF was observed to induce Akt phosphorylation in SAS cells, but not in HSC4 cells (Fig. 2C). These results indicate that the Akt signaling pathway is required for the migration of these cell lines, and that EGF induces Akt phosphorylation in SAS cells, but not in HSC4 cells. The MEK inhibitor PD98059 was identified to significantly inhibit the migration of SAS cells (5 µM, P=0.000039; 10 µM, P=8.9×10−9), but not of HSC4 cells (Fig. 2B). The phosphorylation of ERK was highly and moderately induced by EGF in HSC4 and SAS cells, respectively (Fig. 2C). These data indicate that EGF is able to activate the ERK signaling pathway in certain OSCC cell lines; however, the ERK signaling pathway is only involved in the migration of SAS cells.
Figure 2.

The EGF-EGFR signaling pathway induces the migration of SAS cells via Akt phosphorylation. The effects of (A) MK2206 or (B) PD98059 treatment on the migration of HSC4 and SAS cells. Phase-contrast micrographs and graphs are as described in Fig. 1. (C) Representative western blot demonstrating that EGF promotes ERK phosphorylation in HSC4 cells, and Akt phosphorylation in SAS cells. Protein extracts from each cell sample were probed with anti-ERK, anti-phospho-ERK, anti-Akt and anti-phospho-Akt, and anti-α-tubulin as the loading control. Error bars, SEM; n=3; *P<0.05, **P<0.01, ***P<0.001; EGF, epidermal growth factor; EGFR, EGF receptor; Akt, protein kinase B; ERK, extracellular-regulated kinase; p, phosphorylated.
EGFR inhibitors promote, and EGF suppresses, the accumulation of actin filaments in SAS cells
As the remodeling of the actin network is considered to be involved in cell migration (31), the effects of cetuximab or AG1478 treatment on actin filament levels were examined using phalloidin staining. The degree of staining observed was markedly increased following the treatment of SAS cells with cetuximab (P=0.000020) and AG1478 (P=0.00036), as compared with untreated cells (Fig. 3A). By contrast, the EGFR inhibitors cetuximab (P=0.73) and AG1478 (P=0.89) were not observed to affect the degree of phalloidin staining in HSC4 cells (Fig. 3A). EGFR is induced by EGF binding, it was then examined whether EGF affects actin organization in HSC4 and SAS cells. EGF treatment reduced the degree of palloidin staining in SAS cells (P=0.00014), but not in HSC4 cells (P=0.94; Fig. 3B). These results indicate that the EGF-EGFR signaling pathway regulates actin filament turnover, and that it is necessary for cell motility. Thus, we examined effect of EGFR signaling on profiling and cofilin levels, because these proteins play key roles in actin polymerization and depolymerization, respectively. However, following treatment of the HSC4 and SAS cells with EGFR inhibitors, profiling-1 expression levels were not observed to be altered and cofilin-1 phosphorylation increased in both cells (data not shown).
Figure 3.
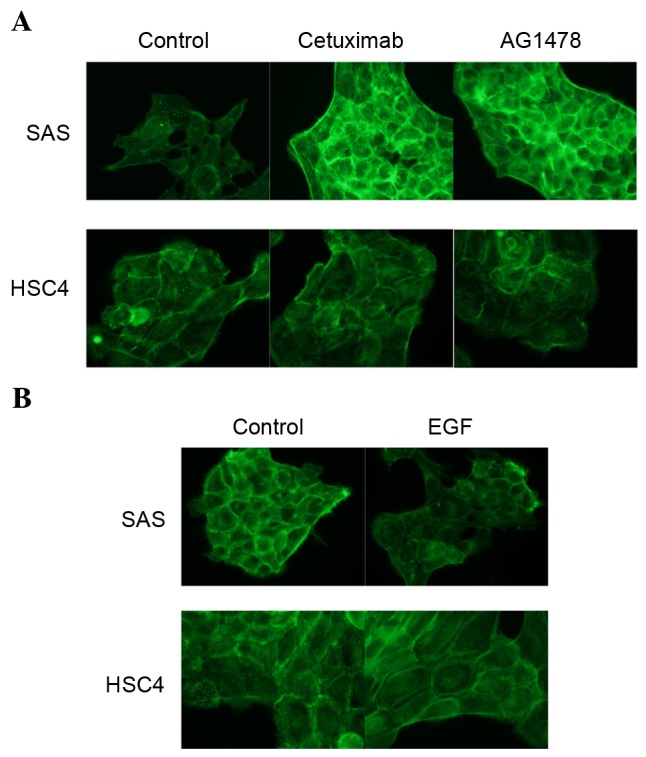
The EGF-EGFR signaling pathway regulates the actin network. Fluorescent images of phalloidin staining of the actin filaments in HSC4 and SAS cells treated with (A) cetuximab or AG1478 and (B) EGF. Fixed cells were permeabilized and stained with Acti-stain™ 488. EGF, epidermal growth factor; EGFR, EGF receptor.
Discussion
The directed migration of tumor cells into the surrounding tissues and blood vessels promotes the tissue invasion of metastatic cancer cells (32). This directed cell migration is often regulated by responses to extracellular stimuli (33–35). Notably, signal transduction via EGFR has an important role in cell motility in various types of cancer (36,37). In the present study, EGF-EGFR signaling was identified to be required for the migration of SAS OSCC cells, but not for HSC4 OSCC cells. In our previous study, it was demonstrated that EGFR activity is essential for the proliferation of HSC4 cells, but not SAS cells (30). The Akt inhibitor MK2206 and ERK inhibitor PD98059, which did not affect HSC4 cell migration, were observed to inhibit SAS cell migration. EGF stimulated the phosphorylation of ERK in HSC4 cells and Akt in SAS cells. These results indicate that the EGF-EGFR signaling pathway induces fundamental phenomena, including cell proliferation and migration, in a cell type-dependent manner. Furthermore, it is possible that the migration of SAS cells is regulated by EGF-EGFR signal transduction via Akt, and that the proliferation of HSC4 cells is regulated by EGF-EGFR signal transduction via the MEK-ERK signaling pathway.
The migration of HSC4 cells was promoted by the addition of FBS; however, the addition of EGF did not affect their migration, indicating that HSC4 cell migration requires certain extracellular stimuli present in serum other than EGF. At present, ~60 RTKs have been identified and their activities have been demonstrated to affect various physiological functions, including cell migration (38). In addition to EGF, previous studies have revealed that the following RTKs are also involved in regulating the migration of various cancer cells: growth arrest-specific 6 (GAS6) receptor (AXL); hepatocyte growth factor receptor (MET); fibroblast growth factor; and discoidin domain receptor family, member 1 (39–43). Therefore, further examination of how these candidate RTKs are involved in the regulation of OSCC cell migration is required.
The cytoskeleton, which is composed of actin filaments, is a highly organized network that enables cellular motion (44). In the present study, it was demonstrated that the degree of phalloidin staining in SAS cells treated with EGFR inhibitors was markedly increased, as compared with the control cells. Untreated control cells exhibited a normal arrangement of actin filaments near the outer layer of the cell periphery. Although this staining pattern was fundamentally unchanged following treatment of the control cells with EGFR inhibitors, intense overall cortical staining was observed. By contrast, the degree of phalloidin staining of the actin filaments was reduced in SAS cells by the addition of EGF; however, the staining pattern was not altered. It was hypothesized that the accumulation of actin filaments may produce irregular tension in the cytoskeletal system that is able to disrupt the orchestrated generation of force and interfere with directional cell migration. Therefore, it was concluded that the effects of EGFR inhibitors on the motility of SAS cells are possibly due to the accumulation of actin filaments in the cytoskeleton.
Previous studies have indicated that profilin and cofilin have key roles in regulating the assembly of actin filaments beneath the plasma membrane, in order to promote cell motility and other actin-associated processes (45,46). Profilin-1 is an actin monomer-binding protein considered to be an essential factor in actin polymerization (47,48). In addition, the depolymerization of actin filaments is regulated by cofilin, the phosphorylated form of which cannot sever actin filaments and therefore shows negative regulation (49–51). Therefore, the effects of EGFR inhibitors on the expression levels of profilin-1 and phosphorylated cofilin-1 were examined. However, our unpublished western blot analysis data suggested that profiling-1 and cofilin-1 may not necessarily have direct roles in the actin dynamics regulated by EGFR signaling. It was hypothesized that various other actin-binding proteins involved in the reorganization of the actin cytoskeleton are regulated downstream of the EGFR signaling pathway, and that EGFR inhibition promotes the overexpression of these proteins, resulting in the accumulation of actin filaments and cell dysmotility.
The proliferation of SAS cells, which possess a stem cell-like potency (52), was resistant to cetuximab in monolayer culture conditions, despite the phosphorylation of EGFR; however, the growth of SAS cell aggregates, in which EGFR levels were increased, was sensitive to cetuximab (30). In the present study, it was demonstrated that SAS cell migration is also sensitive to the EGFR inhibitors cetuximab and AG1478. Therefore, EGFR may be a candidate therapeutic target for the prevention of cancer stem cell dissociation from the primary tumor, migration into the surrounding tissues during the early stage and colonization at distant sites in OSCC metastasis.
Acknowledgements
This manuscript has been edited for English language by Textcheck English consultants. Funding for the present study was provided by Osaka University (grant no. 1508000001) and Osaka Dental University (grant no. 217006).
Glossary
Abbreviations
- EGF
epidermal growth factor
- EGFR
EGF receptor
- OSCC
oral squamous cell carcinoma
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
- RTK
receptor tyrosine kinase
- ERK
extracellular-regulated kinase
- PBS
phosphate-buffered saline
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Cohen EE, Lingen MW, Vokes EE. The expanding role of systemic therapy in head and neck cancer. J Clin Oncol. 2004;22:1743–1752. doi: 10.1200/JCO.2004.06.147. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 4.Kramer R, Shen X, Zhou H. Tumor cell invasion and survival in head and neck cancer. Cancer Metastasis Rev. 2005;24:35–45. doi: 10.1007/s10555-005-5046-2. [DOI] [PubMed] [Google Scholar]
- 5.Ziober AF, Falls EM, Ziober BL. The extracellular matrix in oral squamous cell carcinoma: Friend or foe? Head Neck. 2006;28:740–749. doi: 10.1002/hed.20382. [DOI] [PubMed] [Google Scholar]
- 6.De Hert MJ, de Jong RJ Baatenburg. HGF and c-MET as potential orchestrators of invasive growth in head and neck squamous cell carcinoma. Front Biosci. 2008;13:2516–2526. doi: 10.2741/2863. [DOI] [PubMed] [Google Scholar]
- 7.Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24:2666–2672. doi: 10.1200/JCO.2005.04.8306. [DOI] [PubMed] [Google Scholar]
- 8.Rorth P. Collective cell migration. Annu Rev Cell Dev Biol. 2009;25:407–429. doi: 10.1146/annurev.cellbio.042308.113231. [DOI] [PubMed] [Google Scholar]
- 9.Neiva KG, Zhang Z, Miyazawa M, Warner KA, Karl E, Nör JE. Cross talk initiated by endothelial cells enhances migration and inhibits anoikis of squamous cell carcinoma cells through STAT3/Akt/ERK signaling. Neoplasia. 2009;11:583–593. doi: 10.1593/neo.09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedl P, Wolf K. Plasticity of cell migration: A multiscale tuning model. J Cell Biol. 2010;188:11–19. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seiwert TY, Jagadeeswaran R, Faoro L, Janamanchi V, Nallasura V, El Dinali M, Yala S, Kanteti R, Cohen EE, Lingen MW, et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009;69:3021–3031. doi: 10.1158/0008-5472.CAN-08-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, Raben D, Baselga J, Spencer SA, Zhu J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11:21–28. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 13.Fung C, Grandis JR. Emerging drugs to treat squamous cell carcinomas of the head and neck. Expert Opin Emerg Drugs. 2010;15:355–373. doi: 10.1517/14728214.2010.497754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp Cell Res. 2003;284:31–53. doi: 10.1016/S0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 16.Laimer K, Spizzo G, Gastl G, Obrist P, Brunhuber T, Fong D, Barbieri V, Jank S, Doppler W, Rasse M, Norer B. High EGFR expression predicts poor prognosis in patients with squamous cell carcinoma of the oral cavity and oropharynx: A TMA-based immunohistochemical analysis. Oral Oncol. 2007;43:193–198. doi: 10.1016/j.oraloncology.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 17.Citri A, Yarden Y. EGF-ERBB signalling: Towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 18.Huang C, Jacobson K, Schaller MD. MAP kinase and cell migration. J Cell Sci. 2004;117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- 19.Bunney TD, Katan M. Phosphoinositide signalling in cancer: Beyond PI3K and PTEN. Nat Rev Cancer. 2010;10:342–352. doi: 10.1038/nrc2842. [DOI] [PubMed] [Google Scholar]
- 20.Kölsch V, Charest PG, Firtel RA. The regulation of cell motility and chemotaxis by phospholipid signaling. J Cell Sci. 2008;121:551–559. doi: 10.1242/jcs.023333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loesch M, Chen G. The p38 MAPK stress pathway as a tumor suppressor or more? Front Biosci. 2008;13:3581–3593. doi: 10.2741/2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye M, Hu D, Tu L, Zhou X, Lu F, Wen B, Wu W, Lin Y, Zhou Z, Qu J. Involvement of PI3K/Akt signaling pathway in hepatocyte growth factor-induced migration of uveal melanoma cells. Invest Ophthalmol Vis Sci. 2008;49:497–504. doi: 10.1167/iovs.07-0975. [DOI] [PubMed] [Google Scholar]
- 24.Zuo JH, Zhu W, Li MY, Yi H, Zeng GQ, Wan XX, He QY, Li JH, Qu JQ, Chen Y, Xiao ZQ. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J Cell Biochem. 2011;112:2508–2517. doi: 10.1002/jcb.23175. [DOI] [PubMed] [Google Scholar]
- 25.Wheeler SE, Suzuki S, Thomas SM, Sen M, LeemanNeill RJ, Chiosea SI, Kuan CT, Bigner DD, Gooding WE, Lai SY, Grandis JR. Epidermal growth factor variant III mediates head and neck cancer cell invasion via STAT3 activation. Oncogene. 2009;29:5135–5145. doi: 10.1038/onc.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin WL, Lin YS, Shi GY, Chang CF, Wu HL. Lewisy promotes migration of oral cancer cells by glycosylation of epidermal growth factor receptor. PLoS One. 2015;10:e0120162. doi: 10.1371/journal.pone.0120162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holz C, Niehr F, Boyko M, Hristozova T, Distel L, Budach V, Tinhofer I. Epithelial-mesenchymal-transition induced by EGFR activation interferes with cell migration and response to irradiation and cetuximab in head and neck cancer cells. Radiother Oncol. 2011;101:158–164. doi: 10.1016/j.radonc.2011.05.042. [DOI] [PubMed] [Google Scholar]
- 28.Thomas SM, Bhola NE, Zhang Q, Contrucci SC, Wentzel AL, Freilino ML, Gooding WE, Siegfried JM, Chan DC, Grandis JR. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006;66:11831–11839. doi: 10.1158/0008-5472.CAN-06-2876. [DOI] [PubMed] [Google Scholar]
- 29.Liebmann C. EGF receptor activation by GPCRs: An universal pathway reveals different versions. Mol Cell Endocrinol. 2011;331:222–231. doi: 10.1016/j.mce.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Ohnishi Y, Yasui H, Kakudo K, Nozaki M. Cetuximab-resistant oral squamous cell carcinoma cells become sensitive in anchorage-independent culture conditions through the activation of the EGFR/AKT pathway. Int J Oncol. 2015;47:2165–2172. doi: 10.3892/ijo.2015.3215. [DOI] [PubMed] [Google Scholar]
- 31.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/S0092-8674(03)00120-X. [DOI] [PubMed] [Google Scholar]
- 32.Wyckoff JB, Jones JG, Condeelis JS, Segall JE. A critical step in metastasis: In vivo analysis of intravasation at the primary tumor. Cancer Res. 2000;60:2504–2511. [PubMed] [Google Scholar]
- 33.Seppa H, Grotendorst G, Seppä S, Schiffmann E, Martin GR. Platelet derived growth factor in chemotactic for fibroblasts. J Cell Biol. 1982;92:584–588. doi: 10.1083/jcb.92.2.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yip SC, ElSibai M, Coniglio SJ, Mouneimme G, Eddy RJ, Drees BE, Neilsen PO, Goswami S, Symons M, Condeelis JS, Backer JM. The distinct roles of Ras and Rac in PI 3-kinase-dependent protrusion during EGF-stimulated cell migration. J Cell Sci. 2007;120:3138–3146. doi: 10.1242/jcs.005298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrientos J, Stojadinoric O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16:585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 36.Yue P, Zhang X, Paladino D, Sengupta B, Ahmad S, Holloway RW, Ingersoll SB, Turkson J. Hyperactive EGFR receptor, Jaks and Stat3 signaling promote enhanced colony-forming ability, motility and migration of cisplatin-resistant ovarian cancer cells. Oncogene. 2012;31:2309–2322. doi: 10.1038/onc.2011.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu SV, Subramaniam D, Cyriac GC, AbdoulKhalek FJ, Giaccone G. Emberging protein kinase inhibitors for non-small cell lung cancer. Expert Opin Emerg Drugs. 2014;19:51–65. doi: 10.1517/14728214.2014.873403. [DOI] [PubMed] [Google Scholar]
- 38.Potratz J, Tillmanns A, Berning P, Korsching E, Schaefer C, Lechtape B, Schleithoff C, Unland R, Schäfer KL, Müller-Tidow C, et al. Receptor tyrosine kinase gene expression profiles of Ewing sarcomas reveal ROR1 as a potential therapeutic target in metastatic disease. Mol Oncol. 2016;10:677–692. doi: 10.1016/j.molonc.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birchmeier C, Birchmeier W, Gherardi E, Woude GF Vande. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 40.Gherardi E, Birchmeier W, Birchmeier C, Woude G Vande. Targeting MET in cancer: Rationale and progress. Nat Rev Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 41.Penzes K, Baumann C, Szabadkai I, Orfi L, Kéri G, Ullrich A, Torka R. Combined inhibition of AXL, Lyn and p130Cas kinase block migration of triple negative breast cancer cells. Cell Biol Ther. 2014;15:1571–1582. doi: 10.4161/15384047.2014.956634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sie M, den Dunne WF, Lourens HJ, Meeuwsen-de Boer TG, Scherpen FJ, Zomerman WW, Kampen KR, Horing EW, de Bant ES. Growth-factor-driven rescue to receptor tyrosine kinase (RTK) inhibitors through Akt and Erk phosphorylation in pediatric low grade astrocytoma and ependymoma. PLoS One. 2015;10:e0122555. doi: 10.1371/journal.pone.0122555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Favreau AJ, Vary CP, Brooks PC, Sathyanarayana P. Cryptic collagen IV promotes cell migration and adhesion in myeloid leukemia. Cancer Med. 2014;3:265–272. doi: 10.1002/cam4.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Small JV, Resch GP. The comings and goings of actin: Coupling protrusion and retraction in cell motility. Curr Opin Cell Biol. 2005;17:517–523. doi: 10.1016/j.ceb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Kang F, Purich DL, Southwick FS. Profilin promotes barbed-end actin filament assembly without lowering the critical concentration. J Biol Chem. 1999;274:36963–36972. doi: 10.1074/jbc.274.52.36963. [DOI] [PubMed] [Google Scholar]
- 46.Konakahara S, Ohashi K, Mizuno K, Itoh K, Tsuji T. CD29 integrin-β and LIMK1/cofilin-mediated actin reorganization regulates the migration of haematopoietic progenitor cells underneath bone marrow stromal cells. Genes Cells. 2004;9:345–358. doi: 10.1111/j.1356-9597.2004.00726.x. [DOI] [PubMed] [Google Scholar]
- 47.Yun SP, Ryu JM, Jang MW, Han HJ. Interaction of profiling-1 and F-actin via a β-arrestin-1/JNK signaling pathway involved in prostaglandin E(2)-induced human mesenchymal stem cells migration and proliferation. J Cell Physiol. 2011;226:559–571. doi: 10.1002/jcp.22366. [DOI] [PubMed] [Google Scholar]
- 48.Pantalon D, Carlier MF. How profiling promotes actin filament assembly in the presence of thymosin beta 4. Cell. 1993;75:1007–1014. doi: 10.1016/0092-8674(93)90544-Z. [DOI] [PubMed] [Google Scholar]
- 49.Kobayashi M, Nishita M, Mishima T, Ohashi K, Mizuno K. MAPKAPK-2 mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 2006;25:713–726. doi: 10.1038/sj.emboj.7600973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGough A, Pope B, Chlu W, Weeds A. Cofilin changes the twist of F-actin: Implications for actin filament dynamics and cellular function. J Cell Biol. 1997;138:771–781. doi: 10.1083/jcb.138.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773:642–652. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohnishi Y, Minamino Y, Kakudo K, Nozaki M. Resistance of oral squamous cell carcinoma cells to cexuximab is associated with EGFR insensitivity and enhanced stem cell-like potency. Oncol Rep. 2014;32:780–786. doi: 10.3892/or.2014.3258. [DOI] [PubMed] [Google Scholar]