

Abstract
Twenty strains (including eight phase variant pairs) of nematode-symbiotic and insect-pathogenic Photorhabdus bacteria were examined for the production of proteolytic enzymes by using a combination of several methods, including gelatin liquefaction, zymography coupled to native and sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and activity measurement with two chromogen substrate types. Four protease activities (∼74, ∼55, ∼54, and ∼37 kDa) could be separated. The N-terminal sequences of three of the proteases were determined, and a comparison with sequences in databases allowed identification of these proteases as HEXXH metallopeptidases. Thus, the 74-kDa protease (described formerly as Php-B [J. Marokházi, G. Kóczán, F. Hudecz, L. Gráf, A. Fodor, and I. Venekei, Biochem. J. 379:633-640, 2004) is an ortholog of OpdA, a member the thimet oligopeptidase family, and the 55-kDa protease is an ortholog of PrtA, a HEXXH+H peptidase in clan MB (metzincins), while the 37-kDa protease (Php-C) belongs to the HEXXH+E peptidases in clan MA. The 54-kDa protease (Php-D) is a nonmetalloenzyme. PrtA and Php-C were zymographically detected, and they occurred in several smaller forms as well. OpdA could not be detected by zymography. PrtA, Php-C, and Php-D were secreted proteases; OpdA, in contrast, was an intracellular enzyme. OpdA activity was found in every strain tested, while Php-D was detected only in the Brecon/1 strain. There was significant strain variation in the secretion of PrtA and Php-C activities, but reduced activity or a lack of activity was not specific to secondary-phase variants. The presence of PrtA, OpdA, and Php-C activities could be detected in the hemolymph of Galleria melonella larvae 20 to 40 h postinfection. These proteases appear not to be directly involved in the pathogenicity of Photorhabdus, since strains or phase variants lacking any of these proteases do not show reduced virulence when they are injected into G. melonella larvae.
Secreted microbial enzymes may function as virulence factors that are essential for survival and spread in the host through, e.g., neutralization of the host's defense system and ensuing tissue invasion. However, the roles of only few of these enzymes in the strategies of pathogens have been documented. For example, inhibitor A of Bacillus thuringiensis and the proteases of Serratia marcescens specifically cleave insect immune proteins, cecropins, and attacins (11, 15), and the zinc metalloproteases of Bacteroides fragilis and Clostridium spp. have direct toxin activity (19, 21, 27).
Two gram-negative entomopathogenic genera in the family Enterobacteriaceae, Photorhabdus and Xenorhabdus (4, 14, 26), which are known to be symbiotic partners of the insect-pathogenic nematodes Heterorhabditis and Steinernema, respectively, have been intensively studied in order to understand the molecular basis of symbiosis and insect toxicity and to improve the use of these symbiotic complexes against insect pests. With the exception of a dominant protease activity, no secreted enzyme of these microorganisms has been isolated so far. Although a Xenorhabdus nematophila protease was recently found to destroy antibacterial activity in insect hemolymph (7), the secreted enzymes of these microorganisms are generally assumed to have a role only in the decomposition of the insect cadaver.
In the early studies of enzyme secretion by Photorhabdus and Xenorhabdus strains, protease activity was assayed by nonspecific and relatively insensitive methods, which were based on substrate protein (e.g., gelatin) hydrolysis around colonies on agar plates. Later, the hydrolysis of stained proteins (azocoll, azocasein, Hide Powder Azure) as nonselective protease substrates was used for quantitative assays, and gelatin zymography was employed for sensitive detection of proteolytic activity following sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) analysis of proteins. Both one- and two-dimensional PAGE revealed one strong proteolytic activity, which occurred in various molecular sizes in the 55- to 61-kDa range (5, 10, 23, 25). The enzymes isolated from some primary-phase Photorhabdus strains (Photorhabdus luminescens subsp. luminescens Hm, P. luminescens subsp. laumondii HP88, and Photorhabdus temperata subsp. temperata K122) and X. nematophila proved to be alkaline metalloproteases (5-7, 32). The gene for the protease in P. luminescens W14 was found to encode an enzyme that belongs to the RTX (repeats-in-toxin) family and was designated PrtA after a similar protease in Erwinia chrysanthemi (6, 13).
Photorhabdus and Xenorhabdus strains may occur in two phase variant forms. Primary or natural forms are capable of establishing symbiosis with the nematode and are exclusively retained by the infective juvenile nematodes from a mixed bacterial population, while secondary forms occur with different frequencies in laboratory cultures and insect cadavers (2, 16). Comparative studies of the hydrolysis of various proteolytic substrates with a number of phase variant pairs generated the view that a reduced or missing activity is one of the features of secondary-phase variants (16, 17), although the data are rather contradictory. For 18 Xenorhabdus strains, the missing or reduced proteolytic activity was specific in only 10 of the secondary-phase variants (3). At the same time, of the four secondary-phase Photorhabdus strains (Hm, HP88, W14, and K122) that have been tested so far for protease production, only strain Hm was not found to secrete protease activity (1). Later, however, this strain and the other three secondary-phase variants were reported to produce at least some proteolytic activity (6, 23, 32).
A 60-kDa serine protease from X. nematophila has recently been shown to have an immune suppression effect (7). However, in the case of Photorhabdus strains the available data are insufficient to answer whether the secreted proteases might have a role other than bioconversion (e.g., a role in the establishment of infection) for the following reasons. (i) The number of secreted proteases is not known yet. The very similar properties of proteases detected and isolated from different Photorhabdus strains (5, 6, 10, 23, 25) indicate that these enzymes are probably the orthologs of each other and the enzyme PrtA, which was cloned from the W14 and K122 strains (6). However, it is very unlikely that a pathogenic microorganism secretes only one protease. Indeed, the genomes of the TT01 and W14 strains contain numerous sequences that could be identified as protease- or peptidase-encoding genes (12, 13). The fact that only one proteolytic enzyme has been identified might be due to the low sensitivity of detection, to the use of nonspecific substrates, and to the treatment of samples during preparation and analysis. Thus, proteases which are sensitive to SDS or cannot cleave casein or gelatin, the substrates that are most frequently used in zymography, might have escaped detection. (ii) In most of the cases the reported protease activities were detected in and purified from 5- to 30-day-old cultures, a time obviously too late to be significant in the establishment of infection.
The aim of this study was to explore protease production by Photorhabdus through a comparison of 20 strains (including eight phase variant pairs) by a combination of various detection techniques. We focused on the proteases that could be detected in the first 48 h of culture and infection and could, therefore, have a role in virulence.
MATERIALS AND METHODS
Strains and cell culturing.
The identities, taxonomic positions, and origins of strains used in this study are summarized in Table 1. The strains were obtained from the entomopathogenic nematode-bacterium strain collection maintained at the Department of Genetics, Eötvös Loránd University, Budapest, Hungary. Single colonies were used as the starting material, which was grown on Luria-Bertani (LB) medium plates for 48 h at 28°C and replica tested on NBTA plates (nutrient agar supplemented with 25 mg of bromothymol blue per liter and 40 mg of triphenyl-2,3,5-tetrazolium chloride per liter) to confirm the phase variation status. Liquid cultures were grown in LB medium without antibiotics at 30°C in a rotary shaker.
TABLE 1.
Photorhabdus strains used in this study
| Entomopathogenic symbiosis natural partners
|
Origin of nematode or bacteriumb,c
|
||||
|---|---|---|---|---|---|
| Bacterium
|
Nematode
|
||||
| Species or subspecies | Straina | Hetererhabditis species | Strain | Geographic area | Laboratory |
| P. luminescens subsp. laumondii | TT01/1c | H. bacteriophora | TT01 | Trinidad | N. Boemare |
| Brecon/1b | H. bacteriophora | Brecon | Australia | L. Gerritsen | |
| RH/1c | H. bacteriophora | RH | United States | R. Hurlbert | |
| RH/2c | Secondary-phase variant | R. Hurlbert | |||
| P. luminescens subsp. akhurstii | Indicusb | H. indicus | LN2 | India | A. Burnell |
| P. luminescens subsp. luminescens | Hm/1c | Unknown, lost | United States | N. Boemare | |
| Hm/2c | Secondary phase variant | N. Boemare | |||
| Hb/1c | H. bacteriophora | Hb | United States | N. Boemare | |
| Hb/2c | Secondary-phase variant | N. Boemare | |||
| P. temperata | NC1/1c | H. bacteriophora | NC1 | North Carolina | J. Ensign |
| NC1/2c | Secondary-phase variant | J. Ensign | |||
| P. temperata | WX6c | Unknown, lost | Wisconsin | K. H. Nealson | |
| WX6b | Secondary phase variant | K. H. Nealson | |||
| P. temperata | WX8/1c | Unknown, lost | Wisconsin | K. H. Nealson | |
| WX8/intd | Intermediate variant | B. D. Fodor | |||
| WX8/2d | Secondary-phase variant | B. D. Fodor | |||
| P. temperata subsp. temperata | HSH-2/1c | H. megidis NWE | HSH-2 | Germany | R.-U. Ehlers |
| HSH-2/2c | Secondary-phase variant | R.-U. Ehlers | |||
| K122/1c | H. downesii | K122 | Ireland | D. Clarke | |
| K122/2c | Secondary-phase variant | D. Clarke | |||
The numbers after the slashes indicate the phase variants. Only primary-phase variants are known for the TT01, Brecon, and Indicus strains.
Bacteria were isolated from the nematodes by Emilia Szállás in our laboratory by using the technique of A. Lucskai (unpublished data).
Bacterial strains were rechecked after their arrival and were then stored at −80°C.
Variant isolated in our laboratory by Barna Fodor (1996).
Infection experiments and hemolymph sample preparation.
Serial dilutions of overnight Photorhabdus cultures (Brecon/1, K122/1, K122/2, Hm/1, Hm/2, NC1/1, and NC1/2) were prepared with sterile phosphate-buffered saline (PBS) (137.9 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.5 mM KH2PO4) to obtain 30 to 60 cells in 5.0 μl. The actual numbers of cells were estimated from the number of colonies after plating 50-μl portions of the final dilutions onto LB agar plates. Five microliters of each final cell dilution was injected into 10 fifth-instar larvae of Galleria mellonella (greater wax moth, Lepidoptera), and the larvae were kept at 25°C. Death was checked hourly by observing reflexive movement upon prodding of the head, and the time of death was recorded. The experiment was performed with three replicates.
For detection of proteolytic activity in the hemolymph, 50 to 100 Brecon/1 cells were injected into larvae. Hemolymph samples were taken at various times postinjection (see below) from a leg and diluted immediately 10-fold with ice-cold PBS that contained 0.25 μg of phenylthiourea per ml. Then the cellular fraction was sedimented by centrifugation in an Eppendorf centrifuge at 16,000 × g for 20 min, and the supernatants were used for experiments.
Preparation of Galleria homogenates, culture supernatants, and bacterial cell lysates.
G. mellonella homogenates were prepared 20 and 40 h postinjection, as well as from healthy, uninfected larvae, by homogenizing chilled larvae (∼0.1 g each) in 1.0 ml of PBS that contained 0.25 μg of phenylthiourea per ml. The homogenates were centrifuged in an Eppendorf centrifuge at 16,000 × g for 20 min, and the supernatants were used for zymographic analysis of enzyme activity.
Culture supernatants and cell lysates were prepared from 1.0-ml cell suspensions. The cells were sedimented by centrifugation at 16,000 × g for 5 min. To prepare cell lysate, the cellular pellet was washed twice in 1.5 ml of LB medium and resuspended in 200 μl of 0.1 M Tris buffer (pH 8.0) containing 20% sucrose. After incubation on ice for 10 min, the suspension was centrifuged as described above, the supernatant was discarded, and the cells were resuspended in 1 ml of distilled water. Enzyme activity was analyzed after the cells were allowed to lyse on ice for 2 h.
Gelatin hydrolysis assay on plates.
For the gelatin hydrolysis assay on plates as described by Frazier (18), gelatin nutrient agar plates (12 g of gelatin per liter in nutrient agar) were spot inoculated with 1.0-μl cell suspensions from 24-h cultures (four spots for each strain). After 2 days of incubation at 28°C, the plates were flooded with 5.0 ml of mercuric chloride reagent (12 g of HgCl2 dissolved in 96 ml of a 2.0 M HCl solution), which revealed gelatin hydrolysis as clear, no-precipitation zones around the colonies. The areas of the clear zones (total area minus colony area) were determined densitometrically with the Molecular Analyst software (Bio-Rad).
Gel electrophoresis and zymography.
For native gel electrophoresis, 10% acrylamide-0.26% bisacrylamide gels were made in 0.38 M Tris-HCl buffer (pH 8.8). Gelatin (Bloom 300; Sigma) was copolymerized in the gels at final concentration of 0.025%. Before loading, 5.0 μl of cell culture supernatant, 2.0 μl of 10-fold-diluted hemolymph, or 2.0 μl of G. mellonella body homogenate (see above) was mixed with 2× loading buffer (1.14 M Tris-HCl [pH 8.8], 0.01% bromophenol blue, 20% glycerol) at a 2:1 ratio. Electrophoresis of native gels was continued after the dye front reached the bottom of the gel for an additional 90 min. SDS gel electrophoresis was performed by using a 10% acrylamide-0.26% bisacrylamide separation gel containing 0.025% copolymerized casein (Sigma). Samples were prepared by mixing 10 μl of culture supernatant or 5.0 μl of 10-fold-diluted hemolymph or body homogenate (see above) with 2× loading buffer (3% SDS, 0.16 M Tris-HCl [pH 6.8], 20% glycerol, 0.01% bromophenol blue; nonreducing conditions) at a 2:1 ratio. After electrophoresis, the gels were soaked in two to four changes of 100 ml of a buffer solution containing 50 mM Tris-HCl (pH 8.0), 10 mM CaCl2, and 0.1 M NaCl in order to develop the proteolytic bands. Native gels were incubated in two changes of the solution, the first for 20 min and the second for 60 min. SDS gels were incubated in four changes, the first three for 20 min each and the last one for 120 min. The proteolytic bands were visualized by Coomassie blue R250 staining. When zymography was performed in the presence of EDTA, the EDTA concentration was 5.0 mM during sample treatment and incubation of gels.
Enzyme activity measurement.
The enzyme activities in the culture supernatant and hemolymph samples (see above) were measured at 30°C in 1.0-ml (final volume) mixtures containing 50 mM Tris-HCl (pH 7.0), 10 mM CaCl2, and 0.1 M NaCl (assay buffer). The final substrate concentrations were 50 μM for the 2-furylacryloyl (Fua)-blocked, photometric substrates Fua-Leu-Gly-Pro-Ala (Fua-LGPA) (BACHEM) and Fua-Ala-Leu-Val-Tyr (Fua-ALVY) (20) and 10 and 50 μM for the thiobenzyl ester, photometric substrates succinyl-Ala-Ala-Pro-Phe-thiobenzyl and Z-Lys-thiobenzyl, respectively (Sigma). The reactions were started by addition of 150 μl of culture supernatant or 20 μl of 10-fold-diluted hemolymph (see above). In the case of Fua-blocked substrates, the decrease in absorbance at 324 nm was monitored until the end of the reaction. The catalytic activity (kobs) was calculated by fitting the final portion of the curves (where the remaining substrate concentration was less than 1/10 the Km [∼40 μM for Php-B] [20]) to first-order kinetics by using the Origin 5.0 software (Microcal).
To determine the nature of Php-D activity, 50 μl of the partially purified enzyme was incubated in 1.0 ml of assay buffer (see above) at room temperature for 10 to 15 min prior to measurement of the activity (before addition of the substrate) in the presence of one of the following substances at a concentration of 1.0 mM: EDTA, phenylmethylsulfonyl fluoride, 1,4-dithiothreitol, or Cys.
Isolation and N-terminal sequencing of proteases.
Protease separation and sample preparation for N-terminal analysis were performed by using the Brecon/1 strain. Purification of Php-A will be reported elsewhere, while isolation and characterization of Php-B were carried out as described by Marokházi et al. (20).
Php-C was partially purified from the supernatant of an 80-ml stationary-phase (40 h; optical density at 600 nm, ∼8.0) Brecon/1 culture. Following centrifugation for 20 min at 39,000 × g, the remaining cells were removed from the supernatant by filtration using a 0.2-μm-pore-size membrane. The filtrate was dialyzed against 10 liters of 20 mM Tris buffer (pH 8.0) containing 50 mM NaCl and 10 mM CaCl2 and then applied to a Matrex Silica PEI-300 anion-exchange column (16 by 60 mm; Amicon) equilibrated with the same buffer. Php-C (unlike the other three proteases) did not bind to the column under these conditions, so the flowthrough was collected, dialyzed against 10 liters of 20 mM Tris buffer (pH 8.0) containing 10 mM CaCl2 (but not NaCl), and applied to a PEI-300 column, which was equilibrated with the same buffer. Bound proteins were eluted by using a 0 to 0.4 M NaCl gradient in the same buffer. The protein composition of the fractions was analyzed by SDS-PAGE, and the protein band corresponding to Php-C was identified by parallel zymography.
Php-D was separated during Php-B purification in a Sephadex G-100 gel filtration step (for details see reference 20). Fractions with the highest Fua-ALVY-ase activity (at an elution volume of 120 to 140 ml from a column [100 cm by 16 mm]) were pooled and concentrated to a volume of 3 ml by using a Centricon concentrator (Millipore). The concentrate was purified further on a Hi-Prep 16/60 Sephacryl S-200 gel filtration column (Amersham Biosciences) and 20 mM Tris buffer (pH 8.0) containing 10 mM CaCl2 (flow rate, 0.5 ml/min). SDS-PAGE analysis of the Fua-ALVY-ase activity-containing fractions showed the presence of a ∼54-kDa protein band and a faint 55-kDa band (Php-A, as confirmed by zymography).
For N-terminal sequencing, samples of purified Php-A and Php-B, as well as partially purified Php-C and Php-D, were electrophoresed in 10% acrylamide-SDS gels under reducing conditions. After electrophoresis, the gels were soaked for 10 min in transfer buffer (10 mM 3-cyclohexylamino-1-propanesulfonic acid [pH 11; Sigma], 10% methanol) and then blotted onto an Immobilon-P polyvinylidene difluoride transfer membrane (Millipore) at 200 mA for 2 h. Proteins on the membrane were visualized by Coomassie blue staining. The bands corresponding to the proteases were cut out and subjected to Edman sequencing with a Microtec protein sequencer (Applied Biosystems). The proteins encoded in the genome of P. luminescens subsp. laumondii strain TT01, which had amino-terminal sequences similar to those of our proteins, were identified by using the FASTA algorithm at the website of the Pasteur Institute (http://genolist.pasteur.fra/PhotoList).
RESULTS
Gelatin liquefaction in agar plates.
The gelatin liquefaction assay, a quick method for comparison of protease secretion, was performed with 20 strains of Photorhabdus in a semiquantitative manner (see Materials and Methods). Colonies of 16 strains exhibited gelatinase activity (Table 2). The difference between the strongest (Rh/2) and weakest (Wx6/1) activities was almost fourfold. With the exception of HSH-2/1, all of the primary-phase variants were active. Four of the secondary-phase variants, strains K122/2, HSH-2/2, Wx-6/2, and Hm/2, were inactive, while strains Hb/2, NC1/2, Rh/2, and Wx8/2 were as good protease (gelatinase) secretors as the primary-phase variants.
TABLE 2.
Protease secretion by 20 Photorhabdus strains and phase variants: summary of results obtained with four detection methods
| Straina | Detection method
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Gelatin liquefaction assayb | Zymography following:
|
Fua-LGPA-ase activity (kobs)c,d | ||||||
| SDS-PAGEc
|
Native-PAGEc
|
|||||||
| Php-A1 | Php-A2 | Php-C | Php-A | Php-C1 | Php-C2 | |||
| Brecon/1 | + | ++ (ml) | + (ll) | + (ll) | ++ (ml) | ++ (ll) | ++ (ml) | 11.5 (ml) |
| TT01/1 | + | (+) (s) | − | − | ++ (ml) | + (ml) | − | 21.2 (ml) |
| Rh/1 | + | + (ll) | − | − | + (ll) | ++ (ml) | − | 8.6 (ml) |
| Rh/2 | + | (+) (s) | − | − | (+) (s) | + (ml) | − | 3.8 (ml) |
| Indica/1 | + | ++ (ml) | (+) (ll) | − | + (ml) | ++ (ml) | − | 17.0 (ml) |
| Hm/1 | + | + (s) | − | − | + (ll) | + (ml) | + (ml) | 4.5 (ml) |
| Hm/2 | − | − | − | − | − | − | − | 9.3 (ml) |
| Hb/1 | + | ++ (ml) | (+) (ll) | − | + (ml) | − | − | 1.7 (s) |
| Hb/2 | + | (+) (s) | − | − | (+) (ml) | (+) (s) | + (s) | 3.8 (s) |
| NC1/1 | + | + (ml) | ++ (ll) | − | ++ (ml) | ++ (ll) | ++ (s) | 9.7 (ml) |
| NC1/2 | + | + (ml) | ++ (ll) | − | ++ (ml) | (+) (s) | − | 1.5 (ml) |
| Wx6/1 | + | (+) (s) | − | − | + (ml) | + (ml) | − | 4.3 (ll) |
| Wx6/2 | − | − | − | − | − | − | − | 2.2 (ll) |
| Wx8/1 | + | + (ml) | (+) (s) | − | + (ml) | (+) (ml) | ++ (ml) | 3.4 (II) |
| Wx8/int | + | ++ (ml) | (+) (s) | − | ++ (ml) | (+) (s) | − | 3.6 (ll) |
| Wx8/2 | + | + (ml) | (+) (s) | − | + (ml) | (±) (s) | − | 3.1 (ll) |
| HSH-2/1 | − | − | − | − | − | − | − | 2.1 (s) |
| HSH-2/2 | − | − | − | − | − | − | − | 3.2 (s) |
| K122/1 | + | (+) (ll) | − | + (s) | ++ (ml) | + (s) | ++ (ll) | 9.9 (ll) |
| K122/2 | − | − | − | − | (±) (s) | − | (±) (s) | 0.4 (ll) |
The identities of the strains are shown in Table 1.
+, clearing zone visible; −, clearing zone not visible.
Intensity of the highest activity: −, not detectable; (±), very weak; (+), weak; +, well easily detectable; ++, strong. The times of first detection are indicated in parentheses, as follows: ml, mid-logarithmic phase (optical density at 600 nm between 1.0 and 4.8; h 12 to 21); ll, late logarithmic phase (optical density at 600 nm between 4.8 and 5.3; h 21 to 40); s, early stationary phase (optical density at 600 nm between 5.3 and 6.6; h 40 to 50).
The values were determined in supernatants having optical densities at 600 nm between 4.9 and 8.1 (43-h cultures). kobs is the first-order rate constant and is expressed in seconds−1 (see Materials and Methods).
SDS-PAGE- and native PAGE-coupled zymography.
Zymographic detection of proteases allows analysis of the number of activities after separation of proteins on the basis of their molecular properties. However, if the protein samples are electrophoresed in the presence of SDS (this assay has been used in previous analyses of Photorhabdus protease secretion), although the resolution is better, SDS-sensitive activities may be lost. In order to detect such activities, we employed zymography after native PAGE as well. Since the sensitivity of detection might also depend on the substrate, we tested fibrinogen and casein as potential substrate alternatives to gelatin. Based on both the intensity and the number of activity bands, the highest sensitivity in SDS-PAGE was observed with casein and fibrinogen, while gelatin proved to be the best substrate in native PAGE and permitted separation of high- and low-gelatin-affinity activities. The activities summarized in Table 2 were observed with substrates that proved to be the best ones for zymographic detection after the gel electrophoresis method used. The proteases were provisionally designated Php (for Photorhabdus protease) and were distinguished by letters and numbers according to the major activities and the putative molecular variants, respectively (e.g., Php-A1).
The most prominent activity, which was observed after SDS-PAGE in almost every primary-phase variant and in most of the secondary-phase variant strains (Table 2), was the activity of a ∼55-kDa protease, Php-A (Fig. 1A). Since this protease also proved to be a metalloenzyme (see below), we supposed that it was the same as the proteases that have been detected, cloned, purified, and partially characterized previously by other authors (see above). There were slight variations in the level of Php-A activity and the time of occurrence between strains and between growth conditions (data not shown). The activity appeared from h 12 to 24 of culture (mid-logarithmic to late logarithmic growth phase) in the case of the most intensive Php-A producers (Brecon/1 and NC1/2) (Table 2 and Fig. 1A) and remained detectable for the 44 h of culturing. With the striking exception of Hm/1, there were at least two activity bands in the molecular mass range from 50 to 55 kDa. The smaller species, which were thought to be the molecular variants of Php-A (see Discussion), were not present at the beginning of secretion (Fig. 1A). In each case, two dominant forms, a larger ∼55-kDa form (Php-A1) and a smaller 2- to 4-kDa form (Php-A2), could be distinguished. The ratio of these forms and the occurrence of other forms varied slightly for different cultures of the same strain, but the Php-A1 form was always the most abundant (or most active).
FIG. 1.

Detection of proteolytic activity in culture supernatants. Protease activity was analyzed in representative experiments by SDS-PAGE-coupled zymography (with casein as the substrate) (A), by native PAGE-coupled zymography (with gelatin as the substrate) (B), and with the chromogen substrate (Fua-LGPA-ase activity of the Brecon/1 strain) (C). In panels A and B, the samples analyzed were the same. The strain designations and culture times (in hours) are indicated above the gels. Note that both the Php-A and Php-C bands are fainter and expressed later in the K122/1 culture than in the Brecon/1 culture. (C) Symbols: ▴, optical density of the culture at 600 nm (OD600); •, enzyme activity (kobs). For the method used to determine kobs, see Materials and Methods.
Another major activity, that from a ∼37-kDa protease, referred to as Php-C, was also found with SDS-PAGE-coupled zymography in the cultures of strains Brecon/1 and K122/1 but not in the cultures of the other strains (Table 2 and Fig. 1A). The detection of this activity was less sensitive if the gelatin substrate was used. Depending on the strain, it occurred from h 21 to 40 of culturing (in the late logarithmic and early stationary phases). In strain Brecon/1, Php-C also showed size variation and was resolved by SDS-PAGE as a larger (37-kDa) form and a fainter, smaller (35-kDa), and substantially more active (or abundant) form (Php-C1 and Php-C2, respectively). The zymographic detection of Php-A and Php-C was sensitive to EDTA, indicating that these enzymes are metalloproteases (data not shown).
Zymography following native PAGE also resulted in detection of two major proteolytic bands (Table 2 and Fig. 1B); the band that migrated more slowly was Php-A, and the band that migrated faster was Php-C, as confirmed by zymography of the isolated enzymes (data not shown). Compared to SDS-PAGE, native PAGE revealed three additional features of Php-A and Php-C. (i) Php-A, but not Php-C, had a strong affinity for gelatin; it bound to this zymographic substrate even during electrophoresis. This was evident from the correlation between band intensities and migration distances in the case of Php-A but not in the case of Php-C (see the zymography of Brecon/1 samples in Fig. 1B). Furthermore, if casein was used as the substrate, only one broad proteolytic band was observed, which migrated much faster than Php-A in a gelatin-containing gel and presumably contained both Php-A and Php-C (data not shown). (ii) Php-C appeared to be more sensitive to SDS than Php-A; in numerous cases the (weaker) activities of Php-C were detected only by native PAGE-coupled zymography. For example, this activity was found earlier and in more strains with this method than with SDS-PAGE-coupled zymography (Fig. 1A and B and Table 2). (iii) Not only Brecon/1 (see above) but also a number of other strains produced two molecular forms, Php-C1 and Php-C2 (Table 2).
The observation that both Php-A and Php-C activities remain detectable after nonreducing SDS-PAGE, which is known to destroy (noncovalent) molecular complexes, is not consistent with the interpretation that the supernatants of some cultures lack protease activity because the enzyme molecules are locked in inhibitor complexes. Furthermore, Php-A or Php-C activities were detected after SDS-PAGE only if they were detected after native PAGE too. Therefore, these activities might be missing from the culture supernatants of some strains because the genes of these proteases are not expressed or (less likely) because the expressed proteins cannot be secreted. To check these possibilities, we used zymography coupled to SDS-PAGE to compare the culture supernatants and cytoplasm fractions (cell lysates) of 24-h cultures of the Hm/1, Hm/2, K122/1, K122/2, and Brecon/1 strains. In support of the first alternative, no proteolytic activity was detected in the cytoplasm fraction in either case (data not shown). However, some of the differences in the Php-A activity could be attributed to the presence of an inhibitor(s), as suggested previously (6). For example, according to SDS-PAGE-coupled zymography there was a decrease in Php-A activity between h 21 and 40 of incubation in the supernatant of the K122 culture, while according to native PAGE-coupled zymography there was an increase (Fig. 1A and B). Since the latter detection method can be influenced by the presence of an inhibitor(s), one explanation for the discrepancy might be a faster decrease in the amount of inhibitor(s) (resulting in an increase in activity after native PAGE) than in the amount of Php-A (resulting in a decrease in activity after SDS-PAGE). The comparison of supernatants and cytoplasm fractions also proved that both Php-A and Php-C are secreted enzymes.
Activity measurements with chromogen oligopeptide substrates.
The advantages of analyzing protease production by hydrolysis of synthetic substrates include the quantitative determination and the typification of activities if the cross-reactivity between substrates is limited (e.g., if the hydrolysis of the nonspecific substrate is less than 0.1% of the hydrolysis of the specific substrate).
Two substrates in our study, succinyl Ala-Ala-Pro-Phe-thiobenzyl and Z-Lys-thiobenzyl (specific for chymotrypsin- and trypsin-like proteases, respectively), belong to the group of substrates which are cleaved best by proteases with primary preferences for the amino acids that are N terminal to the scissile bond (e.g., serine proteases). Hydrolysis of the thioester bonds, which are C terminal to Phe or Lys in these substrates, releases a thiobenzyl group, which allows very sensitive detection of protease activity through a coupled reaction with 4,4′-dithiodipyridine. Within 48 h of culturing, no activity was found with these substrates.
Two other substrates, Fua-Leu-Gly-Pro-Ala and Fua-Ala-Leu-Val-Tyr, represented the group of substrates which are used to measure the activity of proteases that have primary preferences for amino acids that are C terminal to the scissile bond (e.g., collagenolytic enzymes and peptidases). Hydrolysis of these substrates at the Leu-Gly and Ala-Leu bonds causes a shift in the absorption spectrum of the Fua chromophore group (29). All 20 strains tested exhibited Fua-LGPA-ase activity, which was first detected usually in the late logarithmic and early stationary growth phases (Table 2). Later in the stationary phase (40 to 50 h, depending on the strain) the production of activity did not increase further (Fig. 1C). The 10- to 50-fold variation in the Fua-LGPA-ase activity of 43-h culture supernatants (Table 2) might be partially attributed to differences in cell density. The Fua-LGPA-ase enzyme, designated Php-B, was purified and proved to be a ∼74-kDa intracellular metalloenzyme (20), which could not be detected by either SDS-PAGE- or native PAGE-coupled zymography.
Cleavage of the Fua-Ala-Leu-Val-Tyr substrate was observed only in the culture supernatant of the Brecon/1 strain. Although this observation in itself indicates that the Fua-Ala-Leu-Val-Tyr-ase enzyme is different from the other three enzymes, to obtain more evidence, the culture supernatant was subjected to a rough separation procedure (see Materials and Methods). The activity seemed to copurify with a ∼54-kDa protein band in reducing SDS-PAGE. The Fua-Ala-Leu-Val-Tyr-ase-enriched fractions from gel filtration chromatography did not contain activity that cleaved Fua-Leu-Gly-Pro-Ala. Furthermore, the Fua-Ala-Leu-Val-Tyr-ase activity was not sensitive to a metal ion chelator (EDTA) or to a serine protease inhibitor (phenylmethylsulfonyl fluoride), but it could be completely inhibited by a disulfide reagent (1,4-dithiothreitol) and could be partially inactivated by cysteine (Table 3). Therefore, this activity cannot be the activity of Php-A, Php-B, or Php-C but is the activity of a fourth enzyme, designated Php-D. The activity of Php-D was detectable in the culture supernatant but not in the cell lysate. Due to the presence of Php-A activity in the Php-D-containing fractions (see below), we could not decide if Php-D is detectable by zymography. The isolated activity proved to be quite unstable, disappearing from the Php-D-containing fraction within several weeks during storage (data not shown).
TABLE 3.
Effects of various protease inhibitors on the Fua-Ala-Leu-Val-Tyr-ase (Php-D) activity
| Enzyme treatmenta | Fua-Ala-Leu-Val-Tyr hydrolysis (Absorbance/min, 10−5) |
|---|---|
| None | 6.08 |
| EDTA (1 mM) | 6.45 |
| PMSF (1 mM) | 4.50 |
| Cysteine (1 mM) | 0.76 |
| DTT (1 mM) | 0.00 |
PMSF, phenylmethylsulfonyl fluoride; DTT, 1,4-dithiothreitol.
Proteolytic activity during infection.
In order to investigate the presence of protease activities during infection, hemolymph samples were taken from strain Brecon/1-infected G. mellonella larvae for analysis by zymography (detection of Php-A and Php-C) and for testing the Fua-Leu-Gly-Pro-Ala-ase activity (Php-B). The Php-B activity was measurable from the ∼33rd hour of infection, when larvae were motionless but still alive (Fig. 2C). The activities of both Php-A and Php-C in the hemolymph were observed 20 to 40 h postinfection (Fig. 2A and B). Since 25 to 40 h after infection hemolymph cannot be separated from the disintegrating tissue debris, contamination of samples with proteases from Galleria might be expected, which could interfere with the detection of activities from Photorhabdus. To check the presence of such activities, whole-body homogenates prepared from both infected and uninfected larvae were also analyzed along with hemolymph samples (Fig. 2A and B). The proteolytic activities of Galleria homogenates were much stronger when SDS-PAGE-coupled zymography was used than when native PAGE-coupled zymography was used. These activities, however, did not influence the detection of Php-A and Php-C activities, because by the time that they occurred in the hemolymph, the proteolytic activities from Galleria were significantly reduced.
FIG. 2.

Detection of proteolytic activity during infection of G. mellonella larvae with the Brecon/1 strain. Protease activity in the hemolymph of infected larvae and in larva homogenates was analyzed by SDS-PAGE-coupled zymography (A), by native PAGE-coupled zymography (B), and by chromogen substrate hydrolysis (Fua-LGPA-ase activity) in hemolymph samples (C). In panels A and B, the same samples were analyzed, and the lanes in each panel represent samples from different larvae. The type of sample (cell culture supernatant, hemolymph, or body homogenate) and the time that the sample was taken (in hours postinfection) are indicated above the panels. In panel C, the activities in 1.0-μl hemolymph samples are plotted as a function of infection time. For the method used to determine kobs, see Materials and Methods.
With regard to the substantial differences between the Photorhabdus strains in both the composition and the intensity of protease secretion, we were interested to see whether there was any correlation between these differences and the pathogenicity of strains. This question has implications for the role of proteases in the establishment of the infection. Therefore, we conducted infection experiments with several strains that had markedly different levels of Php-A, Php-C, and Php-D production (Brecon, NC1/1, NC1/2, Hm/1, Hm/2, K122/1, and K122/2). Thirty to 60 bacterial cells were injected into the hemocoel of G. mellonella larvae, and the time of the death of each animal was recorded (Fig. 3). The average survival, which was used as an approximate indicator of pathogenicity, showed that secondary variants were always more pathogenic (causing death 1 to 6 h earlier), although the difference was not statistically significant in the case of K122/1 and K122/2. The maximal difference between the strains in average survival times of larvae was 10 h (21.8 h for Hm/2 and 31.9 h for Brecon/1). Since the most pathogenic strain, strain Hm/2, did not secrete Php-A, Php-C, and Php-D, whereas the least pathogenic strains, strains Brecon/1 and NC1/1, were intensive secretors of these activities, it appears unlikely that pathogenicity (for G. mellonella) is dependent on secretion of these enzymes.
FIG. 3.
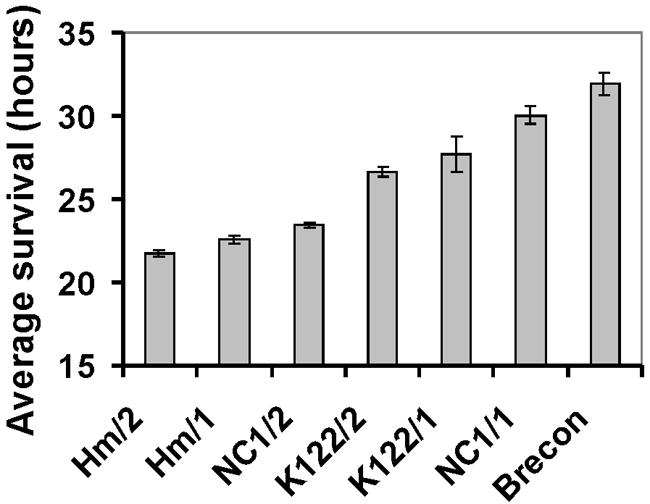
Pathogenicity of several selected Photorhabdus strains for G. mellonella larvae. The results of three replicate experiments were combined for each strain, since there was no significant difference among the replicates (P > 0.05, as determined by a t test). The differences between the strains were significant (P < 0.05, as determined by a t test) except for the difference between K122/1 and K122/2.
Determination of the N-terminal sequence of Php-A, Php-B, and Php-C.
We prepared Php-A, Php-B, and Php-C samples for N-terminal sequencing from a Brecon/1 culture by blotting the partially purified protein on a polyvinylidene difluoride membrane after SDS-PAGE (see Materials and Methods). After sequencing, we identified the enzymes by sequence comparison, taking advantage of the previously published genomic sequence of P. luminescens subsp. laumondii TT01 (12). Figure 4 shows the sequences of Php-A, Php-B, and Php-C aligned with similar sequences encoded in the TT01 genome and with some other related sequences from several microorganisms. Unfortunately, due to the relatively small amount and the instability of Php-D and to the molecular properties that were very similar to those of Php-A, we could not prepare a Php-D sample which was sufficiently devoid of Php-A. This fact and the probably blocked N terminus made it impossible to determine the N-terminal sequence of Php-D.
FIG. 4.

Comparison of the sequences of the amino termini of Php-A, Php-B, and Php-C with similar sequences in the database. Amino acids (single-letter code) that are identical to those in P. luminescens subsp. laumondii strain TT01 sequences are indicated by boldface type; a dash indicates a gap in the alignment. X in the Php-B sequence shows sites where the residue could not be identified. The numbering of positions is the numbering in the open reading frames of the TT01 genome. An asterisk indicates an enzyme translated from a genomic survey sequence (GenBank accession number AQ990436) (13). The database search, sequence alignment, and E value calculation were performed by using the FASTA algorithm with TT01 sequences.
DISCUSSION
In this study, the production of proteolytic activity by members of the nematode symbiont insect-pathogenic bacterial genus Photorhabdus was examined in 20 strains, including eight phase variant pairs, in culture and in seven strains during infection of G. mellonella larvae.
By using a combination of several detection methods, four major activities were found by the early stationary phase of growth in cultures, and these activities were provisionally designated Php-A, Php-B, Php-C, and Php-D. Php-A and Php-C, but not Php-B, could be detected with zymography following both native PAGE and SDS-PAGE with using gelatin, casein, or fibrinogen as the substrate. As judged from their sensitivities to EDTA, all of the activities except the Php-D activity are from metalloenzymes. When they were tested in G. mellonella larvae the activities of Php-A, Php-B, and Php-C were detectable.
The enzyme responsible for the most prominent zymographic activity, which was also the enzyme that was secreted earliest in culture, was Php-A. On the basis of its properties (namely, its sensitivity to EDTA and its molecular mass [∼55 kDa]), Php-A is the same enzyme as PrtA, which has been cloned from the K122 and W14 strains (5, 6), and the proteases that have previously been zymographically detected and partially characterized from the Hm and HP88 strains (23, 25, 32). The strong similarity of the N-terminal sequence of Php-A, purified from the Brecon/1 strain, to the PrtA sequences of TT01 (12), K122, and W14 (Fig. 4) also shows that Php-A is a Brecon/1 ortholog of PrtA, a metzincin metallopeptidase in HEXXH+H clan MB (24).
The molecular and enzymatic properties of Php-C, which is also a secreted enzyme, are distinct from those of Php-A (PrtA), because the two enzymes differ in their molecular masses and N-terminal sequences and because Php-C has a higher sensitivity for SDS and a lower affinity for gelatin. Based on N-terminal amino acid sequence similarity, Php-C is a Brecon/1 strain ortholog of the protein encoded by the plu1382 gene of TT01 (12), which indicates that it, in accordance with our findings, is a metalloenzyme that belongs to HEXXH+E clan MA of metallopeptidases (24).
Php-B, the third metalloenzyme that we found, is an intracellular peptidase (20) which, based on the N-terminal sequence similarity with the plu0124 gene-encoded protein of TT01 (12) (Fig. 4), is the Brecon/1 ortholog of OpdA (oligopeptidase A) and belongs to the HEXXH thimet family of metallopeptidases (24). Our enzymatic analysis of Php-B (OpdA) (20) showed that it is a collagen peptidase because it exhibited the highest activity with the small peptides (e.g., Fua-Leu-Gly-Pro-Ala) that contain the collagen repeat sequence -Pro-Xaa-Gly-Pro- (where Xaa is almost any amino acid). Studies of enzymes from Salmonella enterica serovar Typhimurium and Escherichia coli indicated a physiological role for OpdA in the degradation of cleaved signal peptides (8, 9, 22, 30). Previous results (20) suggest that this enzyme might also have a nutritional role in pathogenic microorganisms by degrading short peptides from the proteins of infected hosts.
Although sequence information is not available for Php-D, which is also a secreted protease of the Brecon/1 strain, the fact that it is not a metalloenzyme but is a thiol-dependent protease distinguishes it from the other three peptidases. Furthermore, Php-D, but not the other three enzymes, cleaves the Fua-Ala-Leu-Val-Tyr peptide.
The zymographic activities of Php-A (PrtA) and Php-C were often found as two to five seemingly associated bands, which we distinguished by numbers (Php-A1 and Php-C1 referred to the species that migrated most slowly). The activities that migrated faster (the most prominent of which were Php-A2 and Php-C2) were thought to be derived from the activities that migrated most slowly. For example, Php-A2, which appeared 5 to 10 h later than Php-A1, could be a variant form expressed from a second copy of the gene encoding Php-A (PrtA) or by (auto)proteolytic processing. Our observation that Php-A2 (among other Php-A variants) began to form in the solution of purified Php-A1 after 6 to 8 months of storage (J. Marokházi and I. Venekei, unpublished data) supports the second alternative.
The congruence of the results obtained by zymography and Frazier's gelatin hydrolysis assay on plates shows that the latter method detects primarily the activity of Php-A (PrtA). It is surprising that, despite the very sensitive substrates that were used, we were unable to detect serine protease secretion. This is at variance with a previous observation that a 56-kDa protease of the HP88 strain was active on such a substrate (32). The difference in culture time might be an explanation for this discrepancy; we tested protease activity in 12- to 44-h cultures, while the 56-kDa enzyme was isolated from a 7-day culture. Another difference between our findings and previous reports is that we detected more than one activity by SDS-PAGE-coupled zymography. Again, the different culture time might be a reason; an alternative explanation is that the sample preparation was dissimilar, since unlike other workers, we did not precipitate the culture supernatant or hemolymph samples before addition to gel loading buffer. This could have resulted in substantially higher sensitivity for our procedure, which also permitted observation of Php-C and earlier detection of Php-A (PrtA) in culture supernatant and during infection. (The reduction of the samples, another difference in the sample treatments, did not influence the sensitivity for Php-A and Php-C [data not shown].)
Php-B (OpdA) was produced by all strains (although at slightly different levels). Php-D production, in contrast, seems to be the property of only the Brecon strain. Php-A (PrtA) and Php-C production exhibited substantial and seemingly strain-specific differences; the findings ranged from a strain (HSH-2) that did not produce these activities to a strain (NC1) that intensively produced Php-A and Php-C in both phase variants (Table 2). The slight variations between strains in both the intensity and the timing of protease production might be attributed to culture conditions (e.g., the cell density or the age of the inocula). However, this cannot be the explanation for larger differences or the absence or presence of activity. The formation of an enzyme-inhibitor complex (e.g., between PrtA and the Inh protein of strain W14 [28, 32]) has been proposed as a potential cause of the reduced protease activity of some Photorhabdus strains or secondary-phase variants. While this might be the interpretation for differences between measured enzyme activities and between the results of native PAGE-coupled zymography and the results of SDS-PAGE-coupled zymography (see Results), it cannot be used for differences between zymographic activities observed after SDS-PAGE because the presence of SDS during gel electrophoresis disrupts (noncovalent) complexes and the inhibitor is well separated from the enzyme by the time of zymogram development. Therefore, despite the essentially qualitative nature of enzymographic techniques, not only the complete absence but also the differences in the intensities of proteolytic bands can be interpreted as due to different levels of secretion. Since in none of the strains tested that did not produce Php-A and Php-C was there any indication of nonfunctioning secretion (i.e., accumulation of activities in the cytosol), it is reasonable to believe that these activities are absent because of missing or inactive genes. However, another possibility, the accumulation of Php-A and Php-C in the cytosol of nonsecreting strains in an inactive (zymogen) form due to different posttranslational regulation (31), cannot be excluded either. A genome analysis could clarify this question.
In previous studies, the secondary-phase variants of several strains of Photorhabdus were found to secrete protease activity (see Introduction), which is not consistent with the generally accepted assumption that missing or reduced protease secretion is a feature of secondary-phase variants (17). Our comparison of eight phase variant pairs of Photorhabdus by using the gelatin liquefaction assay and zymography showed that during the first 48 h of culturing, the lack of or the lower level of secretion of Php-A (PrtA) and Php-C was specific to the secondary-phase variants for only three strains (Hm, K122, and Wx6). Based on these results, the use of proteolytic enzyme secretion for characterization of phase variants of Photorhabdus is not reasonable.
Finally, an interesting question is the physiological role that proteases may play. An answer would require enzyme purification, enzymatic characterization, and identification of the possible natural substrates. Due probably to a milder sample preparation procedure, we found the activities of PrtA (Php-A) and Php-C earlier (well before insect death) than other authors found these activities (during or after insect death). However, as far as the injection of Photorhabdus cells into the hemocoel of Galleria is a relevant model for infection of natural insect hosts via nematodes, our finding that there was no correlation between the production of Php-A and Php-C and the pathogenicity of strains indicates that these proteases are not immediately involved in the establishment of infection.
Acknowledgments
We express our thanks to Byron Adams (United States), Noel Boemare (Montpellier, France), David Clarke (Bath, United Kingdom), Ann Burnell (Maynooth, Ireland), Ralf-Udo Ehlers (Kiel, Germany), Jerry Ensign (Madison, Wis.), Ronald Hurlbert (Washington State University, Pullman), and K. H. Nealson (Milwaukee, Wis.) for providing Photorhabdus and Heterorhabditis strains. We thank Attila Lucskai for allowing us to use his unpublished technique, as well as Emília Szállás and Barna D. Fodor for providing their primary and secondary Photorhabdus strain isolates.
This work was supported by research grants T037907 and T035010 from the National Research Foundation (OTKA), Hungary, to I.V. and A.F.
REFERENCES
- 1.Bleakley, B. H., and K. H. Nealson. 1988. Characterization of primary and secondary forms of Xenorhabdus luminescens strain Hm. FEMS Microbiol. Ecol. 53:241-246. [Google Scholar]
- 2.Boemare, N., A. Givaudan, M. Brehelin, and C. Laumond. 1997. Symbiosis and pathogenicity of nematode-bacterium complexes, p. 21-45. In Nematode symbiosis, vol. 22, no. 1/2. International Science Services, Zeist, The Netherlands. [Google Scholar]
- 3.Boemare, N. E., and R. J. Akhurst. 1988. Biochemical and physiological characterization of colony form variants in Xenorhabdus ssp. (Enterobacteriaceae). J. Gen. Microbiol. 134:751-761. [DOI] [PubMed] [Google Scholar]
- 4.Boemare, N. E., R. J. Akhurst, and R. G. Mourant. 1993. DNA relatedness between Xenorhabdus spp.(Enterobacteriaceae), symbiotic bacteria of entomopathogenic nematodes, and a proposal to transfer Xenorhabdus luminescens to a new genus, Photorhabdus gen. nov. Int. J. Syst. Bacteriol. 43:249-255. [Google Scholar]
- 5.Bowen, D., M. Blackburn, T. Rocheleau, C. Grutzmacher, and R. H. ffrench-Constant. 2000. Secreted proteases from Photorhabdus luminescens: separation of the extracellular proteases from the insecticidal Tc toxin complexes. Insect Biochem. Mol. Biol. 30:69-74. [DOI] [PubMed] [Google Scholar]
- 6.Bowen, D. J., T. A. Rocheleau, C. K. Grutzmacher, L. Meslet, M. Valens, D. Marble, A. Dowling, R. ffrench-Constant, and M. A. Blight. 2003. Genetic and biochemical characterization of PrtA, an RTX-like metalloprotease from Photorhabdus. Microbiology 149:1581-1591. [DOI] [PubMed] [Google Scholar]
- 7.Caldas, C., A. Cherqui, A. Pereira, and N. Simoes. 2002. Purification and characterization of an extracellular protease from Xenorhabdus nematophila involved in insect immunosuppression. Appl. Environ. Microbiol. 68:1297-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conlin, C. A., and C. G. Miller. 1992. Cloning and nucleotide sequence of opdA, the gene encoding oligopeptidase A in Salmonella typhimurium. J. Bacteriol. 174:1631-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conlin, C. A., N. J. Trun, T. J. Silhavy, and C. G. Miller. 1992. Escherichia coli prlC Encodes an Endopeptidase and is Homologous to the Salmonella typhimurium opdA Gene. J. Bacteriol. 174:5881-5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daborn, P. J., N. Waterfield, M. A. Blight, and R. H. ffrench-Constant. 2001. Measuring virulence factor expression by the pathogenic bacterium Photorhabdus luminescens in culture and during insect infection. J. Bacteriol. 183:5834-5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalhammar, G., and H. Steiner. 1984. Characterization of inhibitor A, a protease from Bacillus thuringiensis which degrades attacins and cecropins, two classes of antibacterial proteins in insects. Eur. J. Biochem. 139:247-252. [DOI] [PubMed] [Google Scholar]
- 12.Duchaud, E., C. Rusniok, L. Frangeul, C. Buchrieser, A. Givaudan, S. Taourit, S. Bocs, C. Boursaux-Eude, M. Chandler, J. F. Charles, E. Dassa, R. Derose, S. Derzelle, G. Freyssinet, S. Gaudriault, C. Medigue, A. Lanois, K. Powell, P. Siguier, R. Vincent, V. Wingate, M. Zouine, P. Glaser, N. Boemare, A. Danchin, and F. Kunst. 2003. The genome sequence of the entomopathogenic bacterium Photorhabdus luminescens. Nat. Biotechnol. 21:1307-1313. [DOI] [PubMed] [Google Scholar]
- 13.ffrench-Constant, R. H., N. Waterfield, V. Burland, N. T. Perna, P. J. Daborn, D. Bowen, and F. R. Blattner. 2000. A genomic sample sequence of the entomopathogenic bacterium Photorhabdus luminescens W14: potential implications for virulence. Appl. Environ. Microbiol. 66:3310-3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer-Le Saux, M., V. Viallard, B. Brunel, P. Normand, and N. E. Boemare. 1999. Polyphasic classification of the genus Photorhabdus and proposal of new taxa: P. luminescens subsp. luminescens subsp. nov., P. luminescens subsp. akhurstii subsp. nov., P. luminescens subsp. laumondii subsp. nov., P. temperata sp. nov., P. temperata subsp. temperata subsp. nov., and P. asymbiotica sp. nov. Int. J. Syst. Bacteriol. 49:1645-1656. [DOI] [PubMed] [Google Scholar]
- 15.Flyg, C., and K. G. Xanthopoulos. 1983. Insect pathogenic properties of Serratia marcescens. Passive and active resistance to insect immunity studied with protease-deficient and phage-resistant mutants. J. Gen. Microbiol. 129:453-464. [DOI] [PubMed] [Google Scholar]
- 16.Forst, S., B. Dowds, N. Boemare, and E. Stackebrandt. 1997. Xenorhabdus and Photorhabdus spp.: bugs that kill bugs. Annu. Rev. Microbiol. 51:47-72. [DOI] [PubMed] [Google Scholar]
- 17.Forst, S., and K. Nealson. 1996. Molecular biology of the symbiotic-pathogenic bacteria Xenorhabdus spp. and Photorhabdus spp. Microbiol. Rev. 60:21-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frazier, W. C. 1926. A method for the detection of changes in gelatin due to bacteria. J. Infect. Dis. 39:302-306. [Google Scholar]
- 19.Kling, J. J., R. L. Wright, J. S. Moncrief, and T. D. Wilkins. 1997. Cloning and characterization of the gene for the metalloprotease enterotoxin of Bacteroides fragilis. FEMS Microbiol. Lett. 146:279-284. [DOI] [PubMed] [Google Scholar]
- 20.Marokházi, J., G. Kóczán, F. Hudecz, L. Gráf, A. Fodor, and I. Venekei. 2004. Enzymic characterization with progress curve analysis of a collagen peptidase from an enthomopathogenic bacterium, Photorhabdus luminescens. Biochem. J. 379:633-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moncrief, J. S., R. Obiso, Jr., L. A. Barroso, J. J. Kling, R. L. Wright, R. L. Van Tassell, D. M. Lyerly, and T. D. Wilkins. 1995. The enterotoxin of Bacteroides fragilis is a metalloprotease. Infect. Immun. 63:175-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novak, P., and I. K. Dev. 1988. Degradation of a signal peptide by protease IV and oligopeptidase A. J. Bacteriol. 170:5067-5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ong, K. L., and F. N. Chang. 1997. Analysis of proteins from different phase variants of the entomopathogenic bacteria Photorhabdus luminescens by two-dimensional zymography. Electrophoresis 18:834-839. [DOI] [PubMed] [Google Scholar]
- 24.Rawlings, N. D., and A. J. Barrett. 1995. Evolutionary families of metallopeptidases. Methods Enzymol. 248:183-228. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt, T. M., B. H. Bleakley, and K. H. Nealson. 1988. Characterization of an extracellular protease from the insect pathogen Xenorhabdus luminescens. Appl. Environ. Microbiol. 54:2793-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szállás, E., C. Koch, A. Fodor, J. Burghardt, O. Buss, A. Szentirmai, K. H. Nealson, and E. Stackebrandt. 1997. Phylogenetic evidence for the taxonomic heterogeneity of Photorhabdus luminescens. Int. J. Syst. Bacteriol. 47:402-407. [DOI] [PubMed] [Google Scholar]
- 27.Tonello, F., S. Morante, O. Rossetto, G. Schiavo, and C. Montecucco. 1996. Tetanus and botulism neurotoxins: a novel group of zinc-endopeptidases. Adv. Exp. Med. Biol. 389:251-260. [PubMed] [Google Scholar]
- 28.Valens, M., A. C. Broutelle, M. Lefebvre, and M. A. Blight. 2002. A zinc metalloprotease inhibitor, Inh, from the insect pathogen Photorhabdus luminescens. Microbiology 148:2427-2437. [DOI] [PubMed] [Google Scholar]
- 29.Van Wart, H. E., and D. R. Steinbrink. 1981. A continuous spectrophotometric assay for Clostridium histolyticum collagenase. Anal. Biochem. 113:356-365. [DOI] [PubMed] [Google Scholar]
- 30.Vimr, E. R., L. Green, and C. G. Miller. 1983. Oligopeptidase-deficient mutants of Salmonella typhimurium. J. Bacteriol. 153:1259-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang, H., and B. C. Dowds. 1993. Phase variation in Xenorhabdus luminescens: cloning and sequencing of the lipase gene and analysis of its expression in primary and secondary phases of the bacterium. J. Bacteriol. 175:1665-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wee, K. E., C. R. Yonan, and F. N. Chang. 2000. A new broad-spectrum protease inhibitor from the entomopathogenic bacterium Photorhabdus luminescens. Microbiology 146:3141-3147. [DOI] [PubMed] [Google Scholar]