

Abstract
Although dozens of common variants have been associated with increased risk of type 2 diabetes (T2D), the mechanisms by which these variants increase disease susceptibility are largely unknown. A new study mapping the human pancreatic islet cistrome provides a roadmap for exploring the effects of these variants and suggests that altered enhancer function might be a common contributor to the genetic risk of T2D.
T2D affects over 300 million people worldwide, and its incidence is predicted to double by 2035. Although well-known behavioral factors such as overeating and a sedentary lifestyle are major contributors to the diabetes epidemic, a heritable component has long been recognized to contribute to an individual’s risk of becoming diabetic. Large-scale genetic mapping efforts over the past 10 years have identified more than 60 loci encompassing up to 500 genes as contributing to the disease, but the mechanisms by which these variants act are largely unknown1–3. On page 136 of this issue, Jorge Ferrer and colleagues4 have laid the groundwork for an improved molecular understanding of these T2D-associated variants and of human islet biology in general. In a truly heroic effort combining the work and expertise of laboratories in five countries, the group mapped the locations of five key transcriptional regulators known to function in hormone-producing cells of the endocrine pancreas and integrated these data with multiple histone marks—indicating transcriptionally active and repressed areas in the genome—and measures of accessible or ‘open’ chromatin. The resulting human islet ‘cistrome’, a genome-wide map of chromatin features and transcription factor binding sites, represents a valuable resource that will yield new insights for many years to come.
Enhancer clusters
Detailed analysis of the rich experimental data compiled by Ferrer and colleagues uncovered several important general themes. First, although transcription factors were bound to the promoters of many genes expressed in human islets, this binding occurred regardless of whether a gene was expressed only in islets or in multiple tissues. Instead, islet-enriched gene expression correlated with transcription factor occupancy at distal enhancers, and these enhancers were often found to exist in clusters. Second, computational analyses of islet-specific enhancer clusters not only confirmed the presence of the DNA binding motifs recognized by well-known islet-expressed transcription factors such as FOXA2 and PDX1 but also identified several entirely new motifs, suggesting the existence of additional DNA-binding proteins yet to be discovered as regulators of gene expression in islets. Islet biologists will now have the opportunity to pursue the identification of these potentially new regulatory proteins. Third and perhaps most critical to our understanding of T2D genetics, many of the sequence variants known to be associated with risk of developing T2D are located in or near the aforementioned enhancer clusters.
Pinpointing functional variants
The discovery that islet enhancer clusters frequently harbor genetic variants found to be associated with T2D risk in genome-wide association studies has far-reaching implications. First, altered disease risk conferred by previously mapped ‘diabetes genes’ is only rarely, if ever, caused by altered protein function. Rather, altered gene expression, correlating with variation at distal enhancers, is likely to be a common mechanism explaining altered disease susceptibility (Fig. 1). Second, the extraordinary resource provided by Ferrer and colleagues will greatly facilitate the determination of the molecular mechanisms by which T2D risk loci affect islet function. Pasquali et al.4 have provided an example of this approach by showing that a T2D risk allele—present only in Asian but not in European populations—affects binding of the transcription factor NEUROD1 to an enhancer for the ZFAND3 gene, previously identified as a T2D risk locus through a meta-analysis of genome-wide association studies5. Thus, the islet chromatin and transcription factor maps are powerful tools for locating cis-regulatory variants likely to underlie altered disease risk.
Figure 1.
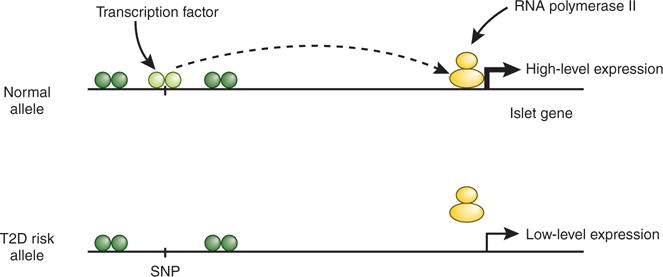
Model of how genetic variants in enhancers of islet-expressed genes contribute to islet cell dysfunction and increased risk of T2D. Top, an allele with an intact transcription factor binding site in an enhancer cluster far distal to the promoter of an islet-expressed gene. Interaction of the enhancer with the promoter leads to high levels of gene transcription. Bottom, a T2D risk allele that disrupts transcription factor binding in the enhancer cluster. The interaction of the enhancer with the promoter is weakened, and transcriptional activity is reduced. Over many decades, reduced levels of the corresponding protein contribute to islet dysfunction.
In the future, it will be interesting to determine whether and to what degree transcription factor occupancy and chromatin state are altered in endocrine cells from individuals with T2D. To this end, it might be necessary to perform analyses on material obtained from cell populations enriched for α and β cells, the two major endocrine cell types in pancreatic islets, which produce the glucose-raising hormone glucagon and the glucose-lowering hormone insulin, respectively. Although most researchers have focused on the role of relative insulin deficiency and impaired β cell function in the etiology of T2D, β cells make up only about 50% of endocrine cells in the islet6, and it is clear that glucagon-producing α cells are major contributors to the disease phenotype7. Given their shared developmental origin and related functions in stimulus-secretion coupling, it is not surprising that the transcriptomes and epigenomes of human α and β cells are quite similar8,9. Nevertheless, important difference also exist—in particular, the fact that many so-called ‘β cell genes’ exist in a poised chromatin state in α cells—suggesting that partial or full phenotypic interconversion of islet cell types, with the associated functional consequences, is possible and might occur in individuals with T2D8. In addition, it is important to remember that not all alterations in islet function are predetermined by risk-associated alleles. In particular, it is clear that the diabetic state itself has a major impact on the transcriptome of human pancreatic islets. For instance, a previous study found, through microarray expression analysis, that ARNT was misexpressed in diabetic islets10. More recently, it was discovered that an imprinted locus on chromosome 14, encoding the MEG3 long non-coding RNA and 54 microRNAs, was strongly downregulated in β cells from individuals with T2D11. Thus, the final phenotype of diseased β cells is determined both by germline predisposition, through the combined effects of dozens or hundreds of risk alleles, and by the changes to the epigenome and transcriptome that occur during an individual’s lifetime.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Bramswig NC, Kaestner KH. Diabetologia. 2013 Dec 21; doi: 10.1007/s00125-013-3150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris AP, et al. Nat Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voight BF, et al. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pasquali L, et al. Nat Genet. 2014;46:136–143. doi: 10.1038/ng.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho YS, et al. Nat Genet. 2012;44:67–72. [Google Scholar]
- 6.Brissova M, et al. J Histochem Cytochem. 2005;53:1087–1097. doi: 10.1369/jhc.5C6684.2005. [DOI] [PubMed] [Google Scholar]
- 7.Unger RH, Cherrington AD. J Clin Invest. 2012;122:4–12. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bramswig NC, et al. J Clin Invest. 2013;123:1275–1284. doi: 10.1172/JCI66514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorrell C, et al. Diabetologia. 2011;54:2832–2844. doi: 10.1007/s00125-011-2283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunton JE, et al. Cell. 2005;122:337–349. doi: 10.1016/j.cell.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 11.Kameswaran V, et al. Cell Metab. 2014;19:135–145. doi: 10.1016/j.cmet.2013.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]