

INTRODUCTION
Cancer treatments have been transformed with recent advances in cancer immunotherapy.1 As monotherapies, these agents have demonstrated clinical activity across many tumor types. Further advances in the effectiveness of cancer immunotherapies will require targeting antitumor immune response at multiple levels, which may be accomplished through combination approaches. This review discusses the current landscape of cancer immunotherapy, combinations in clinical development, strategies for dose selection and trial design, and clinical pharmacology and regulatory considerations.
HISTORY OF CANCER IMMUNOTHERAPY AND APPROVED THERAPIES
Cancer kills over 8 million people worldwide every year and the number of diagnosed cases is expected to almost double in the next two decades.2 Surgery, radiation, chemotherapy, and targeted agents are commonly used to treat these patients, but many patients either relapse or are refractory to treatment. In addition, patients and physicians must manage a variety of side effects that have a significant impact on patients’ quality of life, which limits the use of these agents.
It is well established that cancer cells can be recognized by the immune system, and it is hypothesized that the defeat of the immune surveillance system underlies the development of malignancies and the lack or loss of response to treatment.3, 4 Under normal circumstances, a functioning immune surveillance system will recognize and eliminate transformed cells. Ironically, this Darwinian process ultimately results in the selection of tumor cells resistant to processing by the immune system through loss of antigenicity, defects in antigen presentation, and decreased immunogenicity (e.g., through upregulation of PD1, a negative regulator of the immune system).5 Immune escape is also accomplished by the alteration of the tumor microenvironment,5 whereby tumor cells recruit immune‐suppressive cells to promote conditions for their survival. Immuno‐oncology approaches attempt to restore the immune surveillance system and activate the patient's immune system to fight their cancer. These approaches have garnered significant attention and are projected to be the new standard of care for diverse tumor types. Indeed, the clinical data of recent regulatory approvals of immunotherapy treatments including blinatumomab (BLINCYTO), ipilimumab (Yervoy), nivolumab (Opdivo), and pembrolizumab (Keytruda) across multiple cancer types demonstrate the clinical feasibility of this approach.
The promise of immunotherapy to treat cancer was first realized over 100 years ago (Figure 1). In 1890, Emil von Behring and Erich Wernicke found that animals infected with diphtheria could be cured by injection of sera produced by animals immunized with an attenuated form of diphtheria, and this treatment was successfully used to treat a child with diphtheria in 1891.6 This introduced the use of serum as therapy and for the first time showed that immunity could be transferred, thereby demonstrating the clinical utility of passive immunity. The first application of immunotherapy in oncology also occurred in 1891, when William B. Coley (known as the father of immunotherapy) injected bacteria into a patient with cancer as a means of stimulating the immune system to shrink the patient's tumor, a strategy that was successful.7
Figure 1.
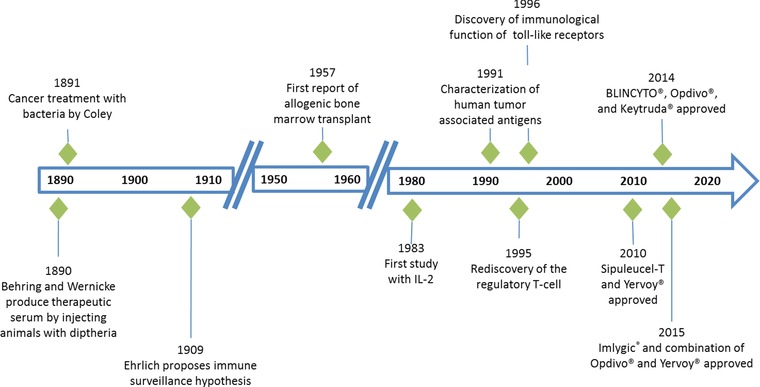
History of immunotherapy. Key events leading to the development of currently marketed immunotherapies including sipuleucel‐T (Provenge), ipilimumab (Yervoy), blinatumomab (BLINCYTO), nivolumab (Opdivo), pembrolizumab (Keytruda), and talimogene laherparepvec/T‐Vec (Imlygic).
Since then, significant progress has been made in the understanding and application of immunotherapy as monotherapy for cancer treatment. These agents can be classified as either active therapies that induce an immune response in otherwise nonresponsive patients or passive therapies that stimulate a patient's intrinsic immune response8 (Table 1). Active therapies include cytokines, immunomodulatory monoclonal antibodies (mAbs), and cancer vaccines, and passive therapies include BiTE antibody constructs, bispecific and multispecific antibodies, oncolytic viruses, cell‐based therapies, and tumor‐targeting mAbs. The checkpoint inhibitors (e.g., PD‐1, PD‐L1, CTLA‐4, and LAG‐3) are immunomodulatory mAbs that address immune escape by tumor cells that leverage normal immune‐suppressive mechanisms to prevent autoimmunity and tissue damage in response to acute infection in otherwise healthy individuals, but promote tumor progression in cancer patients.9 BiTE antibody constructs have dual specificity for T cells and cancer cells and bind to an invariant component of a T‐cell receptor and a specific surface antigen on a cancer cell (e.g., CD19), forcing them into proximity.10 Because they do not require a T‐cell clone with a specific T‐cell receptor or an MHC class I or peptide antigen for T‐cell recognition, BiTE antibody constructs can overcome immune escape. Oncolytic viruses selectively kill cancer cells and stimulate the immune system (e.g., Imlygic), while dendritic cell vaccines (e.g., sipuleucel‐T) involve the extraction of dendritic cells from the patient, exposure of those cells to cancer cells or antigens, and reintroduction of these now active immune cells to the patient (alternative approaches to vaccination against cancer are also under investigation).11 Adoptive T‐cell therapies including CAR‐T cell approaches depend on the genetic alteration of T cells to express particular antigen receptors on their surface that can recognize and kill cancer cells.12 The therapeutic use of neoantigens to stimulate T‐cell responses in cancer patients also have potential, with data from mouse models showing that vaccination with neoantigens can be effective.13 Immune system modulation by antibody‐dependent cellular cytotoxicity and complement‐dependent cytotoxicity mechanisms has also been successfully achieved with agents targeting CD20, CD52, SLAMF7, and CD38 showing clinical efficacy.
Table 1.
Classes of immunotherapy agents in oncology
| Active Immunotherapies | |
|---|---|
| Classes | Examples |
| Cancer vaccines | Sipuleucel‐T |
| Cytokines | Interleukin‐2, interferon‐α |
| Immunomodulatory mAbs | Nivolumab, ipilimumab, pembrolizumab |
| Passive immunotherapies | |
| Classes | Examples |
| Cell‐based therapies | Adoptive T‐cell therapy (e.g., TIL, TCR, CAR‐T) |
| Oncolytic viruses | T‐Vec |
| Bi‐ and multispecific antibodies | Blinatumomab |
| Tumor‐targeting mAbs | Rituximab |
mAbs, monoclonal antibodies; TIL, tumor infiltrating lymphocytes; TCR, T‐cell receptor; CAR‐T, chimeric antigen receptor T‐cell therapy; T‐Vec, Talimogene laherparepvec.
More than 15 cancer immunotherapies were approved for use as monotherapies in various solid and liquid tumor indications and three combination immunotherapies were approved as of 2015 (Table 2; refer to the United States product inserts [USPIs] for specific approved indications). Currently, the majority of approved cancer immunotherapies and those in development are biologics or cell‐based therapies, as these are ideal modalities to target protein–protein interactions and signaling pathways. However, there is a significant opportunity for the development of small molecule immuno‐oncology therapeutics,14 due to their unique ability to modulate the activity of intracellular targets (e.g., indoleamine 2,3‐dioxygenase [IDO1]), which are not easily accessible by a biologic. Through leveraging multiple mechanisms of action and modalities of traditional cytotoxic agents, targeted agents and biologic, and small molecule immunotherapies, various tumor types are treatable.
Table 2.
FDA‐approved immunotherapy agents
| Checkpoint inhibitors | |||
| MOA | Agent | Year | Indication c |
| Anti‐PD1 | Nivolumab | 2014 | Melanomaa, b |
| 2015 | Lung cancer, RCC | ||
| Anti‐PD1 | Pembrolizumab | 2014 | Melanomaa, b |
| 2015 | Lung cancera, b | ||
| Anti‐CTLA‐4 | Ipilimumab | 2011 | Melanoma |
| BiTE antibody constructs ; Bi‐ and multispecific antibodies | |||
| MOA | Agent | Year | Indication c |
| CD3/CD19 | Blinatumomab | 2014 | ALLa, b |
| Vaccines and oncolytic viruses | |||
| MOA | Agent | Year | Indication c |
| Dendritic cell | Sipuleucel‐T | 2010 | Prostate cancer |
| Oncolytic virus | T‐Vec | 2015 | Melanoma |
| Cytokines | |||
| MOA | Agent | Year | Indication c |
| Cytokine | IL‐2 | 1992 | RCC |
| 1998 | Melanoma | ||
| Cytokine | IFN‐α | 1986 | HCL |
| 1988 | AIDS‐related Kaposi's Sarcoma Melanoma | ||
| 1995 | Melanoma | ||
| 1997 | NHL | ||
| mAbs | |||
| MOA | Agent | Year | Indication c |
| CD52 | Alemtuzumab | 2001 | CLL |
| CD20 | Ofatumumab | 2009 | CLL |
| CD20 | Rituximab | 1997 | NHL |
| 2010 | CLL | ||
| CD38 | Daratumumab | 2015 | Multiple Myelomab |
| HER2 | Trastuzumab | 1998 | Breast cancer |
| 2010 | Gastric cancer | ||
| EGF | Cetuximab | 2004 | Colorectal cancer |
| 2011 | Head/neck cancer | ||
| CD20 ADC | 90Y‐Ibritumomab tiuxetan | 2002 | NHL |
| CD30 ADC | brentuximab vedotin | 2011 | Hodgkin lymphoma, ALCL |
| Cell‐based therapies | |||
| No TIL, TCR, or CAR‐T cell therapies are FDA approved | |||
| Combination Immunotherapies | |||
| MOA | Agent | Year | Indication c |
| Cytokine + VEGF | IFN‐α+ bevacizumab | 2009 | Renal cancer |
| Anti‐PD1 + anti‐CTLA‐4 | Nivolumab + ipilimumab | 2015 | Melanomaa, b |
| SLAMF7 + SOC | Elotuzumab + lenalidomide + dexamethasone | 2015 | Multiple Myelomab |
Denotes approval via the FDA's accelerated approval pathway
Denotes priority review status. RCC, renal cell carcinoma; ALL, acute lymphoid leukemia; MA, malignant ascites; HCL, hairy cell leukemia; NHL, Non‐Hodgkin's Lymphoma; IL‐2, interleukin 2; IFN‐α, interferon alpha
Refer to the USPIs for specific information of each approved indication (e.g., histological and molecular subtypes, line of treatment).
Combination immunotherapy landscape
Combination immunotherapies that involve various phases of the cancer–immunity cycle may enhance the ability to prevent immune escape by targeting multiple mechanisms by which tumor cells avoid elimination by the immune system, with synergistic effects that may offer improved efficacy in broader patient populations (Figure 2). Current immunotherapy combinations involve combining multiple immunotherapies or other cancer therapies such as chemotherapy, radiation, and targeted therapies (Table 3). The US Food and Drug Administration (FDA) approvals of bevacizumab and interferon‐alpha for the treatment of renal cancer in 2009 and nivolumab and ipilimumab for the treatment of melanoma in 2015 reveal the potential for combination immunotherapies. The combination of bevacizumab and interferon‐alpha resulted in a median progression‐free survival (PFS) of 10.2 months vs. 5.4 months in the control group receiving interferon‐alpha monotherapy.15 The combination of nivolumab and ipilimumab was also more effective than the respective single agents. As monotherapies, these agents provided a meaningful improvement in PFS and/or overall survival (OS) vs. standard of care. However, in combination, these agents were even more effective, with an overall median PFS of 11.5 months for the combination of nivolumab and ipilimumab, compared with 2.9 months and 6.9 months for ipilimumab and nivolumab alone.16 Although this increased efficacy came at the cost of increased adverse events,16 which is not an unexpected result for immunotherapy combination given the potential for overlapping toxicities, the benefit/risk assessment resulted in the accelerated approval of this combination. There was a strong mechanistic basis for testing the combination of nivolumab and ipilimumab. While both are checkpoint inhibitors targeting negative regulators of the immune system, they act on nonredundant regulatory pathways. Moreover, CTLA‐4 acts in the lymphoid system, while PD‐1 and PD‐L1 act downstream in the tumor microenvironment.17 The mechanisms of resistance to PD‐1 and CTLA‐4 inhibitors may therefore be addressed by combination therapy (e.g., the CTLA‐4 blockade mitigates the CTLA‐4 upregulation that may partially underlie resistance to PD‐1 blockade). These mechanisms are not well understood and combination therapy does not completely resolve nonresponse, suggesting additional mechanisms of resistance that need to be addressed with agents with other mechanisms of action (MOAs). While many of the current combination trials are expansions of the nivolumab and ipilimumab combination into other indications, novel combinations of checkpoint inhibitors with vaccines, cytokines, and molecularly targeted agents are also ongoing (Table 3).
Figure 2.
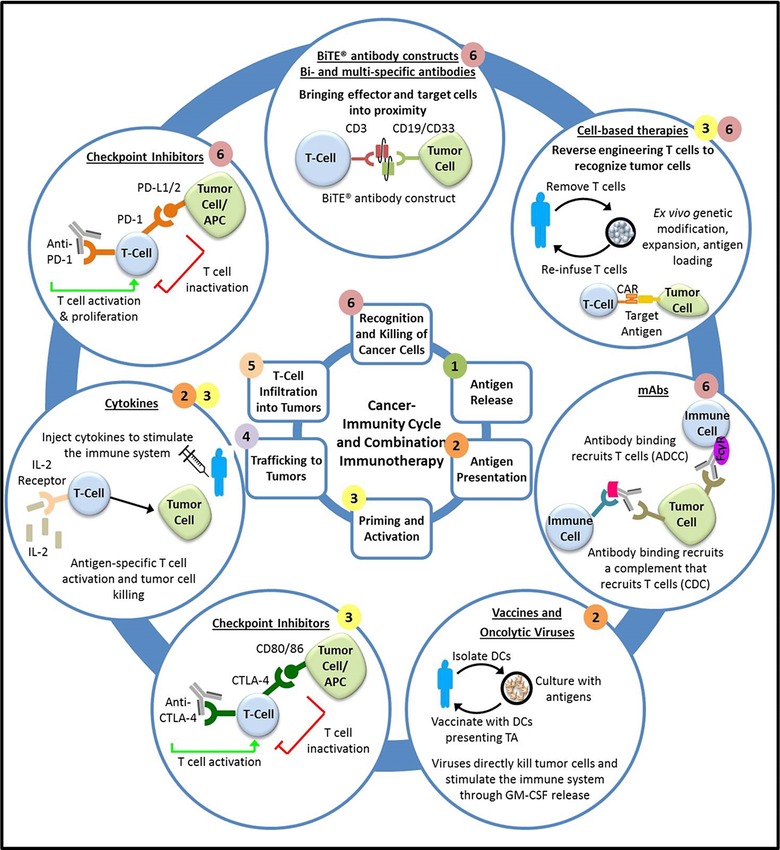
Intervention in the cancer‐immunity cycle by immunotherapy agents. Overcoming resistance and restoring a functional immune‐surveillance system requires leveraging multiple, complementary mechanisms of action and agents that acts in multiple phases of the cancer‐immunity cycle (numbers denote the phases at which each type of immunotherapy acts).
Table 3.
Selected list of combination immunotherapies in clinical development
| Immunotherapy + Immunotherapy | |||
|---|---|---|---|
| Combination therapy | Mechanisms of action | Phase | Indication |
| Nivolumab + ipilimumab | Anti‐PD1 + anti‐CTLA‐4 | I/II | Gastric, TNBC, PA, SCLC, Bladder, Ovarian |
| II/III | Melanoma, RCC | ||
| III | SCLC, GBM, NSCLC | ||
| Nivolumab + BMS‐986016 | Anti‐PD1 + anti‐LAG3 | I | Solid tumors |
| Nivolumab + viagenpumatucel‐L | Anti‐PD1 + vaccine | I | NSCLC |
| Nivolumab + urelumab | Anti‐PD1 + anti‐4‐1BB | I/II | Solid tumors, B‐cell NHL |
| Atezolizumab + MOXR0916 | Anti‐PDL1 + anti‐OX40 | I | Solid tumors |
| Atezolizumab + varlilumab | Anti‐PDL1 + anti‐CD27 | II | RCC |
| Atezolizumab + GDC‐0919 | Anti‐PDL1 + IDO inhibitor | I | Solid tumors |
| Epacadostat + atezolizumab, durvalumab, or pembrolizumab | IDO inhibitor + anti‐PDL1 or anti‐PD1 | I/II | Solid tumors |
| Pembrolizumab + T‐Vec | Anti‐PD1 + vaccine | III | Melanoma |
| Durvalumab + tremelimumab | Anti‐PDL1 + anti‐CTLA‐4 | I/II | Melanoma |
| I/II/III | SCCHN | ||
| II | Mesothelioma, UBC, TNBC, PA | ||
| III | NSCLC, Bladder | ||
| Pidilizumab + dendritic cell/RCC fusion cell vaccine | Anti‐PD1 + vaccine | II | RCC |
| Immunotherapy + Targeted Therapy | |||
| Combination therapy | Mechanisms of action | Phase | Indication |
| Atezolizumab + bevacizumab | Anti‐PDL1 + anti‐VEGF | II/III | RCC |
| Atezolizumab + cobimetinib | Anti‐PDL1 + MEK inhibitor | I | Solid tumors |
| Atezolizumab + vemurafenib | Anti‐PDL1 + BRAF inhibitor | I | Melanoma |
| Atezolizumab + erlotinib or alectinib | Anti‐PDL1 + EGFR or ALK inhibitor | I | NSCLC |
| Nivolumab + bevacizumab | Anti‐PD1 + anti‐VEGF | II | RCC |
| Pembrolizumab + pazopanib | Anti‐PD1 + tyrosine kinase inhibitor | I | RCC |
| Pembrolizumab + dabrafenib + trametinib | Anti‐PD1 + BRAF inhibitor + MEK inhibitor | I/II | Melanoma |
| Durvalumab + dabrafenib + trametinib | Anti‐PDL1 + BRAF inhibitor + MEK inhibitor | I/II | Melanoma |
| Nivolumab + sunitinib, pazopanib, or ipilimumab | Anti‐PD1 + RTK inhibitor, RTK inhibitor, | I | RCC |
| Immunotherapy + Chemotherapy | |||
| Combination therapy | Mechanisms of action | Phase | Indication |
| Nivolumab + platinum doublet chemoa | Anti‐PD1 + chemotherapy | III | NSCLC |
| Pembrolizumab + cisplatin | Anti‐PD1 + chemotherapy | III | Gastric |
| Pidilizumab + lenalidomide | Anti‐PD1 + chemotherapy | I/II | Multiple myleloma |
| Pidilizumab + sipuleucel‐T + cyclophosphamide | Anti‐PD1 + vaccine + chemotherapy | II | Prostate |
| Atezolizumab + carboplatin/paclitaxel +/– bevacizumab | Anti‐PDL1 + chemotherapy +/– anti‐VEGF | III | NSCLC |
TNBC, triple negative breast cancer; PA, pancreatic adenocarcinoma; SCLC, small‐cell lung cancer; RCC, renal cell carcinoma; GBM, glioblastoma multiforme; NSCLC, non‐smallcell lung cancer; NHL, non‐Hodgkin's lymphoma; SCCHN, squamous cell carcinoma of the head and neck; UBC, urothelial bladder cancer. aSquamous: gemcitabine + cisplatin or carboplatin; Nonsquamous: pemetrexed + cisplatin or carboplatin.
Combination therapies may dramatically improve the outcome for cancer patients, and indeed it is expected that such therapies will eventually become the standard of care for cancer treatment, but the discovery of effective combinations is a challenging endeavor. With nearly 200 molecules approved by the FDA for the treatment of cancer, including over 15 immunotherapy agents, experimentally testing every possible combination of these drugs would be unfeasible, even with high‐throughput experimental methods and a mechanistic basis for the selection of agents with complementary MOAs that target multiple mechanisms of resistance and immune escape. Therefore, new systems approaches are needed to reduce the search space and prioritize combinations for experimental testing. In addition, immunotherapies have unique properties that complicate their clinical development, and are magnified in the context of combination therapies. The regulatory environment is also evolving, particularly for novel agents including cell‐based therapies, for which many of the traditional development paradigms are not applicable.
Preclinical development and clinical translation of safety/efficacy
The objectives for preclinical evaluation of cancer immunotherapies are identical to other anticancer drugs and substantial progress has been made in the field of translational cancer research. These objectives include the (i) identification of the pharmacological characteristics of a development molecule, (ii) determination of an initial safe and starting dose for human studies, and (iii) understanding of the toxicological profile of the development molecule. However, for cancer immunotherapies there are unique challenges in translating preclinical data to the clinic, including cell lines or animal models that do not adequately mimic the tumor, tumor microenvironment, human immune response, or the propensity to develop resistance.
The starting dose in first‐in‐human (FIH) trials of anticancer agents should be carefully selected using all available nonclinical data (e.g., pharmacokinetics, pharmacodynamics [PK/PD], and toxicology) prior to the initiation of clinical studies. Specifically, both the estimated highest recommended starting dose from the most sensitive species from toxicology results and the minimal anticipated biological effect level (MABEL) from preclinical PK/PD results are important data sets to inform FIH dose selection.18 For cancer immunotherapies, MABEL appears to be the most common approach for FIH dose selection. For example, the no‐observed‐adverse‐effect‐level (NOAEL) of nivolumab (as determined in repeat‐dose toxicology studies in cynomolgus monkeys) was 50 mg/kg;19 however, the FIH dose was 0.1 mg/kg, presumably on the basis of preclinical pharmacology data that suggested a much lower MABEL than NOAEL. For ipilimumab, the NOAEL was 10 mg/kg20 and the FIH dose was 0.3 mg/kg. Similarly, the NOAEL for pembrolizumab was 200 mg/kg/day,21 yet the starting dose in patients with NSCLC was 1 mg/kg.
To define the MABEL from preclinical studies for FIH trials, knowledge of the PK/PD relationship is essential. Human PK prediction is an essential component in predicting the human dose corresponding to MABEL. For small molecules, the prediction of human PK is typically achieved using an empirical allometric scaling approach of data obtained from multiple preclinical species. For biologics that demonstrate linear PK, single‐species allometric scaling can generally provide reasonable estimates of human PK.22 In theory, this single‐species approach could also apply to mAbs with nonlinear clearance that have high therapeutic doses where the target‐mediated clearance is saturated and the overall clearance is in the linear range. However, in this situation the animal data used for the human PK projection would need to be in the linear range as well.
In terms of the prediction of human PD (either biomarkers or exploratory end points in early development) and human efficacious dose projection, PKPD modeling holds a unique position in translational research to integrate diverse sets of preclinical information. However, animal models for cancer immunotherapy present very unique challenges. Not only is there a time delay in tumor response, but the interaction between the tumor and mouse immune system is uniquely complex due to the involvement of multiple cell types across different tissue compartments.23 For combination therapy, a synergistic effect of the combination further increases the complexity of the correlation between drug exposure and antitumor response. In this situation, mechanistic‐based physiologically based pharmacokinetic (PBPK) and quantitative systems pharmacology (QSP) models may have a significant advantage in helping to capture the complex dynamics observed in preclinical models to predict human PD.
In both monotherapy and combination settings, it is challenging to identify suitable preclinical models that predict human efficacy and safety. It may be necessary to utilize all relevant information, including novelty of the agent, mechanism of action, nature of the target, relevance of animal species and models, potential pharmacological impact of genetic polymorphisms, dose–response, and exposure–response.24 Since a combination of a cancer immunotherapy with other immunotherapies or anticancer agents may cause unexpected toxicities, the starting dose of the agents in combination should generally be lower than the maximum tolerated dose (MTD) or the highest tested safe dose in monotherapy to avoid severe adverse events by overlapping or unexpected toxicities. Preclinical studies may also be useful in the evaluation of potential additive or synergistic toxicities of the combination.
For effector cell therapy, the traditional approaches used to select the FIH starting dose may not be applicable. Since it is a highly individualized therapy, multiple variables, including disease, treatment conditions, and potential for immune reactions need to be considered during dose selection for adoptive cell‐based immunotherapies. Currently there are no clear standards in place to guide the selection of a starting dose for adoptive cell‐based therapies. Published clinical trial results to date indicate that most treatment doses for CAR‐T cell therapies were falling within a similar range of roughly 106–107 cells/kg.25, 26 To date, there is not an apparent correlation between T‐cell dose and clinical response or between T‐cell dose and various toxicities.27 However, it appears that lower T‐cell dosing may achieve effective clinical outcomes with a reduced potential for uncontrolled cellular proliferation associated with cell‐based therapies.27
In order to begin clinical studies, global health authorities require the submission of adequate information from preclinical pharmacological and toxicological studies that form the sponsor's position that it is reasonably safe to conduct human studies. To support sponsors in the generation of the preclinical data package needed for clinical evaluation, there are various guidances available from the International Conference on Harmonization (ICH), the FDA, European Medicines Authority (EMA), and Japan Pharmaceuticals and Medical Devices Agency (PMDA),18 , 28, 29, 30, 31, 32 including specific guidelines for cancer immunotherapies.33, 34, 35 For combinations of new investigational drugs (including, but not limited to, cancer immunotherapies), the FDA has recently published a guidance to assist sponsors in the codevelopment of two or more new investigational drugs;36 referred to here as 2+ new molecular entity (NME) combinations. Briefly, the FDA believes that codevelopment is appropriate when there is a strong biological rationale for the combination and the nonclinical characterization of activity of each agent alone and in combination suggests that the combination would provide activity superior to each individual agent and available therapies. In general, with the rapid advances in technology and increasing number of combinations within and between cancer immunotherapeutic products, it is anticipated that additional guidance documents will be available in the future. However, as each clinical trial application is evaluated on a case‐by‐case basis, close and frequent interactions with health authorities are encouraged and advised, particularly for gene‐ and cell‐based products.
CLINICAL DEVELOPMENT AND TRIAL DESIGN
Strategies to identify efficacious dose(s)/schedule(s) for late‐stage clinical development
In the current oncology drug development paradigm, trials are designed to maximize the chance of an effective therapy and often include patients with advanced disease and a limited life expectancy. Based on the strategies that were originally developed for cytotoxic chemotherapies, dose selection for the clinical development of targeted agents and cancer immunotherapies are typically based on the MTD, which is commonly identified in phase I studies and oftentimes with a limited knowledge of long‐term tolerability. For traditional oncology molecules (e.g., chemotherapy, targeted agents), it is also common for the recommended phase II dose (RP2D) to also be the MTD. The selection of the MTD as the RP2D for cancer therapeutics is commonly based on the assumption that greater efficacy is associated with a higher dose; however, this approach has multiple limitations.37 For example, identification of the MTD from early clinical studies (e.g., phase I) is commonly confounded by the study designs typically employed. Phase I studies are commonly small, with large interindividual variability (due to the large diversity of tumor types and disease burden), and individualized treatment regimens. Phase II studies, while larger than phase I studies, with only select tumor types, do not typically explore dose levels lower than the MTD or RP2D identified from the phase I study/studies that could help to better define dose–response (PK/PD) relationships. Despite its limitations, this approach has long been successfully employed for single‐agent development, but it may not be suitable for immunotherapies developed in combination.
For cancer immunotherapies, the MTD‐based approach to identify the RP2D is particularly challenging. The assumption of a monotonic increase in efficacy with increasing dose may not be appropriate for immunotherapies that require a balance of a boosting of the immune system to combat cancer while avoiding overstimulation.38 In addition, the identification of an MTD for immunotherapies may not be achievable. For example, in single‐agent phase I studies with nivolumab39, 40 (up to 10 mg/kg), pembrolizumab (up to 10 mg/kg),41 and ipilimumab42 (up to 20 mg/kg) an MTD was not identified. An ipilimumab dose of 10 mg/kg was used in subsequent clinical trials based on preclinical data that suggested the concentrations achieved at this dose provided a maximal effect; ultimately, a dose of 3 mg/kg was approved. The inability to identify an MTD is also commonly the case with cancer vaccines due to their flat dose–toxicity curve.33 A single MTD was identified for the combination of nivolumab and ipilimumab, but only four combinations of doses were tested, and it is likely that multiple MTDs exist for combination therapies. For example, in a phase I study of neratinib and temsirolimus (nonimmunotherapy cancer agents), 12 dose combinations were explored and the combinations of 200 mg neratinib with 25 mg temsirolimus and 160 mg neratinib with 50 mg temsirolimus were identified as MTDs.
Clearly, the difficulty in either establishing an MTD for an immunotherapy administered as a single agent or establishing a single MTD for combination therapies complicates the selection of the RP2D dose, and prevents the historically straightforward approach of selecting the MTD as the RP2D. In cases such as this, the RP2D should be based on a benefit/risk assessment and comprehensive exploration of the surface contour describing the relationship between exposure, safety and tolerability, and response; such an assessment has always been the basis for selecting the RP2D dose, it has simply been a relatively straightforward assessment when an MTD is easily identified. Indeed, because the benefit/risk comparison at different doses levels during clinical development of ipilimumab was inadequately characterized, the FDA issued a postmarketing requirement to compare the efficacy at the approved dose (3 mg/kg, Q3W) to efficacy at a higher dose (10 mg/kg, Q3W) for patients with unresectable stage III or stage IV melanoma. The sheer number of “tunable” variables in a combination regimen, including dose level, administration frequency, the length of the dosing holiday, the duration of treatment for each dose, and the sequence of administration for each drug, and the inability to explore all possible combinations of these variables in clinical trials, complicates the situation further for combination immunotherapies. To overcome these challenges, novel trial designs that explore the dose–response surface for combination immunotherapies can be implemented and complemented by model‐based analyses to better understand the therapeutic window of these combinations.
The use of patient‐reported outcomes (PRO) to inform dose selection are also recommended in early clinical development, given the tolerability and adherence issues associated with these drugs, although PROs are rarely included in the labeling of oncology drugs in the US (although they are more common in Europe and for nononcology drugs in the US).43 While patient tolerance for side effects of anticancer drugs may be higher due to the severity of the disease, many patients discontinue cancer therapy as a result of adverse events, and this issue may be more prevalent for immunotherapies as the duration of treatment and response to these molecules is prolonged. For example, in clinical trials 9% of patients on nivolumab discontinue treatment due to adverse events, 26% experience a dose delay, mainly due to adverse events, and 42% experience a serious adverse reaction,44 while discontinuation rates (due to adverse events) up to 50% have been reported for the highest approved dose of ipilimumab of 10 mg/kg.45 Approximately 50% of patients receiving a combination of nivolumab and ipilimumab discontinued treatment due to adverse events.46 Adherence to the dosing regimen may also negatively impact efficacy and is commonly a result of adverse events, with adherence rates as low as 52% reported for anticancer therapeutics.47
Application of modeling and simulation to inform clinical development
To identify the optimal dose and schedule of a cancer immunotherapy, it is advantageous to apply a more strategic approach that integrates multiple data sources throughout various stages of development (Figure 4). Specifically, a quantitative, model‐based approach that integrates exposure–response (i.e., biomarker and/or efficacy) analyses with exposure–safety analyses may provide useful information on the benefit/risk profile of a drug candidate and inform dose selection. In early development, exposure–response analyses typically involve the assessment of tumor growth inhibition, with specific guidelines for assessment outlined in the Response Evaluation Criteria in Solid Tumors (RECIST) and the Immune‐Related Response Criteria (irRC).48 The predictive value of tumor assessments with respect to overall survival has been established for traditional cytotoxic drugs, and data from the development of nivolumab and ipilimumab suggest that there is also a relationship between tumor size assessments in early development and overall survival for immunotherapies.23 It may also be of value to assess other biomarkers of response that leverage the effects of immunotherapies on the immune system, such as cytokine elevation and markers of T‐cell activation.
Figure 4.
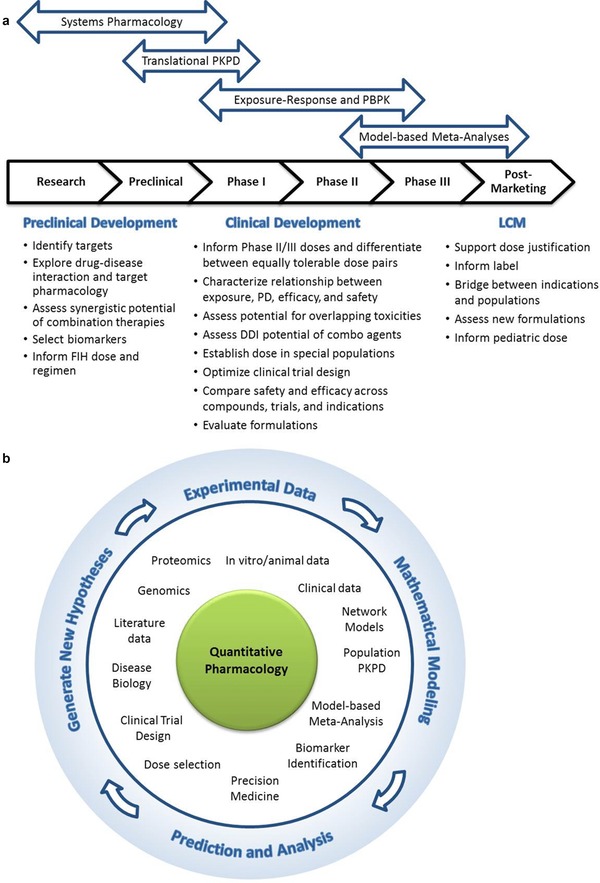
The role of modeling and simulation in combination immunotherapy development. (a) Modeling and simulation approaches used to answer questions during preclinical and clinical development and life‐cycle management. (b) Iterative cycle between experimentation, modeling, prediction, analysis, and the generation of new, testable hypotheses. LCM, life‐cycle management; FIH, first‐in‐human; PD, pharmacodynamics; PKPD, pharmacokinetics/pharmacodynamics; PBPK model, physiologically based pharmacokinetic model; DDI, drug–drug interaction.
The application of exposure–response analyses may have a significant impact on development and approval. In a phase I study of nivolumab in multiple solid tumor indications, for example, the same dose escalation scheme was used for each indication, but different doses were chosen for the expansion phases.40 However, exposure–response modeling, in which the relationship between dose/exposure and receptor occupancy, adverse events leading to discontinuation, objective response rate, progression‐free survival, and tumor growth dynamics was assessed, supported the selection of a common dose of nivolumab for melanoma, non‐smallcell lung cancer (NSCLC), and renal cell carcinoma, with no postmarketing commitment to evaluate additional dose levels.49 Exposure–response modeling also supported the selection of the dose for pembrolizumab; doses of 2 mg/kg Q3W or 10 mg/kg Q3W were studied and the 2 mg/kg Q3W was approved on the basis of the exposure–response analyses, which showed that the higher dose level was not associated with additional treatment benefit.
Systems pharmacology approaches can also elucidate the underlying mechanisms of cancer immunotherapies,50 and provide useful information on the schedule and sequencing of administration of various agents within a combination. Novel clinical trial designs complement these approaches, as they allow for the collection of the necessary data to inform them, which is otherwise too sparse to allow these approaches to be applied effectively. The success of these approaches, particularly early in development, also depends on the identification of biomarkers of efficacy and safety signals (see Biomarkers section below).
The lessons from ipilimumab are also informative and speak to the value of quantitative modeling. A pooled exposure–response analysis including almost 500 patients from four studies showed exposure‐dependent efficacy and safety at doses of 3 mg/kg (approved dose for unresectable or metastatic melanoma) and 10 mg/kg51 The approved dose of ipilimumab in unresectable or metastatic melanoma (3 mg/kg) is currently being compared with the 10 mg/kg dose in a phase III postmarketing requirement (PMR) clinical trial to better characterize the risk/benefit profile, given the model predictions of increased efficacy and adverse events with higher doses.51, 52 The activity of pembrolizumab was unaffected by dose or schedule in randomized trials,53 and exposure–response analysis also provided support for the approved dose of 2 mg/kg Q3W.49, 54
Quantitative systems pharmacology (QSP) modeling offers another promising approach to elucidate the complex biology of combination immunotherapies.55, 56 Translating the clinical experience with single‐agent immunotherapies into phase I, phase II, and phase III combination doses is not straightforward, particularly if the clinical trials are not designed to support exposure–response analyses. Quantitative systems pharmacology takes a network‐centric, holistic view of biology, and by quantitatively describing the pathophysiology of disease has the potential to help address these needs.
Since QSP models capture a drug's mechanism of action, they can help identify and prioritize targets, explore biomarkers of response, and identify potential characteristics by which to stratify patients. Mechanistic QSP models can be interrogated to assess mechanisms of tumor immunosuppression and means to circumvent them and the effect of disease severity and progression on treatment outcomes. Such models are particularly useful in combination immunotherapy development. Unlike traditional exposure–response analyses, which are top‐down approaches dependent on the availability of sufficient clinical data, these bottom‐up and middle‐out approaches could be used for prior predictions of synergistic interactions and their effect on efficacy and safety.
PBPK modeling is another quantitative, mechanistic approach that has proven useful in the development of immunotherapies, as demonstrated by its application to blinatumomab development. Drug–drug interactions (DDIs) are not common for biologics, but immunotherapies are a special case due to their potential impact on cytokine‐mediated changes in cytochrome P450 activity (see “Clinical Pharmacology Considerations,” below).57 Indeed, transient elevations in cytokine levels are observed after blinatumomab administration. In this case, the potential for cytokine‐mediated DDI was predicted based on data from in vitro hepatocytes incubated with blinatumomab or cytokines and the clinical cytokine profiles.58 The model predicted little potential for DDI and, as a result of this prediction, no clinical DDI studies were conducted.
Model‐based meta‐analyses are another tool to compare treatments without head‐to‐head clinical trials, link biomarker response to clinical efficacy, and bridge across populations and indications.59 This approach leverages both internal and external data and complements the exposure–response modeling, quantitative systems pharmacology, and PBPK modeling approaches. All have the potential to inform and even accelerate the development of immunotherapies, but successful implementation is not without its obstacles. Without prospective planning to include modeling in the clinical development plan, the data available to inform these models may be insufficient. Such challenges are magnified in the context of combination immunotherapy development, where uncertainties are magnified and therefore require even more data to develop reliable models.
Novel clinical trial design to support model‐based dose selection for combination regimens
A modified 3+3 approach to trial design is frequently employed for combination studies, in which one agent is administered at its approved dose and schedule and the dose of the other agent(s) is/are gradually increased until a combination MTD is found. The phase I study of nivolumab (Dose B) and ipilimumab (Dose A) used the traditional 3+3 design (as outlined in Figure 3, with the addition of a cohort treated at a lower dose of ipilimumab),60 and the combination of nivolumab with lirilumab also used this scheme, studying increasing doses of lirilumab combined with 3 mg/kg nivolumab.61 However, this approach offers only a narrow exploration of the dose–toxicity contour (Figure 3), even when variations on this study design are employed. It is also challenging to know which agent should be considered the main driver of the response and thus would be appropriate to be dosed at its MTD, if established, or the approved dose and schedule, an even more challenging task for bispecific antibodies in which the dose titration of the individual pharmacophores cannot be carried out independently.
Figure 3.
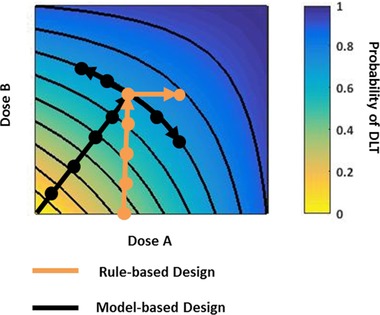
Dose–response contour for rule‐based and model‐based clinical trial designs. Doses explored relative to the dose–toxicity contour using a traditional, rule‐based method vs. a model‐based design. DLT, dose‐limiting toxicity. Trials based on rule‐based designs typically maintain the dose of one agent as a constant while escalating the dose of the other agent, resulting in a narrow exploration of the dose–toxicity contour. Trials utilizing model‐based designs may vary the doses of both agents to more comprehensively explore the dose–toxicity contour.
Rather, alternative rule‐based strategies, such has the 3+3+3 design, the accelerated titration design, and nonparametric up‐and‐down designs, and model‐based approaches (reviewed by Le Tourneau et al.62) may provide value by expanding the characterization of the dose–toxicity contour (Figure 3).63 Moreover, through the identification of multiple MTDs of the combination and integrating that data with the exposure–response data, an RP2D with a greater likelihood of achieving maximum efficacy with acceptable safety may be determined. Simulation studies suggest that model‐based trial designs may also treat more patients at optimal or near‐optimal doses and expose fewer patients to subtherapeutic doses. Although criticized for potentially long trial durations, simulation studies suggest that trials employing the CRM method may be of comparable length to trials employing traditional designs.
However, model‐based designs are almost never used.62 This is likely due to the difficulty of implementing such designs, with their dependence on reliable biomarkers, real‐time modeling and sample assessments, the lack of familiarity with the designs, and the potential for regulatory hurdles when employing such designs. The rule‐based, nonparametric up‐and‐down trial design employed to study the combination of neratinib and temsirolimus represents a reasonable compromise between fully adaptive, model‐based designs and the traditional designs like the 3+3 (for specific details on the trial design, refer to references 64 and 65).64, 65 It was straightforward to operationalize, but still offered superior performance and exploration of many more dose levels than would typically be studied.64
Patient selection
One of the promises of immunotherapies lies in reinitiating a self‐sustaining cycle of cancer immunity that results in potentially deep and durable clinical response.4 Favorable efficacy was observed in patients treated with checkpoint inhibitors, including ipilimumab, where ∼20% of melanoma patients appear to receive long‐term clinical benefits (with >10 years survival).66 For combinations of cancer immunotherapies, an important question is whether it is possible to further expand the “responder” population by combining checkpoint inhibitors with agents with a complementary mechanism of action in the cancer‐immunity cycle.4 As there is large variability in tumor biology across patients and tumor types, the strategy to develop combination therapy and patient selection needs to take into account the underlying variability in tumor biology. A rational patient selection strategy for cancer drug development will rely on a thorough understanding and characterization of tumor biology and cancer‐immunity interactions. While clinical trials for traditional cytotoxic agents typically include a heterogeneous patient population, patient stratification on the basis of such factors as tumor type, target expression on tumor cells and T cells, and tumor mutational load may increase the probability of success for combination immunotherapies.
It is hypothesized that the major barriers to therapeutic efficacy differ for “inflamed” and “noninflamed” tumors, and thus the patient stratification strategy should also consider the condition of the tumor microenvironment. In general, tumors can be considered as “inflamed” or “noninflamed” based on the level of preexisting immune response in the tumor microenvironment.17 For inflamed tumors, the cancer‐immunity cycle is likely intact up until the point of tumor cell killing by T cells, which can be subdued by PD‐L1:PD‐1 interaction. aPD‐1/aPD‐L1 treatments (e.g., nivolumab, pembrolizumab, atezolizumab) can restore the effector function of preexisting anticancer T cells rapidly, and thus, preexisting immune response has been shown to be desirable for efficacy. Similarly, the expression of PD‐L1 on immune and tumor cells has shown to be correlated with efficacy and thus was used as a patient selection strategy for aPD‐1/PD‐L1 treatments in multiple tumor types.67, 68, 69 Recently, pembrolizumab was approved for use with a companion diagnostic, the PD‐L1 IHC 22C3 pharmDx test, the first test designed to detect PD‐L1 expression in non‐smallcell lung tumors.70 In contrast, antitumor responses were observed in patients treated with the combination therapy of ipilimumab and nivolumab regardless of tumor cell expression of PD‐L1 at baseline.46, 60 Similarly, the combination of tremelimumab (aCTLA‐4) and durvalumab (aPD‐L1) shows clinical activity regardless of PD‐L1 status at baseline.71 These data suggest that it may not be appropriate to use baseline PD‐L1 expression as a selection criteria for all cancer immunotherapies, particularly in combinations with multiple modes of actions. While the mechanism for the combination effect remains an active area of research, it is hypothesized that aCTLA‐4 treatment may drive T cells into the tumor microenvironment, leading to a more favorable microenvironment for aPD‐1 efficacy.72 It is also possible that PD‐L1 expression on tumor‐infiltrating immune cells may be of relevance in addition to expression on tumor cells.69
In contrast, noninflamed tumors represent microenvironments that lack a preexisting immune response. In these cases, additional interventions are likely required to enable immune recognition and T‐cell infiltration.17 While the underlying mechanisms responsible for the lack of T‐cell infiltration are not well understood, combination strategies aim to increase the frequency of antitumor T cells by various approaches including tumor vaccine, adoptive T‐cell transfer, and augmenting the activity of costimulating pathways.17 The FDA and EMA recently approved the first‐of‐its‐kind oncolytic virus, Imlygic, for the local treatment of unresectable melanoma lesions, which highlights the clinical significance of inducing immune inflammation within the tumor microenvironment.73, 74 The importance of tumor mutational heterogeneity in cancer is an active topic of research and may play a significant role in patient selection for combination therapies.75 Tumor mutational load has been shown to correlate with clinical response to checkpoint blockade in human cancers.76 Research by Snyder et al. suggests that somatic neoepitopes were associated with a prolonged benefit in responders to aCTLA4 treatment in melanoma.77
From a practical perspective of the clinical development of combination therapies, it is useful to think about whether the combination therapy is expected to provide the most benefit in responders vs. nonresponders to prior immunotherapy, or in diagnostic‐positive vs. diagnostic‐negative populations. For combinations with immune checkpoint inhibitors, prior‐responders to checkpoint inhibitors or patients with high PD‐L1 expression may represent a population with a generally intact cancer‐immunity cycle, and thus, in this patient population, there may be a reasonable probability of success in identifying an effective combination therapy that aims to address the potential resistance mechanisms. In contrast, as there is less known about the underlying mechanisms that account for nonresponders to immune checkpoint inhibitors, it may be rather difficult to identify effective combination therapies that can potentially overcome multiple suppression barriers in the cancer‐immunity cycle. However, as multiple cancer immunotherapies are now approved therapies, it becomes more difficult to demonstrate superior efficacy in randomized studies where the comparator has favorable efficacy. As such, there may be an interest to target a nonresponder to prior immunotherapy or PD‐L1‐low expresser population as a potentially attractive development option, especially if accelerated approval (FDA), conditional marketing authorization (EMA), or SAKIGAKE “pioneer/forerunner” (PMDA) is a potential development strategy of interest.
Biomarkers
Biomarker data can be useful in guiding dose and regimen selection in early clinical development. For nivolumab, peripheral blood receptor occupancy and its associated time course of PD‐1 on circulating CD3+ T cells were evaluated as potential PD markers and compared across multiple dosing regimens in refractory solid tumors.39 It was inferred from the data that shorter dosing intervals might increase occupancy and tissue penetration and should be explored in later development. For blinatumomab, peripheral cytokine levels were used as a PD marker for acute safety assessment and contributed to the stepwise dosing recommendation for blinatumomab, in which the dose is increased after the first week of treatment to reduce the potential for acute cytokine release in patients with non‐Hodgkin's lymphoma.78 In addition, the exposure–response relationship for peripheral B‐cell depletion was modeled and compared with that for tumor size change. The difference in the two exposure–response curves provided potential insight into translating peripheral B‐cell response to tumor response, including the accessibility of tumor to the drug. As mentioned earlier, for pembrolizumab higher baseline tumor expression of PD‐L1 has been associated with better efficacy in multiple tumor types and, as a result, a companion diagnostic tool was recently approved for pembrolizumab in 2L NSCLC.67, 68, 69, 70 Therefore, it is useful to consider including baseline PD‐L1 expression status as a covariate in the exposure–response analyses, as appropriate.
Similar principles of biomarker‐informed dose selection could be applicable to combination drug development, with an added component that accounts for the interplay between the combined therapeutic agents. For example, combined blockade of PD‐1:PD‐L1 with other coinhibitors, such as TIM‐3, CTLA‐4, and LAG‐3, has a synergistic effect in reversing T‐cell exhaustion and restoring CD8+ effector function.79 Engagement of costimulatory receptors such as OX40, CD137, or CD27 can affect T‐cell activation, proliferation, migration, and development of memory T cells.80 Thus, the “combined effects” and associated biomarkers of therapeutic agents will require a deep understanding of the underlying biology of the mechanism of action of each therapy and their respective role in the cancer‐immunity cycle.
CLINICAL PHARMACOLOGY CONSIDERATIONS
The majority of approved immunotherapies are biologics (e.g., mAbs, vaccines, viruses) or cellular‐based therapies (Table 3). For biologic cancer immunotherapies, the clinical pharmacology considerations are consistent with other types of biologic therapeutics (for review, see Zhao et al.).81 Similarly, for small molecule immunotherapies, the clinical pharmacology characteristics and assessments follow the traditional small molecule development paradigms for other therapeutic areas.82 For vaccines and cell‐based therapies, there is currently limited knowledge of their clinical pharmacology properties. Depending on the construct and the drug delivery vehicle, the absorption, distribution, metabolism, and elimination (ADME) properties can differ widely between different cancer vaccines.83
Dedicated studies to evaluate the impact of renal or hepatic impairment on the pharmacokinetics of a biologic are not typically required, nor are they typically conducted for biologic immunotherapies. However, population PK analyses are sometimes used to support the lack of an impact of hepatic/renal impairment on the PK of a cancer immunotherapy biologic. For example, population PK analyses for ipilimumab evaluated the impact of mild/moderate renal impairment, baseline aspartate transaminase (AST), total bilirubin, and alanine transaminase (ALT) levels on the ipilimumab PK and none of these variables were demonstrated to have a clinically significant impact on ipilimumab exposure.52
As with all biologics, cancer immunotherapy biologics also have the potential for immunogenicity, which may have an impact on their efficacy and safety. Briefly, the formation of antidrug antibodies (ADAs) can impact efficacy by altering the pharmacokinetics of the biologic by impacting clearance mechanisms and by targeting domains critical for efficacy. In addition, potential safety consequences are variable and unpredictable, and may include anaphylaxis, cytokine release syndrome, and/or crossreactivity to endogenous proteins.84 Assessments of ADAs should follow the normal paradigms for biologics.84 ADAs were assessed in clinical studies with blinatumomab and nivolumab in which no association was observed between the development of ADAs and adverse events.85, 86 Immunogenicity of the combination of nivolumab and ipilimumab was also assessed and included as a covariate in the population PK model, which suggested that ADAs of these therapies do not have a clinically meaningful impact on exposure.87, 88 It should be noted that these results are not generalizable and ADAs should be evaluated in the clinical development programs for each cancer immunotherapy biologic.
Drug–drug interaction (DDI) studies, which are not typically conducted for biologics, may be required for biologic immunotherapies due to the potential for cytokine‐mediated alterations in drug metabolism. It has been observed that cancer immunotherapies influence cytokine levels, and it has also been established that the activities of select cytochrome P450 enzymes (the major metabolic pathway for small molecules) are altered in the presence of cytokines.57 Therefore, there is a potential for a clinically relevant DDI to occur, and thus both the FDA and EMA recommend assessing the potential for a cytokine‐mediated DDI. Unfortunately, currently available in vitro and in vivo models are not reliable predictors of a cancer immunotherapy‐mediated DDI. Therefore, clinical evaluation of the DDI potential is the recommended approach by health authorities. Due to the long terminal half‐life of biologics, crossover study designs are challenging. In addition, due to toxicity concerns and the potential for immunogenicity, studies often need to be conducted in cancer patients. As an alternative to dedicated clinical DDI studies, several sponsors have used population PK approaches or PBPK modeling approaches to characterize the potential for their drug to be a victim of DDI and to support their product approval without conducting a dedicated drug–drug‐interaction study.88, 89 Through the collection of proinflammatory cytokine data in early clinical development of immuno‐oncology agents, the risk of a cytokine‐mediated DDI can be evaluated without the need for dedicated DDI studies.
SAFETY AND TOLERABILITY CONSIDERATIONS FOR CANCER IMMUNOTHERAPY COMBINATIONS
Although cancer immunotherapies offer significant antitumor benefits, they are associated with a unique side effect profile. Adverse events associated with immune checkpoint inhibitors (e.g., ipilimumab, nivolumab) are autoimmune in nature and are commonly referred to as “immune‐related adverse events” (irAE). In general, these irAEs of checkpoint inhibitors include rash, diarrhea, colitis, autoimmune hepatitis, arthritis, pneumonitis, and endocrinopathy.90 In contrast, the most common treatment‐emergent adverse events of CD19 CAR T‐cell therapy and the bispecific T‐cell‐engaging (BiTE) antibody, blinatumomab, are cytokine release syndrome and neurological toxicity.26, 86 However, with careful monitoring and early intervention, both the irAEs of checkpoint inhibitors and cytokine release syndrome and neurological toxicity of CD19 CAR T‐cell therapy and blinatumomab are treatable and reversible with established management guidelines.91, 92, 93
As combinations of cancer immunotherapeutics have the potential for enhanced efficacy, they also have the potential for increased toxicity. This was recently observed in randomized, double‐blind phase III study of combined nivolumab and ipilimumab or monotherapy in previously untreated metastatic melanoma.16 Briefly, grade 3/4 treatment‐related adverse events (trAEs) were observed in 16% and 27% of patients treated with nivolumab or ipilimumab monotherapy, respectively. However, when treated in combination, grade 3/4 trAEs were observed in 55% of patients. These trAEs lead to treatment discontinuation in 9%, 15% and 38% of patients treated with nivolumab alone, ipilimumab alone, and nivolumab/ipilimumab combination, respectively. While the rate and severity of the trAEs increased with combination treatment relative to each monotherapy, there were no new safety signals and the AEs appeared to be immune‐related and manageable with established treatment guidelines.
The potential for increased toxicity was also observed with combinations between a cancer immunotherapy and a targeted therapy, as was the case with the combination of ipilimumab and the BRAF inhibitor, vemurafenib, two agents approved for the treatment of advanced melanoma. This combination was supported by nonclinical data suggesting that BRAF inhibitors may enhance immune‐cell function and antigen presentation and clinical data suggesting minimal overlapping single‐agent toxicities of these two therapies.94 However, in a phase I study evaluating the concurrent administration of vemurafenib and ipilimumab, dose‐limiting hepatotoxicity was observed, halting further enrollment.94 The results of this study highlight the importance of optimization of combination regimens of cancer immunotherapy and molecularly targeted agents, despite their distinct mechanism of action and individual safety profiles.
While it is more common for AEs to increase with cancer immunotherapy combinations, there are some examples where the distribution of toxicities changes when a checkpoint inhibitor is combined with chemotherapy. With ipilimumab monotherapy, the rate of treatment‐related diarrhea and colitis (≥grade 3) was ∼8–23%.95 However, when ipilimumab was dosed in combination with dacarbazine or fortumustine, the rate of ≥grade 3 diarrhea/colitis was reduced to 4% and 5%, respectively.96, 97 In contrast, the rate of autoimmune hepatitis increased with each combination, with ∼20% and 14% of patients experiencing a grade 3 or 4 increase in ALT and/or AST with ipilimumab plus dacarbazine or fortumustine treatment, respectively, compared with the typical rate of ipilimumab monotherapy of 3–7%.95, 96, 97
Currently, the majority of experience with cancer immunotherapy combinations has been limited to combinations with ipilimumab. However, recent data suggest that combinations with PD‐1/PDL‐1 immune checkpoint inhibitors may be more tolerable. Epacadostat (INCB024360), a small molecule inhibitor of the immune modulator indoleamine 2,3‐dioxygenase I, is being investigated in multiple phase I studies in combination with ipilimumab, pembrolizumab, durvalumab, or atezolizumab (NCT01604889, NCT02178722, NCT02318277, NCT02298153, respectively). As a single agent, epacadostat was generally well tolerated at doses up to 700 mg b.i.d., with no single‐agent MTD identified.98 An ongoing phase I/II study evaluating epacadostat in combination with ipilimumab showed a favorable response rate; however, ≥grade 3 irAEs occurred in 23% of patients evaluated at doses from 25 mg b.i.d. up to the combination MTD of 50 mg b.i.d. epacadostat.99 In combination with pembrolizumab, there is also an encouraging response rate; however, the toxicity profile is more favorable than the ipilimumab combination, with grade 3 trAEs observed in 11% of patients at doses from 25 mg to 300 mg b.i.d. epacadostat.100
The toxicity profile of immuno‐oncology combinations is just beginning to be understood. From the limited experience thus far, trAEs appear to increase in rate and severity with cancer immunotherapy combinations; however, depending on the combination partner, alterations in the toxicity profile are also possible. Therefore, when combining an immunotherapy with another treatment modality, it is important to determine the optimal dose, schedule, and sequence. In addition, the patient selection strategy should take into account the individual patient's performance status and the overall benefit/risk ratio of the various therapies for the patient, particularly in patients with preexisting autoimmune diseases.
REGULATORY CONSIDERATIONS AND MEANS TO ACCELERATE APPROVAL
Regulatory agencies worldwide have shown significant interest in cancer immunotherapies through the recent approvals of multiple cancer immunotherapies including mAbs administered as monotherapies (e.g., nivolumab, pembrolizumab, ipilimumab) or in combination (e.g., nivolumab/ipilimumab), novel antibody platforms (e.g., blinatumomab), and oncolytic viruses (e.g., T‐Vec) (Table 2). For cancer immunotherapies and other medicines that have the potential to demonstrate substantial improvement over existing therapies for serious illnesses including cancer, the FDA, EMA, and PMDA have each developed programs to support their expedited clinical development and approval. These programs include Fast Track Designation, Breakthrough Designation, Accelerated Approval, and Priority Review through the FDA, Accelerated Assessment, Conditional Marketing Authorization and Priority Medicines (PRIME) through the EMA, and SAKIGAKE “pioneer/forerunner” designation through PMDA.
One of the biggest challenges in obtaining expedited regulatory approval of cancer immunotherapies is the selection of a clinical trial end point that adequately reflects clinical benefit. While an improvement in OS remains the gold standard clinical trial end point for regulatory approval, this end point takes significant time to mature, particularly in the first‐line setting. Thus, surrogate clinical end points that aim to predict OS have been developed and used to obtain early drug approval without waiting to reach an OS end point.101, 102 These surrogate end points include PFS, objective response rate (ORR), and duration of response (DoR). Currently, there is no clear guidance or consensus on which of these surrogate end points is the best early predictor of long‐term survival, and it is highly dependent on tumor type. Notably, the accelerated approvals of both pembrolizumab in PD‐L1+ advanced NCSLC and the combination of nivolumab/ipilimumab in BRAF wildtype unresectable or metastatic melanoma were granted by the FDA based on an ORR end point.85, 103 However, for ipilimumab, the improvement in OS would not have been adequately captured if relying solely on a PFS or ORR end point, since responses to ipilimumab may be delayed and may even occur after apparent disease progression.104
Traditionally, RECIST or modified WHO criteria are used to evaluate antitumor responses. However, these criteria were developed for chemotherapies and may not accurately capture the unique response kinetics of patients treated with immunotherapy, particularly in cases where durable responses are observed postprogression. Therefore, to better capture the unique response kinetics of immunotherapies, Wolchock et al. developed the “Immune‐related Response Criteria” (irRC);48 however, prospective data are required to determine the value of irRC in predicting OS. While OS is likely to remain the primary end point for full approval, new (or modified) surrogate end points that accurately predict OS are needed to facilitate the expedited development of cancer immunotherapies and combinations.
CONCLUSION
The role of the immune system in combating cancer has been recognized for over a century, but immunotherapies are only now coming to fruition, with the recent approvals of the checkpoint inhibitors nivolumab, ipilimumab, and pembrolizumab, the BiTE antibody construct blinatumomab, the oncolytic virus T‐Vec, and the cancer vaccine sipuleucel‐T. These approvals have paved the way for combination approaches of immunotherapies with another immunotherapy, molecularly targeted agents, traditional cytotoxic agents, and/or radiation therapy, which are expected to revolutionize cancer treatment. Already the promise of this approach has been realized with the FDA approval of combination nivolumab and ipilimumab treatment, which leverages the synergistic potential of these two checkpoint inhibitors to achieve an improved response rate compared with monotherapy.
The success of combination immunotherapy is dependent on our ability to manage development hurdles including (i) the design of the right preclinical experiments and the translation of those experiments into the clinic, (ii) optimization of the dose and schedule of the combination regimen, and (iii) management of the overlapping toxicities that can be expected based on the mechanism of action of immunotherapies. The application of quantitative clinical pharmacology approaches in the translational space and throughout clinical development may help to address these challenges. In addition to these development hurdles and due to the growing field of cancer immunology, established guidelines from regulatory agencies to guide immunotherapy development and their combinations are not yet available. As the field continues to evolve, it is anticipated that the historical designation of immunotherapies as breakthrough therapies and the limited development programs required for approval are likely to change as the familiarity with and availability of immunotherapies grow. However, the likelihood that a more comprehensive development is needed for regulatory approval may be balanced by a clearer regulatory strategy for immunotherapies and combination therapies that will likely emerge as more immunotherapies enter the global market.
Combination immunotherapy is the future of cancer treatment and its success is dependent on addressing each of these challenges during development in order to provide the most beneficial treatment to patients.
Conflict of Interest
K.M.M. and C.C.L. are employees of Genentech and stock shareholders of Roche Holding. A.G., T.Y., Y.Z., and S.K. are employees and stock shareholders of Amgen Inc.
Author Contributions
The first two authors contributed equally to this work. T.Y., K.M., C‐C.L., Y.Z., and S.K. wrote the article. ©2016 American Society for Clin. Pharmacol. Ther.
- 1. Couzin‐Frankel, J. Cancer immunotherapy. Science 342, 1432–1433 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Bray, F. , Jemal, A. , Grey, N. , Ferlay, J. & Forman, D . Global cancer transitions according to the Human Development Index (2008–2030): a population‐based study. Lancet Oncol. 13, 790–801. [DOI] [PubMed] [Google Scholar]
- 3. Kim, R. , Emi, M. & Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 121, 1–14 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen, D.S. & Mellman, I. Oncology meets immunology: the cancer‐immunity cycle. Immunity 39, 1–10 (2013). [DOI] [PubMed] [Google Scholar]
- 5. Beatty, G.L. & Gladney, W.L . Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 21, 687–692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nobelprize.org Emil von Behring: The Founder of Serum Therapy. Nobel Media AB Web. 3 Dec 2015 (2014).
- 7. McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft‐tissue sarcomas. Iowa Orthopaed. J. 26, 154–158 (2006). [PMC free article] [PubMed] [Google Scholar]
- 8. Galluzzi, L. et al Classification of current anticancer immunotherapies. Oncotarget 5, 12472–12508 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nagorsen, D. & Baeuerle, P.A. Immunomodulatory therapy of cancer with T cell‐engaging BiTE antibody blinatumomab. Exp. Cell Res. 317, 1255–1260 (2011). [DOI] [PubMed] [Google Scholar]
- 11. Rosenberg, S.A. , Yang, J.C. & Restifo, N.P. Cancer immunotherapy: moving beyond current vaccines. Nat. Med. 10, 909–915 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosenberg, S.A. , Restifo, N.P. , Yang, J.C. , Morgan, R.A. & Dudley, M.E. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 8, 299–308 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schumacher, T.N. & Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 14. Adams, J.L. , Smothers, J. , Srinivasan, R. & Hoos, A . Big opportunities for small molecules in immuno‐oncology. Nat. Rev. Drug Discov. 14, 603–622 (2015). [DOI] [PubMed] [Google Scholar]
- 15. Escudier, B. et al Bevacizumab plus interferon alfa‐2a for treatment of metastatic renal cell carcinoma: a randomised, double‐blind phase III trial. Lancet 370, 2103–2111 (2007). [DOI] [PubMed] [Google Scholar]
- 16. Larkin, J. et al Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Spranger, S. & Gajewski, T. Rational combinations of immunotherapeutics that target discrete pathways. J. Immunother. Cancer 1, 16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. European Medicines Agency (EMA) . Committee for Medicinal Products for Human Use (CHMP), Guideline on Strategies to Identify and Mitigate Risks for First‐in‐Human Clinical Trials with Investigational Medicinal Products. (July 2007).
- 19. European Medicines Agency . Committee for Medicinal Products for Human Use (CHMP), Assessment Report: Opdivo (April 25, 2015).
- 20. European Medicines Agency . Committee for Medicinal Products for Human Use (CHMP), Assessment Report: Yervoy (ipilimumab). (May 19, 2011).
- 21. European Medicines Agency . Committee for Medicinal Products for Human Use (CHMP), Assessment Report: Keytruda. (May 21, 2015).
- 22. Dong, J.Q. et al Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first‐in‐human prediction. Clin. Pharmacokinet. 50, 131–142 (2011). [DOI] [PubMed] [Google Scholar]
- 23. Stroh, M. et al Challenges and opportunities for quantitative clinical pharmacology in cancer immunotherapy: something old, something new, something borrowed, and something blue. CPT Pharmacometrics Syst. Pharmacol. 4, 495–497 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gibbs, J.P. Prediction of exposure–response relationships to support first‐in‐human study design. AAPS J. 12, 750–758 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kochenderfer, J.N. et al B‐cell depletion and remissions of malignancy along with cytokine‐associated toxicity in a clinical trial of anti‐CD19 chimeric‐antigen‐receptor‐transduced T cells. Blood 119, 2709–2720 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grupp, S.A. et al Chimeric antigen receptor‐modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalos, M. et al T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science Tramsl. Med. 3, 95ra 73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use ICH Harmonized Tripartite Guideline: Nonclinical Evaluation for Anticancer Pharmaceuticals S9. (October 29, 2009).
- 29.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use ICH Harmonized Tripartite Guideline: Preclinical Safety Evaluation of Biotechnology‐Derived Pharmaceuticals S6 (R1). (July 16, 1997).
- 30.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use ICH Harmonized Tripartite Guideline: Guidance on Nonclincal Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals M3(R2). (June 11, 2009).
- 31.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research, Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products. (November 2013).
- 32.European Medicines Agency (EMEA) Note for Guidance on Nonclinical Evaluation for Anticancer Pharmaceuticals (December 2008).
- 33.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research (CBER), Guidance for Industry: Clinical Considerations for Therapeutic Cancer Vaccines (October 2011).
- 34. European Medicines Agency (EMA) . Oncology Working Group, Guideline on the evaluation of anticancer medicinal products in man. (December 13, 2012).
- 35.Ministry of Health Labour and Welfare Grant for Expedited Review of Drugs and Other Products Project for Enhanced Practical Application of Innovative Drugs, M.D., and Regenerative Medicine Products Guidance Development Review Committee 2015 Guidance on Cancer Immunotherapy Development Early‐Phase Clinical Studies ‐ For Development of Safe and Effective Immunotherapy (January 30, 2015).
- 36.FDA Codevelopment of Two or More New Investigational Drugs for Use in Combination. (June 2013).
- 37. Mick, R. & Ratain, M.J. Model‐guided determination of maximum tolerated dose in phase I clinical trials: evidence for increased precision. J. Natl. Cancer Inst. 85, 217–223 (1993). [DOI] [PubMed] [Google Scholar]
- 38. Mandrekar, S.J. , Qin, R. & Sargent, D.J. Model‐based phase I designs incorporating toxicity and efficacy for single and dual agent drug combinations: Methods and challenges. Stat. Med. 29, 1077–1083 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brahmer, J.R. et al Phase I study of single‐agent anti–programmed death‐1 (MDX‐1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 28, 3167–3175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Topalian, S.L. et al Safety, activity, and immune correlates of anti–PD‐1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Patnaik, A. et al Phase I study of pembrolizumab (MK‐3475; Anti–PD‐1 monoclonal antibody) in patients with advanced solid tumors. Clin. Cancer Res. 21, 4286–4293 (2015). [DOI] [PubMed] [Google Scholar]
- 42. Callahan, M.K. & Wolchok, J.D. At the bedside: CTLA‐4‐ and PD‐1‐blocking antibodies in cancer immunotherapy. J. Leuk. Biol. 94, 41–53 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Basch, E. et al Patient‐reported outcomes in cancer drug development and us regulatory review: Perspectives from industry, the food and drug administration, and the patient. JAMA Oncol. 1, 375–379 (2015). [DOI] [PubMed] [Google Scholar]
- 44. Information, O.U.S.P. Opdivo Prescribing Information . Princeton, NJ: Bristol‐Myers Squibb Company (Last updated: October 9, 2015).
- 45. Eggermont, A.M.M. et al Adjuvant ipilimumab vs. placebo after complete resection of high‐risk stage III melanoma (EORTC 18071): a randomised, double‐blind, phase 3 trial. Lancet Oncol. 16, 522–530 (2015). [DOI] [PubMed] [Google Scholar]
- 46. Postow, M.A. et al Nivolumab and ipilimumab vs. ipilimumab in untreated melanoma. N. Engl. J. Med. 372, 2006–2017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Puts, M.T.E. et al Factors influencing adherence to cancer treatment in older adults with cancer: a systematic review. Ann. Oncol. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wolchok, J.D. et al Guidelines for the evaluation of immune therapy activity in solid tumors: immune‐related response criteria. Clin. Cancer Res. 15, 7412–7420 (2009). [DOI] [PubMed] [Google Scholar]
- 49. Agrawal, S. , Feng, Y. , Roy, A. , Kollia, G.D. & Lestini, B.J. Nivolumab dose selection: challenges, opportunities and lessons learned for cancer immunotherapy. J. Immunother. Cancer 3, 141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singh, I. et al. A Systems Pharmacology Model to Characterize the Effect of Blinatumomab in Patients With Adult B Precursor Acute Lymphoblastic Leukemia. Poster presented at the ASCPT annual meeting, Atlanta, GA (March 2014).
- 51. Feng, Y. et al Exposure–response relationships of the efficacy and safety of ipilimumab in patients with advanced melanoma. Clin. Cancer Res. 19, 3977–3986 (2013). [DOI] [PubMed] [Google Scholar]
- 52. Feng, Y. et al Model‐based clinical pharmacology profiling of ipilimumab in patients with advanced melanoma. Br. J. Clin. Pharmacol. 78, 106–117 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Khoja, L. , Butler, M.O. , Kang, S.P. , Ebbinghaus, S. & Joshua, A.M. Pembrolizumab. J. Immunother. Cancer 3, 36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chatterjee, M. , Turner, D.C. , Ahamadi, M. , Alwis, Dinesh de , Elassaiss‐Schaap, J. , Freshwater, T. , de Greef, R. , Li, C. , Mayawala, K. , Stone, J. , Kondic, A. Model‐based analysis of the relationship between pembrolizumab exposure and efficacy in patients with melanoma and NSCLC: across indication comparison. PAGE 24 Abstr 3579 [www.page‐meeting.org/?abstract=3579] (2015). [Google Scholar]
- 55. Sorger, Peter K (co‐chair), S.R.B.A.c.‐c., Darrell R. Abernethy, R.B.A., Kim L. R. Brouwer, Andrea Califano, David Z., D'Argenio, R.I., William J. Jusko, Richard Lalonde, Douglas A. Lauffenburger,, Brian Shoichet, J.L.S., Shankar Subramaniam, Piet Van der Graaf and & Vicini, P. Quantitative and Systems Pharmacology in the Post‐genomic Era: New Approaches to Discovering Drugs and Understanding Therapeutic Mechanisms An NIH White Paper by the QSP Workshop Group (October, 2011).
- 56. Klinke, D.J. 2nd. Enhancing the discovery and development of immunotherapies for cancer using quantitative and systems pharmacology: Interleukin‐12 as a case study. J. Immunother. Cancer 3, 27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang, S.M. et al Therapeutic protein‐drug interactions and implications for drug development. Clin. Pharmacol. Ther. 87, 497–503 (2010). [DOI] [PubMed] [Google Scholar]
- 58. Xu, Y. et al Physiologically based pharmacokinetic model to assess the influence of blinatumomab‐mediated cytokine elevations on cytochrome P450 enzyme activity. CPT: Pharmacometrics Syst.Pharmacol. 4, 507–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mandema, J.W. , Gibbs, M. , Boyd, R.A. , Wada, D.R. & Pfister, M. Model‐based meta‐analysis for comparative efficacy and safety: application in drug development and beyond. Clin. Pharmacol. Ther. 90, 766–769 (2011). [DOI] [PubMed] [Google Scholar]
- 60. Wolchok, J.D. et al Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 369, 122–133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sanborn, R. et al A phase I dose‐escalation and cohort expansion study of lirilumab (anti‐KIR; BMS‐986015) administered in combination with nivolumab (anti‐PD‐1; BMS‐936558; ONO‐4538) in patients (Pts) with advanced refractory solid tumors. J. Clin. Oncol. 31 (2013). [Google Scholar]
- 62. Le Tourneau, C. , Lee, J.J. & Siu, L.L. Dose escalation methods in phase I cancer clinical trials. J. Natl. Cancer Inst. 101, 708–720 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Harrington, J.A. , Wheeler, G.M. , Sweeting, M.J. , Mander, A.P. & Jodrell, D.I. Adaptive designs for dual‐agent phase I dose‐escalation studies. Nat. Rev. Clin. Oncol. 10, 277–288 (2013). [DOI] [PubMed] [Google Scholar]
- 64. Gandhi, L. et al Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2‐dependent and other solid tumors. J. Clin. Oncol. 32, 68–75 (2014). [DOI] [PubMed] [Google Scholar]
- 65. Mandrekar, S.J. Dose‐finding trial designs for combination therapies in oncology. J. Clin. Oncol. 32, 65–67 (2014). [DOI] [PubMed] [Google Scholar]
- 66. Schadendorf, D. et al Pooled analysis of long‐term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 33, 1889–1894 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Garon, E.B. et al Pembrolizumab for the Treatment of Non–SmallCell Lung Cancer. N. Engl. J. Med. 372, 2018–2028 (2015). [DOI] [PubMed] [Google Scholar]
- 68. Cha, E. , Wallin, J. & Kowanetz, M. PD‐L1 inhibition with MPDL3280A for solid tumors. Semin. Oncol. 42, 484–487 (2015). [DOI] [PubMed] [Google Scholar]
- 69. Herbst, R.S. et al Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. FDA approves Keytruda for advanced non‐small cell lung cancer . FDA News Release http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm465444.htm (October 2, 2015).
- 71. Rizvi, N. , Chaft, J. , Balmanoukian, A. , Goldberg, S.B. , Sanborn, R.E. , Steele, K.E. , Rebelatto, M.C. , Gu, Y. , Karakunnel, J.J. , Antonia, S. Tumor response from durvalumab (MEDI4736) +tremelimumab treatment in patients with advanced non‐small cell lung cancer (NSCLC) is observed regardless of PD‐L1 status. J. Immunother. Cancer 3, P 193 (2015). [Google Scholar]
- 72. Hamid, O. et al A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Tramsl. Med. 9, 204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. FDA approves first‐of‐its‐kind product for the treatment of melanoma . FDA News Release (October 27, 2015).
- 74. Ledford, H. Cancer‐fighting viruses near market. Nature 526, 622–623 (2015). [DOI] [PubMed] [Google Scholar]
- 75. Lawrence, M.S. et al Mutational heterogeneity in cancer and the search for new cancer‐associated genes. Nature 499, 214–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rizvi, N.A. et al Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Snyder, A. et al Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hijazi, Y. et al Blinatumomab exposure and pharmacodynamic response in patients with non‐Hodgkin lymphoma (NHL). abstract #3051 (ASCO Annual Meeting 2013).
- 79. Chen, L. & Flies, D.B. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat. Rev. Immunol. 13, 227–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gao, J. , Bernatchez, C. , Sharma, P. , Radvanyi, L.G. & Hwu, P. Advances in the development of cancer immunotherapies. Trends Immunol. 34, 90–98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhao, L. , Ren, T.H. & Wang, D.D. Clinical pharmacology considerations in biologics development. Acta Pharmacol. Sin. 33, 1339–1347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pharmacokinetics in Drug Development: Regulatory and Development Paradigms, Vol. Vol 2. (American Association of Pharmaceutical Scientists, Bonate, P. and Howard, D. (eds) (2004)).
- 83. Kramps, T. & Probst, J. Messenger RNA‐based vaccines: progress, challenges, applications. Wiley Interdiscipl. Rev. RNA 4, 737–749 (2013). [DOI] [PubMed] [Google Scholar]
- 84. U.S. Department of Health and Human Services, Food and Drug Administration, & Center for Drug Evaluation and Research (CDER) . C.f.B.E.a.R.C. Guidance for Industry: Immunogenicity Assessment for Therapeutic Protein Products. (August 2014).
- 85.Opdivo(R) United States Prescribing Information [USPI]. (November 2015).
- 86.BLINCYTO(R) United States Prescribing Information [USPI]. (December 2014).
- 87. Statkevich, P. et al Assessment of the immunogenicity of nivolumab (nivo) and ipilimumab (ipi) in combination and potential impact on safety and efficacy in patients with advanced melanoma. Clin. Pharmacol. Ther. 99, 10.1002/cpt.1310 (2016). [Google Scholar]
- 88. Zhu, L. et al Model‐based assessment of drug‐drug interaction and immunogenicity on the pharmacokinetics (PK) of nivolumab and ipilimumab in combination in advanced melanoma patients. Clin. Pharmacol. Ther. 99, doi:10.1002/cpt1310 (2016). [Google Scholar]
- 89. Xu, Y. et al Physiologically based pharmacokinetic model to assess the influence of blinatumomab‐mediated cytokine elevations on cytochrome P450 enzyme activity. CPT Pharmacometrics Syst. Pharmacol. 4, 507–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Postow, M.A. , Callahan, M.K. & Wolchok, J.D . Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gangadhar, T.C. & Vonderheide, R.H. Mitigating the toxic effects of anticancer immunotherapy. Nat. Rev. Clin. Oncol. 11, 91–99 (2014). [DOI] [PubMed] [Google Scholar]
- 92. Villadolid, J. & Amin, A. Immune checkpoint inhibitors in clinical practice: update on management of immune‐related toxicities. Transl. Lung Cancer Res. 4, 560–575 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Maude, S.L. , Barrett, D. , Teachey, D.T. & Grupp, S.A . Managing cytokine release syndrome associated with novel T cell‐engaging therapies. Cancer J. 20, 119–122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ribas, A. , Hodi, F.S. , Callahan, M. , Konto, C. & Wolchok, J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med. 368, 1365–1366 (2013). [DOI] [PubMed] [Google Scholar]
- 95. Weber, J.S. , Kähler, K.C. & Hauschild, A . Management of immune‐related adverse events and kinetics of response with ipilimumab. J. Clin. Oncol. 30, 2691–2697 (2012). [DOI] [PubMed] [Google Scholar]
- 96. Robert, C. et al Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, 2517–2526 (2011). [DOI] [PubMed] [Google Scholar]
- 97. Di Giacomo, A.M. et al Ipilimumab and fotemustine in patients with advanced melanoma (NIBIT‐M1): an open‐label, single‐arm phase 2 trial. Lancet Oncol. 13, 879–886 (2012). [DOI] [PubMed] [Google Scholar]
- 98. Beatty GL et al Phase I study of the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of the oral inhibitor of indoleamine 2,3‐dioxygenase (IDO1) INCB024360 in patients (pts) with advanced malignancies. J. Clin. Oncol. 31, 3025 (2013). [Google Scholar]
- 99. Gibney, G. et al Updated results from a phase 1/2 study of epacadostat (INCB024360) in combination with ipilimumab in patients with metastatic melanoma. The European Cancer Congress Vienna, Austria (September 25, 2015).
- 100. Gangadhar, T.C. et al Preliminary results from a phase I/II study of epacadostat (incb024360) in combination with pembrolizumab in patients with selected advanced cancers. J. Immunother. Cancer 3 (2015). [Google Scholar]
- 101. U.S. Department of Health and Human Services, Food and Drug Administration , Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) Guidance for Industry: Biologics Clinical Trial End points for the Approval of Cancer Drugs and Biologics. (May 2007).
- 102.European Medicines Agency Guideline on the evaluation of anticancer medicinal products in man. (December 2012).
- 103.Keytruda(R) United States Prescribing Information [USPI], . (October 2015).
- 104. Hodi, F.S. et al Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]