

Abstract
In fluorescent in situ hybridization (FISH), the efficiency of hybridization between the DNA probe and the rRNA has been related to the accessibility of the rRNA when ribosome content and cell permeability are not limiting. Published rRNA accessibility maps show that probe brightness is sensitive to the organism being hybridized and the exact location of the target site and, hence, it is highly unpredictable based on accessibility only. In this study, a model of FISH based on the thermodynamics of nucleic acid hybridization was developed. The model provides a mechanistic approach to calculate the affinity of the probe to the target site, which is defined as the overall Gibbs free energy change (ΔG°overall) for a reaction scheme involving the DNA-rRNA and intramolecular DNA and rRNA interactions that take place during FISH. Probe data sets for the published accessibility maps and experiments targeting localized regions in the 16S rRNA of Escherichia coli were used to demonstrate that ΔG°overall is a strong predictor of hybridization efficiency and superior to conventional estimates based on the dissociation temperature of the DNA/rRNA duplex. The use of the proposed model also allowed the development of mechanistic approaches to increase probe brightness, even in seemingly inaccessible regions of the 16S rRNA. Finally, a threshold ΔG°overall of −13.0 kcal/mol was proposed as a goal in the design of FISH probes to maximize hybridization efficiency without compromising specificity.
Fluorescent in situ hybridization (FISH) has proven to be a powerful molecular method for identification, visualization, and quantification of organisms of interest in microbial communities (reviewed in reference 2). Originally introduced by DeLong et al. (11), the use of FISH expanded to the study of bacterial populations in environmental applications (for reviews, see references 1, 2, and 34) and medical applications (22). Despite the wide use of FISH for more than a decade, the design and use of FISH probes remain a highly empirical procedure. Since the rRNA primary chains contain regions of variable conservation, it is straightforward to determine a target site on the rRNA with the desired level of specificity based on sequence comparisons (1, 34). However, a limitation that is difficult to overcome is the inability to predict if the signal intensity of cells hybridized with the fluorophore-labeled probe is high enough to be detectable (2, 13, 34).
Low fluorescent responses in hybridized samples can be related to a variety of factors, such as low ribosome content of cells, difficulty in permeating cell walls, and the inaccessibility of target sites due to either the secondary and tertiary structures of the rRNA (i.e., RNA-RNA interactions) or the effect of ribosomal proteins (i.e., RNA-protein interactions) (2). The former two depend solely on the studied organism and can be potentially circumvented by changes in the experimental protocol, such as using a different fixative to better permeate the cell wall (e.g., the methods of Roller et al. [24]) or by indirect labeling of the oligonucleotide to amplify the signal intensity per probe (e.g., the method of Schonhuber et al. [26]). On the other hand, the inaccessibility of target sites depends on the regional variations in the three-dimensional (3D) structure of the ribosome and, hence, cannot be manipulated effectively without a mechanistic understanding of the hybridization of the probe to the structurally complex rRNA molecule.
The attempts to develop a more thorough understanding of hybridization efficiency in FISH have been empirical. Initially, Frischer et al. (12) demonstrated the importance of the higher-order structure of the in situ 16S rRNA by revealing that there were significant differences in the signal intensity of probes when the target was in situ 16S rRNA (FISH experiments) but not when the same probes were hybridized to denatured 16S rRNA (blot hybridizations). Later, Fuchs et al. (13) made a detailed analysis of the dependence of hybridization efficiency on the location of the target site by using a large set of probes covering the entire 16S rRNA of Escherichia coli. The experiments were standardized, such that the ribosome content and permeability were fairly constant and the theoretical dissociation temperature (Td) of the probes was kept between 48 and 60°C (above the hybridization temperature of 46°C) according to equation 1, which was empirically developed by Suggs et al. (29) based on hybridization of DNA oligonucleotides with blotted DNA targets and has been used for rough estimations of DNA/RNA duplex stability in FISH (7, 13, 14, 28). Consequently, differences in the brightness (fluorescence intensity) of hybridized probes were regarded mainly as a function of the accessibility of their target sites, and the signal intensity data yielded a putative accessibility map of the E. coli 16S rRNA (13). The resulting map showed that accessibility was highly heterogeneous over the 16S rRNA and very sensitive to small shifts in the location of the target site. In addition, some portions of the central domain and the 3′ major domain (domains from reference 37) were consistently inaccessible, while the 5′ domain was more accessible. In a series of publications, taking the same approach as in their initial study (13) Amann and coworkers characterized the accessibility of the large subunit (lsu) rRNA for E. coli (14) and of the small subunit (ssu) rRNA for Pirellula sp., Metallosphaera sedulla, and Saccharomyces cerevisiae, representatives of bacterial, archaeal, and eukaryotic domains, respectively (7). Hence, they were able to compare the hybridization efficiencies of probes targeting homologous sites in the ssu rRNA, unveiling remarkable discrepancies among the different organisms (7). These discrepancies, together with the sensitivity to small shifts in the target site, raise the question of whether hybridization efficiency can be defined as merely a function of accessibility. Moreover, changing the type of fluorophore labeling the probes has also been observed to cause significant differences in the relative brightness of hybridized probes (7, 14). As a result, the dependence of hybridization efficiency on the type of organism, the exact location of the target site, and the type of fluorophore undermines the usefulness of the empirically developed accessibility maps as tools in the design of new probes and new FISH applications.
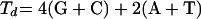 |
(1) |
In this study, we propose a mechanistic approach to simulate hybridizations in FISH. To explore hybridization efficiency, we adopt the notion of the affinity of an oligonucleotide to its folded nucleic acid target as a complementary idea to the accessibility concept. This oligonucleotide affinity is thermodynamically defined and predicted based on equilibrium chemistry and, ultimately, it offers a mechanistic option to understand in situ hybridizations and to design FISH applications.
Thermodynamic definition of probe affinity.
The proposed hybridization mechanism (Fig. 1), adopted from that reported by Matthews et al. (20), incorporates the DNA-RNA, RNA-RNA, and DNA-DNA interactions that take place during FISH. Reaction 1 represents the binding of the probe (Pu) to the available (open) complementary site (Ru). Since the target region on the rRNA is not expected to be in a completely denatured state (12), it must undergo conformational changes for the hybridization to occur, a reaction that is considered in the scheme as the reversible unfolding of Rf (reaction 3). Similarly, the probe itself might have a stable folded structure due to self-complementarity. Thereby, reaction 2 parallels reaction 3 as a folding-unfolding mechanism for the DNA probe.
FIG. 1.

Proposed reaction scheme for FISH. P, R, and PR denote the DNA probe, the target (rRNA), and the DNA/rRNA hybrid, respectively. The subscripts f and u represent folded and unfolded conformations.
We define affinity of the probe to its target as an overall Gibbs free energy change (ΔG°overall) for the reaction scheme in Fig. 1. To obtain ΔG°overall, one needs to estimate the free energy change for the individual reactions (ΔG°i; i = 1, 2, or 3). These values can be predicted from thermodynamic parameters experimentally derived for duplexes and 2D structure motifs of nucleic acids with nearest-neighbor models (reviewed in reference 33). Equilibrium constants for each reaction can be obtained by using the inverse of the fundamental relationship in equation 2, where R is the ideal gas law constant (R = 1.99 × 10−3 kcal/mol K), T is the hybridization temperature, which is typically 46°C in FISH (i.e., 319.15 K), and Ki is the equilibrium constant for reaction i. Then, the equilibrium state of the overall system can be characterized by defining an overall equilibrium constant Koverall according to equation 3, with K1 = [PR]/([Pu] [Ru]), K2 = [Pf]/[Pu], and K3 = [Rf]/[Ru]. Finally, ΔG°overall, the descriptor of probe affinity, can be obtained from Koverall using equation 2.
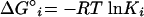 |
(2) |
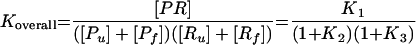 |
(3) |
MATERIALS AND METHODS
Calculation of Gibbs free energy changes.
ΔG°1 was calculated according to equations 4 to 6 using the nearest-neighbor ΔH° and ΔS° values provided by Sugimoto et al. (30) for DNA/RNA duplexes. For a probe that is n nucleotides long there are n − 1 nearest neighbors in the hybrid. The initiation enthalpy and entropy changes are ΔH°initiation = 1.9 kcal/mol and ΔS°initiation = −3.9 kcal/mol (30). The logarithmic term in equation 5 corrects ΔS° for the salt concentration in the hybridization buffer (25). In all hybridizations studied, [Na+] = 0.9 M and T = 319.15 K.
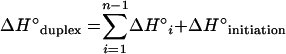 |
(4) |
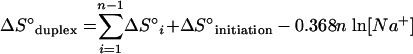 |
(5) |
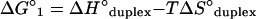 |
(6) |
To calculate ΔG°2 and ΔG°3, mfold version 3.1 (21, 39) was employed for folding the probe and target sequences in silico. When more than one structure was predicted by mfold, the ΔG° output for the optimum folding (i.e., minimum ΔG°) was used. In general, ΔG°2 was directly obtained as the ΔG° value for the probe sequence. When the sequence had no self-complementarity (only about 1% of the probes analyzed), ΔG°2 was indeterminate, as mfold could not predict any structures. In this case, ΔG°2 was assumed to be +4.6 kcal/mol, which is a sufficiently large positive value with a negligible effect on ΔG°overall (equations 2 and 3).
To estimate ΔG°3, two different structures representing the folded (Rf) and unfolded (Ru) conformations at the target site were generated with mfold. Since mfold does not allow salt correction for RNA folding, the default [Na+] of 1 M was used instead of 0.9 M. In the folding of Ru, the bases defining the target site were forced to be unpaired. ΔG°3 was then calculated as follows: ΔG°3 = ΔG°Rf − ΔG°Ru. Four different methods were followed to generate the structures of Rf and Ru with mfold. (i) The input sequence was the domain of the rRNA that included the target site of the probe. In E. coli numbering, there are four different domains of the 16S rRNA, corresponding to residues 1 to 566, 567 to 912, 913 to 1396, and 1397 to 1542 (37), and six different domains of 23S rRNA, with residues 1 to 561, 562 to 1269, 1270 to 1646, 1647 to 2014, 2015 to 2625, and 2626 to 2904 (5). When the target site covered bases on two adjacent domains, the sequence spanning both domains was used. No special constraints were applied (i.e., mfold-predicted structures were used). (ii) The input sequence was defined as the shortest segment that encompassed the helix (or helices) targeted by the probe according to the 16S rRNA secondary structure model of E. coli, which was obtained from the Comparative RNA website (http://www.rna.icmb.utexas.edu/) (10). (This and the following two methods were applied to E. coli 16S rRNA only.) No special constraints were applied. (iii) Watson-Crick and wobble base pairs (not pseudoknots) and loops in the 16S rRNA secondary structure model (10) were imposed on mfold by the appropriate constraint files. In the folding of Ru, the region complementary to the target site was allowed to refold. The input sequences were as in method i, but were adjusted to include complete loops at the end of the domains. (iv) Method iv was the same as method iii, except that the region complementary to the target site was not allowed to make new contacts in the folding of Ru.
The sequences used in ΔG°3 calculations corresponded to the 16S rRNA (GenBank accession number AE000460) and the 23S rRNA (accession number AE000129) of E. coli strain K-12, the 16S rRNA of Pirellula sp. (accession number X81938) and M. sedula (accession number X90481), and the 18S rRNA of S. cerevisiae (accession number J01353). Since the latter three sequences, originally deposited by Behrens et al. (7), were not complete at the 5′ and 3′ ends of the rRNA, the missing bases were assumed complementary to the probes reported to be targeting the terminal segments in the same study (7).
Spatial accessibility of target sites.
The E. coli homology model of the ssu developed by Tung et al. (32) was used for determining the blockage of a probe target site within the 3D structure of the ribosome. Using the atomic coordinates for this model (Protein Data Bank accession code 1M5G) as the input, the solvent-accessible surface area of each residue on the 16S rRNA (ASAi) was obtained with the NACCESS software (16). Then, by removing the protein coordinates from the 3D structure, a hypothetical solvent-accessible surface area was estimated (ASAinucleic acid). The difference between ASAinucleic acid and ASAi defined a parameter representing the hypothetical contact area between proteins and the nucleotide (equation 7).
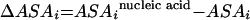 |
(7) |
The equivalent area parameters for the complete target site of a probe (ASAts, ASAtsnucleic acid, and ΔASAts) were calculated by averaging the corresponding values for the target site residues.
Statistical analysis.
Fluorescence intensities reported for the probes used in the development of accessibility maps for 16S rRNA (13) and 23S rRNA (14) of E. coli, 16S rRNA of Pirellula sp. and M. sedula (7), and 18S rRNA of S. cerevisiae (7) were used as hybridization efficiency data sets representing populations of FISH probes sufficiently large to allow correlation analyses. For a sample space of n probes, the degree of association between the array X representing a calculated variable (ΔG°1, ΔG°overall, ΔASAts, etc.) and the array I of the reported fluorescence intensity was quantified by the Pearson product-moment correlation coefficient r(X,I). Thus, r2, the coefficient of determination, indicates what fraction of the variance of I is due to that of X (17). The statistical significance of an r value was evaluated with a two-sided P test, which yielded the number P(X,I). Both r(X,I) and P(X,I) were obtained using MATLAB version 6.5.1. A confidence level of 95% (equivalent to a significance level of 5%, or P = 0.050) was set as the minimum to conclude that there was a significant relation between the calculated variable and fluorescence intensity.
Probe design and synthesis.
DNA probes tested in this study were designed to be complementary to the 16S rRNA of E. coli K-12 (GenBank accession number AE000460). They were synthesized and monolabeled at the 5′ end with fluorescein phosphoramidite at the University of Wisconsin Biotechnology Center. Table 1 presents a complete list of the probes and their specifications.
TABLE 1.
Characteristics of DNA probes targeting specific regions of the 16S rRNA of E. coli
| 16S rRNA regiona | Probe nameb | Position (5′-3′) | Sequence (5′-3′)c | Free energy changes (kcal/mol)
|
Relative probe brightnessd | |||
|---|---|---|---|---|---|---|---|---|
| ΔG°1 | ΔG°2 | ΔG°3 | ΔG°overall-i | |||||
| Helix 9 (144-178) | E148 | 148-165 | CCGTTTCCAGTAGTTATC | −18.1 | 2.0 | −8.7 | −9.4 | 0.59 ± 0.04 |
| E147 | 147-165 | CCGTTTCCAGTAGTTATCC | −20.7 | 2.0 | −8.7 | −12.0 | 0.70 ± 0.03 | |
| E146 | 146-165 | CCGTTTCCAGTAGTTATCCC | −23.3 | 2.0 | −8.7 | −14.6 | 1.00 ± 0.05 | |
| E144 | 144-165 | CCGTTTCCAGTAGTTATCCCCC | −28.5 | 2.0 | −11.2 | −17.3 | 1.11 ± 0.03 | |
| E148 + 2C | 148-165 | CCGTTTCCAGTAGTTATCGG | −18.1 | 0.4 | −8.7 | −9.1 | 0.34 ± 0.06 | |
| E148 + 6C | 148-165 | CCGTTTCCAGTAGTTATCAAACGG | −18.1 | −1.9 | −8.7 | −7.5 | 0.10 ± 0.02 | |
| E148 + 8C | 148-165 | CCGTTTCCAGTAGTTATCGGAAACGG | −18.1 | −5.5 | −8.7 | −3.9 | 0.06 ± 0.01 | |
| Helices 37 and 38 | Eco 1014 | 1014-1031 | GAAGGCACATTCTCATCT | −16.8 | 1.3 | −8.6 | −8.1 | 0.04 ± 0.01 |
| (1006-1036) | E1013 | 1013-1034 | CCCGAAGGCACATTCTCATCTC | −24.5 | −0.7 | −9.8 | −13.8 | 0.66 ± 0.07 |
| E1018 | 1018-1034 | CCCGAAGGCACATTCTC | −18.5 | −0.7 | −9.8 | −7.8 | 0.12 ± 0.02 | |
| Helix 46 (1241-1296) | Eco 1274 | 1274-1291 | ACTTTATGAGGTCCGCTT | −18.4 | 0.4 | −2.3 | −15.8 | 0.49 ± 0.05 |
| E1270 | 1270-1291 | ACTTTATGAGGTCCGCTTGCTC | −24.3 | 0.4 | −3.3 | −20.7 | 0.93 ± 0.06 | |
| Helices 39, 43, and | Eco 1184 | 1184-1201 | TGACTTGACGTCATCCCC | −20.3 | −0.7 | −7.0 | −12.4 | 0.08 ± 0.02 |
| 45 (1158-1199) | E1181 | 1181-1201 | TGACTTGACGTCATCCCCACC | −25.1 | −0.7 | −7.0 | −17.2 | 0.22 ± 0.04 |
| E1177 | 1177-1201 | TGACTTGACGTCATCCCCACCTTCC | −31.3 | −0.7 | −8.0 | −22.4 | 0.40 ± 0.02 | |
| E1172 | 1172-1201 | TGACTTGACGTCATCCCCACCTTCCTCCAG | −38.7 | −0.7 | −12.3 | −25.5 | 0.14 ± 0.01 | |
| E1177 + 5N | 1177-1201 | TGACTTGACGTCATCCCCACCTTCCAAGCC | −31.3 | −0.7 | −8.0 | −22.4 | 0.16 ± 0.02 | |
Helices encompassing the targeted segments (numbered according to reference 13). Numbers in parentheses indicate the positions of the helices in the 16S rRNA.
Probes in italics were first reported by Fuchs et al. (13).
Nucleotides in bold are additions to the first probe of each group; italics identify the stem of hairpin-like probes; underlining indicates nucleotides not complementary to the bases in the target region.
Scale based on defining the brightness of E146 as 1.0. Standard deviations were calculated from triplicate hybridization experiments.
Whole-cell hybridization.
Experiments were carried out with a pure culture of E. coli K-12. Cells were grown overnight at 37°C in Lennox Luria-Bertani broth (Becton Dickinson, Franklin Lakes, N.J.) and harvested during the early stationary growth phase. For fixation, 0.25 ml of cell culture was combined with 0.75 ml of 4% paraformaldehyde in phosphate-buffered saline (PBS; 130 mM NaCl, 10 mM Na2HPO4; pH 7.2). After 30 min, cells were pelleted by a 3-min centrifugation at ∼10,000 × g and resuspended in 1 ml of PBS. The centrifugation-resuspension procedure was repeated three more times to remove the fixative. After the last centrifugation, cells were resuspended in 1 ml of hybridization buffer (0.9 M NaCl, 0.1% sodium dodecyl sulfate, 20 mM Tris [pH 7.2], and a 250 nM concentration [∼1.5 ng/μl] of the probe) and incubated for 3 h at 46°C. For washing excess probe, the solution was replaced with hybridization buffer without probe and the samples were incubated at 46°C for 20 min. Cells were then pelleted and resuspended two times in 1 ml of cold (4°C) buffer with high ionic strength (0.9 M NaCl and 20 mM Tris [pH 8.0]) to prevent the dissociation of hybrids. In experiments involving extended hybridization periods, the cold buffer was changed to a PBS buffer (0.138 M NaCl, 0.0027 M KCl; pH 7.4; Sigma Aldrich Co., St. Louis, Mo.) with 0.01% Tween 20 (Fisher Scientific, Fair Lawn, N.J.) to minimize cell aggregation.
Flow cytometry and data acquisition.
Samples were analyzed on an EPICS XL flow cytometer (Beckman Coulter Co., Fullerton, Calif.) equipped with a 488-nm argon ion laser. Forward-angle light scatter (FS), right-angle light scatter (SS), and fluorescence detectors converted signals to logarithmic scale. Green fluorescence (FL1) was detected using a 525-nm bandpass filter. Commercially prepared saline (IsoFlow; Beckman Coulter) was used as sheath fluid. Prior to measuring the samples, the flow cytometer was calibrated using FlowCheck beads (Fisher Scientific).
A total of 10,000 events falling into a bacterial region loosely defined on the FS-SS plot for E. coli populations were collected for each measurement. Fluorescence was quantified as the mean FL1 of events within a hybridized events gate on the FL1-FS diagram, which was defined with the help of negative controls prepared with a probe that does not bind to rRNA (nonEUB, 5′-ACTCCTACGGAGGCAGC-3′ [35]). In replicate experiments, the variability of the mean FL1 was observed to be proportional to changes in the mean FS (r2 > 0.95 when values covered broad ranges and the regression line was forced to pass through zero), possibly due to a variable extent of cell aggregation (8) in different samples (FS is an indicator of event size). Consequently, all fluorescence measurements were standardized as the mean FL1 over the mean FS.
For each individual experiment, duplicate hybridizations were performed with each probe and duplicate measurements were taken per hybridization. Thus, the result of the experiment was calculated as the average of the four FL1/FS values. Occasionally, a low number of cells (or other unknown factors) would result in a biased FL1 measurement due to excessive background events in the hybridized events gate. These measurements were disregarded as outliers if the FL1/FS value was more than three standard deviations below the average of the remaining FL1/FS values from all replicate experiments (excluding all potential outliers).
Brightness scale.
A scale of probe brightness was defined by assigning a brightness unit of 1.0 to the fluorescence intensity of probe E146 (FL1/FS = 5.25, based on three independent experiments). Thus, all fluorescence intensity results were normalized by dividing the FL1/FS values by 5.25 (Table 1). In this scale, the brightness of all negative controls (i.e., samples hybridized with non-EUB) was generally below 0.02 and, hence, corrections for background fluorescence were not necessary.
In order to link our relative scale to that developed by Fuchs et al. for their E. coli 16S rRNA accessibility map (i.e., class 1 [81 to 100%], class 2 [61 to 80%], class 3 [41 to 60%], class 4 [21 to 40%], class 5 [6 to 20%], and class 6 [0 to 5%], with the percentage based on their brightest probe [13]), probe E146 was tested simultaneously with probes Eco 145 (5′-TTTCCAGTAGTTATCCCC-3′; class 2, 66%), Eco 181 (5′-CTTTGGTCTTGCGACGTT-3′; class 2, 65%), Eco 316 (5′-ACCGTGTCTCAGTTCCAG-3′; class 2, 70%), Eco 1410 (5′-GCAACCCACTCCCATGGT-3′; class 1, 90%), and EUB338 (5′-GCTGCCTCCCGTAGGAGT-3′; class 3, 58%), which are probes that were also used by Fuchs et al. In three independent experiments with log-phase cells as in Fuchs et al.'s protocol, E146 was consistently brighter than Eco 181, Eco 316, Eco 1410, and EUB338 and slightly dimmer (by less than 10%) than Eco 145 (data not shown). In four independent experiments with early stationary phase cells as in the regular protocol of this study, E146 was 1.5 ± 0.1 (mean ± standard deviation) times as bright as EUB338 (3), which is a frequently used probe targeting the bacterial domain and is known to yield moderate to high fluorescence intensity (13). Since these results suggested that E146 is a very bright (class 1 or 2) probe, we assume that the fluorescence intensity of E146 (i.e., 1.0 brightness units in our scale) corresponds to 70% (mid-range of class 2) in Fuchs et al.'s scale. This equivalency was used when a comparison of probe brightness in the two scales was necessary and provided a conservative interpretation of our results.
RESULTS
Evaluation of the proposed hybridization scheme by using the E. coli 16S rRNA accessibility maps of Fuchs et al. (13) and Behrens et al. (7).
The two available 16S rRNA accessibility maps of E. coli were developed using fluorescein (13) and cy3 (7) as the fluorophores. The cy3 map is composed of 176 probes, all of which are also included in the fluorescein map. The fluorescein map has 16 additional probes (hence, a total of 192), since Fuchs et al. analyzed some regions of the 16S rRNA at higher resolution. This database of oligonucleotide probes is ideal for the statistical analysis of the adequacy of the hybridization mechanism shown in Fig. 1. The two data sets covered the 16S rRNA, with at least two probes targeting every nucleotide, and provided brightness of hybridized probes for experiments conducted on a single organism and with appropriate protocols so that cell permeability and ribosome content did not influence the observed variations in probe brightness. In addition, as described below, the data sets offer a wide enough range of ΔG° values for the three reactions in Fig. 1, thus facilitating a statistical analysis of how the brightness of hybridized probes correlates to the free energy changes in each reaction. Table 2 presents a comparison of key statistics describing the ΔG° values, and Table 3 summarizes the correlation coefficients obtained when comparing each ΔG° with probe brightness for the set of probes common to both accessibility maps. A complete table of the ΔG° values calculated for each probe can be found at the website www.cae.wisc.edu/∼noguera/FISH.html.
TABLE 2.
Summary of statistics describing the dispersion of calculated free energy changes and solvent-accessible surface area of the 176 common probes used by Fuchs et al. (13) and Behrens et al. (7) in the generation of the E. coli 16S rRNA accessibility maps
| Calculated parametera | Units | Statistic
|
|||
|---|---|---|---|---|---|
| Mean | Standard deviation | Maximum | Minimum | ||
| ΔG°1 | kcal/mol | −19.6 | 3.1 | −11.4 | −26.1 |
| ΔG°2 | kcal/mol | 0.3 | 1.2 | 4.6b | −2.9 |
| ΔG°3-i | kcal/mol | −7.5 | 2.8 | −0.2 | −13.8 |
| ΔG°3-ii | kcal/mol | −7.7 | 3.2 | −1.0 | −16.1 |
| ΔG°3-iii | kcal/mol | −12.6 | 6.6 | 5.9 | −29.3 |
| ΔG°3-iv | kcal/mol | −15.3 | 8.9 | 5.9 | −35.6 |
| ΔG°overall-i | kcal/mol | −11.6 | 3.5 | −4.0 | −19.5 |
| ΔG°overall-ii | kcal/mol | −11.4 | 3.8 | −1.7 | −18.5 |
| ΔG°overall-iii | kcal/mol | −6.5 | 7.1 | 12.8 | −24.5 |
| ΔG°overall-iv | kcal/mol | −3.9 | 9.2 | 14.4 | −24.5 |
| ΔASAts | Å2/residue | 22.6 | 16.5 | 70.5 | 0.0 |
ΔG°3 was calculated using four different methods (i to iv). ΔG°overall depends on the method used to calculate ΔG°3.
Value corresponds to value assumed when mfold did not predict any probe structure.
TABLE 3.
Correlation coefficients r(X,I) and significance level P(X,I) describing the relation between a calculated parameter (X) and the reported fluorescent intensity (I) of probes used by Fuchs et al. (13) and Behrens et al. (7) for the generation of E. coli 16S rRNA accessibility maps
| Calculated parameter (X)a | Probes common to both maps (n = 176)
|
Probes targeting sites relatively free of protein contact (n = 58)d
|
||||||
|---|---|---|---|---|---|---|---|---|
| Fluorescein labeled
|
Cy3 labeled
|
Fluorescein labeled
|
Cy3 labeled
|
|||||
| r(X,I) | P(X,I) | r(X,I) | P(X,I) | r(X,I) | P(X,I) | r(X,I) | P(X,I) | |
| ΔG°1 | −0.46 | 0.000 | −0.35 | 0.000 | −0.58 | 0.000 | −0.58 | 0.000 |
| ΔG°2 | 0.11 | 0.145 | 0.33 | 0.000 | 0.31 | 0.020 | 0.43 | 0.001 |
| ΔG°3-i | 0.18 | 0.012 | 0.14 | 0.060 | 0.19 | 0.162 | 0.03 | 0.801 |
| ΔG°3-ii | 0.27 | 0.000 | 0.17 | 0.021 | ||||
| ΔG°3-iiib | 0.30 | 0.000 | 0.08 | 0.280 | ||||
| ΔG°3-ivc | 0.30 | 0.000 | 0.03 | 0.733 | ||||
| ΔG°overall-i | −0.57 | 0.000 | −0.47 | 0.000 | −0.72 | 0.000 | −0.61 | 0.000 |
| ΔG°overall-ii | −0.62 | 0.000 | −0.47 | 0.000 | ||||
| ΔG°overall-iii | −0.48 | 0.000 | −0.26 | 0.001 | ||||
| ΔG°overall-iv | −0.43 | 0.000 | −0.17 | 0.036 | ||||
| ΔG°overall-mixed | −0.79 | 0.000 | −0.69 | 0.000 | ||||
| ΔASAts | −0.18 | 0.014 | 0.08 | 0.299 | ||||
ΔG°3 was calculated using four different methods (i to iv). The subscript “mixed” represents results obtained when these methods were combined to maximize the correlation. ΔG°overall depends on the method used to calculate ΔG°3.
Calculations were based on 169 probes, because not all targets allowed usage of method iii.
Calculations were based on 151 probes, because not all targets allowed usage of method iv.
Probes with ΔASAts < 11.3 Å2/residue, equivalent to one half of the mean ΔASAts.
The calculated ΔG°1 values for the 176 probes ranged from −11.4 kcal/mol to −26.1 kcal/mol (Table 2). The correlations (Table 3) were relatively strong, with coefficients (r) of −0.46 for fluorescein-labeled probes and −0.35 for cy3-labeled probes, and were statistically significant above the 99.9% confidence level (i.e., P < 0.001). The negative sign in the correlations agrees with the expectation that a greater stability of a DNA/RNA duplex (i.e., more-negative ΔG°1) would result in higher brightness of the probe.
The free energy change ΔG°2 characterizes the intramolecular reaction of the probe itself. On average, ΔG°2 was 0.3 kcal/mol, with two-thirds of the probes having a positive ΔG°2. A positive ΔG°2 (equivalent to K2 < 1) indicates that the hybridization temperature of 46°C is greater than the melting temperature of the probe structure and, therefore, the folded conformation should not significantly affect the hybridization efficiency. The large fraction of probes with positive ΔG°2 is likely due to the size of probes, which was restricted to between 16 and 19 nucleotides (13), thus minimizing the chances of self-complementarity. The calculated ΔG°2 values were generally low in magnitude compared to ΔG°1 and ΔG°3 (Table 2). The correlation with probe brightness was statistically significant (i.e., confidence at or above 95%) only for the cy3-labeled probes (Table 3). The coefficients were positive in accordance with the hybridization mechanism (Fig. 1), wherein a more negative (lower) ΔG°2 indicates a more stable self-structure of the probe (Pf) and causes a decrease in hybridization efficiency.
The free energy change ΔG°3 depends on the secondary interactions of the target site with the rest of the rRNA. Unlike ΔG°1 and ΔG°2, ΔG°3 cannot be calculated accurately with a straightforward method. Many different ways of estimating ΔG°3 could be conceived, as suggested by Matthews et al. (20) for mRNA and in this study for rRNA (see Materials and Methods). The calculated ΔG°3 values were widely dispersed regardless of the method (Table 2) and generally negative, reflecting the high degree of self-complementarity found in the 16S rRNA. As with ΔG°2, a more negative value of ΔG°3 means more competitive intramolecular folding and should thereby dampen the hybridization, which is consistent with the positive sign of the correlations (Table 3). Taking together the results for the fluorescein- and cy3-labeled probes, method ii produces the best correlations between ΔG°3 and probe brightness, followed by method i.
The overall measure of affinity is obtained by calculating ΔG°overall as a function of ΔG°1, ΔG°2, and ΔG°3 with equations 2 and 3 as explained earlier. Negative correlation coefficients are expected for this variable, since a more negative ΔG°overall represents a greater concentration of the hybrid and, thus, greater brightness from the hybridized probes. The best correlations for ΔG°overall are obtained when ΔG°3 is calculated with methods i and ii (Table 3). Interestingly, if one assumes that different methods of calculating ΔG°3 are valid for hybridizations in different regions of the 16S rRNA molecule, it is possible to increase the strength of the correlation between affinity and brightness, as shown by the ΔG°overall-mixed values in Table 3. These results were obtained by iteratively searching for a combination of ΔG°3 values that maximized the absolute value of the correlation coefficient for ΔG°overall. A regular pattern of which method was best for which region on the 16S rRNA was not evident and, therefore, we selected method i to apply consistently in this study, since it not only provided high correlations but also offered the simplicity of not requiring a priori knowledge of native 16S rRNA secondary structures.
In the end, the efficacy of the proposed mechanism to model hybridization efficiency in FISH was statistically evaluated using the correlations shown in Table 3 for the set of 176 probes. The coefficients were at or above a confidence level of 95% (P ≤ 0.05) with the exception of ΔG°2 in the fluorescein map and some estimates of ΔG°3 in the cy3 map (the confidence level for ΔG°3-i was 94%). Additional confidence in the model comes from the observation that the signs of the coefficients conformed to the hybridization scheme with no exception. Furthermore, the strength of the correlations increased as the variables were combined to estimate ΔG°overall. Finally, the predicted affinity (i.e., ΔG°overall-i) correlates with hybridization efficiency by r values of −0.57 and −0.47 for the fluorescein and cy3 data sets, respectively. This means that the 33% variation in the brightness of fluorescein-labeled probes (r2 = 0.33) and 22% in that of cy3-labeled probes (r2 = 0.22) could be explained by differences in probe affinity.
Importance of the tertiary rRNA structure and ribosomal proteins.
Besides the interactions accounted for in our affinity model (Fig. 1), rRNA-protein and tertiary rRNA-rRNA interactions are molecular associations that can influence hybridization efficiency but are not currently thermodynamically predictable. The recently published high-resolution 3D structure of the ssu of the ribosome (3.0-Å resolution of the ssu of Thermus thermophilus [37]) and the corresponding homology model for E. coli (32) enable the investigation of to what extent rRNA-protein interactions affect hybridization efficiency. Specifically, for each one of the 176 probes in the accessibility map data set, we used the software NACCESS (16) and the E. coli ssu homology model (32) to estimate how much of the water-accessible surface area of a particular target site was covered by proteins, and we correlated these surface area values (ΔASAts) with probe brightness. As shown in Table 2, the resulting ΔASAts values were very dispersed, with some probes targeting completely protein-free sites (ΔASAts = 0.0 Å2/residue) and others targeting sites with intense protein coverage. The mean ΔASAts was 22.6 Å2/residue, which corresponds to 16% of the water-accessible surface area in the target sites (ASAtsnucleic acid) having direct contact with ribosomal proteins. The maximum ΔASAts was 70.5 Å2/residue, equivalent to a maximum protein coverage of 50% in the target site (ΔASAts/ASAtsnucleic acid = 0.5). Table 3 shows that there was a statistically significant (98.6% confidence level) negative correlation between the brightness of the fluorescein-labeled probes and the degree of protein blockage at their target site. However, the correlation was not statistically significant for the cy3-labeled probes. Therefore, proteins surrounding the target site might affect the brightness of fluorescein-labeled probes through specific quenching interactions with the fluorophore rather than by preventing hybridization.
As an additional method to evaluate the effect of ribosomal proteins on hybridization efficiency, we analyzed the correlation between the ΔG° values and probe brightness for a specific subset of probes targeting sites relatively free of proteins (Table 3). These probes made a sample space of 58 elements, which is large enough to be representative of their population. As shown in Table 3, with this subset the strength of the correlation between probe affinity (ΔG°overall-i) and brightness increased from −0.57 to −0.72 for the fluorescein-labeled probes and from −0.47 to −0.61 for the cy3-labeled probes. The magnitude of r values for the other free energy changes also increased, except for the ΔG°3-i of cy3-labeled probes. The general improvement in correlations is consistent with protein coverage having a significant negative effect on hybridization efficiency.
Predictive power of thermodynamic affinity.
Given that the confidence level for the correlations between fluorescence intensity and ΔG°overall-i was greater than 99.9% (P < 0.001) (Table 3), our analysis leaves no doubt that hybridization efficiency is related to probe affinity. This relation is illustrated in Fig. 2a, which shows normalized fluorescence intensity against ΔG°overall-i for the fluorescein-labeled probes used by Fuchs et al. (13). The plot shows an obvious negative slope, which portrays the significance and the sign of the r value. However, the data points are widely scattered, limiting the possibility of using probe affinity as a direct predictor of hybridization efficiency. The large scattering could be due to factors not included in the thermodynamic assessment of probe affinity, such as rRNA-protein or tertiary rRNA-rRNA interactions, quenching of the dye in its microenvironment (19, 23, 31), etc. In particular, the benefit of eliminating the potential influence of proteins is evident by the reduced data scattering when plotting ΔG°overall-i versus probe fluorescence for the subset of probes targeting sites with low protein blockage (Fig. 2b). This reduced data scattering translates into a significant improvement in the r value for this subset of probes (Table 3). Based on the correlation coefficients obtained, thermodynamic affinity can explain 51% of the variations in the brightness of fluorescein-labeled probes (r2 = 0.51) and 37% of that of cy3-labeled probes (r2 = 0.37) when proteins are not of concern. Besides proteins and other factors exterior to the proposed hybridization scheme, a likely source of data scattering is the uncertainty in the estimations of ΔG°3 values. This was demonstrated by the remarkable decrease in scattering and the corresponding increase in correlation obtained when the optimum mixture of ΔG°3 values was used (Table 3), a result that is graphically depicted in Fig. 2c.
FIG. 2.

Relation between normalized fluorescence intensity of fluorescein-labeled probes (13) and ΔG°overall. (a) Complete set of 176 probes; (b) probes targeting sites relatively free of protein contact; (c) all 176 probes when ΔG°3 estimates were mixed to maximize r2. The solid diamond, circle, and square symbols denote probes Eco 1014, Eco 1184, and Eco 1274, respectively.
A potential strategy to reach the actual limits of the predictive power of probe affinity is to focus the correlation analyses on specific regions of the 16S rRNA rather than on the entire molecule. This is because the effects of exterior factors or the uncertainties in the estimation of ΔG°3 are likely to remain relatively constant within a confined region of the 16S rRNA. Taking advantage of the probes used by Fuchs et al. (13) to analyze helices 6 and 18 of 16S rRNA (numbering from reference 13) at high resolution, we evaluated the relation between ΔG°overall-i and fluorescence intensity at these small regions, as shown in Fig. 3. The correlation coefficients were −0.90 (r2 = 0.81) for helix 6 and −0.81 (r2 = 0.68) for helix 18. As anticipated, the slopes of the regression lines differed, probably because one or several factors were affecting the hybridization differently at the two helices. With the plots in Fig. 3, it is not difficult to imagine that the large scattering observed in Fig. 2a could reflect the superimposition of multiple highly correlated region-specific relationships of affinity and brightness having different trends. More importantly, with the region-specific analyses the correlations are high enough to allow the use of probe affinity as an indicator of hybridization efficiency. For instance, to consistently achieve a fluorescence intensity of >40% in the normalized scale of Fig. 3 (equivalent to at least a class 3 probe in the brightness scale defined by Fuchs et al. [13]), the probe should have a ΔG°overall-i of <−13.0 kcal/mol. Likewise, a dim probe that targets these helices is likely to have ΔG°overall-i > −10 kcal/mol.
FIG. 3.

Fluorescence intensity as a function of probe affinity for probes from Fuchs et al. (13) targeting helix 6 (open circles, solid regression line) and helix 18 (solid squares, dashed regression line). The dotted curve represents equation 9 fitted to the data by assuming that a [PR]/[R0] ratio of 1 corresponds to 75% fluorescence and a [PR]/[R0] ratio of 0 represents 3% fluorescence in Fuchs et al.'s normalized scale.
Although simple linear correlations are sufficient to demonstrate the dependence of probe brightness on probe affinity, a linear regression is not necessarily the best model to represent this relation, because it does not capture the thermodynamic relationships in the affinity model. Theoretically, if the total concentration of probe is in excess of the target molecules ([Po] ≫ [Ro], a typical condition in FISH), equation 3 can be rewritten as equation 8 and rearranged to obtain an expression for the hybridized fraction of target molecules as a function of ΔG°overall and [Po] (equation 9). Note that ΔG°overall embraces the GC% and probe length parameters found in classical duplex stability approximations such as equation 1, and [Po] is an additional variable denoting the mechanism by which an increase in probe concentration would force the reaction in Fig. 1 towards the formation of the hybrid. Accordingly, a higher hybrid concentration can be achieved by either increasing [Po] or increasing probe length, with the latter approach resulting in a more negative ΔG°overall. Equation 9 is represented as the dotted line in Fig. 3 when [Po] = 250 nM, which is the approximate probe concentration used by Fuchs et al. (13). Despite the poor fit to the experimental data, the theoretical curve reveals the existence of a transition zone in which hybridization efficiency will be very sensitive to small changes in ΔG°overall. Outside this transition region, plateau zones with low and high hybridization efficiencies are expected, depending on the magnitude of ΔG°overall. The deviations of the data points from the theoretical sigmoidal trend can be attributed to the fact that probes are targeting different locations of the 16S rRNA, albeit concentrated on two helices, and therefore factors other than predicted affinity can influence hybridization efficiency.
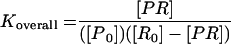 |
(8) |
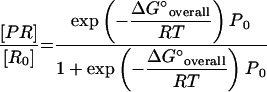 |
(9) |
Figure 4 shows a more rigorous evaluation of the curve represented by equation 9. For this experiment, we selected a region of the 16S rRNA with low protein coverage (helix 9; average ΔASAi = 5.5 Å2) and, starting with E148 as a probe with moderate brightness (Table 1), we modified it to manipulate ΔG°overall-i without significant changes in the target site. First, by elongating the 3′ end of the probe with nucleotides complementary to the target site (probes E147, E146, and E144) we systematically increased probe affinity. Then, to obtain probes with lower affinity we again elongated the 3′ end of probe E148, but this time the additional bases were complementary to those at or near the 5′ end of the probe, thus creating probes with relatively stable hairpin conformations (E148+2C, E148+6C, and E148+8C). Hence, probe affinity was raised by increasing the magnitude of ΔG°1 without a significant change in ΔG°2 or ΔG°3, whereas it was lowered by increasing the magnitude of ΔG°2 with absolutely no change in ΔG°1 or ΔG°3 (see Table 1 for details). In Fig. 4, the theoretical curve was fit to the experimental data by assuming that the average fluorescence intensity of the two brightest probes, E146 and E144, corresponded to saturation of the target sites and that the dimmest probe, E148+8C, showed only background fluorescence.
FIG. 4.

Probe brightness as a function of probe affinity in a subregion of helix 9. The dotted curve represents equation 9 fitted to the data by assuming that [PR]/[R0] values of 1 and 0 corresponded to 1.05 and 0.1 brightness units, respectively.
Use of the affinity model to improve hybridization efficiency.
The examples in Fig. 3 and 4 illustrate the potential use of ΔG°overall-i as a predictor of probe brightness for specific regions of the 16S rRNA. An additional and perhaps more useful application of the correlation between probe affinity and brightness is the use of the affinity concept in conjunction with the accessibility maps to increase the hybridization efficiency of probes targeting potentially inaccessible sites. Figure 5 summarizes the results of experiments carried out at four different regions of the 16S rRNA to illustrate this application. Each experiment is explained in the following sections.
FIG. 5.

Effect of increasing the magnitude of ΔG°overall-i on the fluorescence intensity of probes targeting sites located in helix 9 (circles), helices 37 and 38 (squares), helices 39, 43,and 45 (triangles), and helix 46 (diamonds). The data points (except solid circles) are from Table 1. Solid arrows indicate the trend for each target site. The dotted arrow shows the effect of using 30% formamide in the hybridization of E1177. Data points represented by solid circles are from Fuchs et al. (13), and the dashed line shows the equivalency to the lower limit of class 3 in Fuchs et al.'s scale.
(i) Helix 9.
In the fluorescein accessibility map (13), probes Eco 145, Eco 155, and Eco 163 target only this helix (between positions 144 and 180) and have reported brightnesses in classes 2, 3, and 4, respectively. We estimated the ΔG°overall-i for these probes as −11.1, −10.9, and −7.2 kcal/mol, respectively. In order to find out whether this combined information was sufficient to predict the relative brightness of new probes targeting this region, we designed E148 as a probe with a moderate ΔG°overall-i of −9.4 kcal/mol and expected to reach a hybridization efficiency in between probes Eco 155 and Eco 163. In addition, we elongated E148 to create E146, a probe with a ΔG°overall-i of −14.6 kcal/mol and therefore expected to have a significantly higher hybridization efficiency. As shown in Fig. 5, the resulting probe brightness followed the expected tendency.
(ii) Helices 37 and 38.
Probe Eco 1014 is a class 5 probe in both accessibility maps (7, 13), presumably indicating that this site in the 16S rRNA is inaccessible to FISH probes. As shown in Fig. 2a, its low hybridization efficiency agrees with its relatively low affinity of −8.1 kcal/mol. To create a higher-affinity probe targeting this site, we added three bases to the 5′ end and one base to the 3′ end of Eco 1014 (Table 1). With a ΔG°overall-i of −13.8 kcal/mol, this new probe (E1013) had a substantially greater brightness, about 16-fold higher than that of Eco 1014 (Fig. 5). However, a potential concern with the design method followed was that higher brightness could also arise from the change in the position of the fluorophore. To eliminate this possibility, we used a third probe, E1018, which had an affinity similar to that of Eco 1014, and the position of its fluorophore did not change compared to that of E1013 (Table 1). In this case, lowering the affinity resulted in the expected decrease in brightness (Fig. 5), confirming that the increase in hybridization affinity was indeed caused by the increase in probe affinity.
(iii) Helix 46.
Eco 1274 is reported to be a class 5 probe when labeled with fluorescein (13) and a class 4 probe when labeled with cy3 (7). We selected Eco 1274 as a subject of study because, as shown in Fig. 2a, this probe had low fluorescence intensity even though its affinity was high (ΔG°overall-i = −15.8 kcal/mol). Its low hybridization efficiency must be due to factors other than affinity and, therefore, it is less likely that increasing probe affinity would result in higher probe brightness. By elongating Eco 1274 we created E1270, a probe with very high affinity (ΔG°overall-i = −20.7 kcal/mol). The results for Eco 1274 and E1270 are shown in Fig. 5. Contrary to what Fuchs et al. (13) reported, probe Eco 1274 had almost moderate brightness, which is in agreement with the high ΔG°overall-i and with the cy3 map of Berhens et al. (7). Yet, E1270 was significantly brighter, showing again that a seemingly inaccessible site yielded a higher fluorescent response when the affinity was increased.
(iv) Helices 39, 43, and 45.
Eco 1184 is a class 5 probe in both accessibility maps (7, 13), even though the calculated probe affinity is relatively high (ΔG°overall-i = −12.4 kcal/mol). Similar to Eco 1274, its position in Fig. 2a is at the fringe of the data cluster and, therefore, this is an additional example of a region where factors other than predicted probe affinity may significantly affect hybridization efficiency. By adding three bases to the 3′ end of Eco 1184, we created E1181, which still yielded a fairly low signal intensity despite its relatively high affinity (ΔG°overall-i = −17.2 kcal/mol) (Fig. 5). An additional elongation of this probe by four bases at its 3′ end resulted in E1177, with an even higher affinity of −22.4 kcal/mol, yet a moderate increase in probe brightness (Fig. 5). To find out the limits of what we could achieve by elevating ΔG°overall, we tested E1172, a 30-base-long oligonucleotide with an extraordinary affinity of −25.5 kcal/mol. Interestingly, what we encountered at this extreme was a remarkable reduction in hybridization efficiency compared to E1177, demonstrating that in a site where hybridization is affected by factors other than affinity, there is a limit on the ability to increase probe brightness merely by increasing ΔG°overall.
Is the inaccessibility of some target sites kinetic?
The only data point indicating a decrease in hybridization efficiency upon an increase in affinity was obtained when the probe length was beyond the range of 15 to 25 nucleotides typical for FISH (34). To test whether the size of the probe alone could account for the difference in hybridization, we created E1177+5N, which is a 5-nucleotide elongation of E1177 at the 3′ end, but the added bases were not complementary to the adjacent rRNA or to the rest of the probe (see Table 1 for details). Thus, E1177+5N is equivalent to E1172 in length and to E1177 in ΔG°overall-i. The normalized fluorescence in Table 1 shows that the brightness of E1177+5N was statistically the same as that of E1172 and, therefore, the differences in hybridization between E1177 and E1172 can be attributed to the change in the probe length. A possible mechanism for the size effect would be the limited penetration ability of longer probes through the permeabilized cell wall. Considering this as a potential kinetic limitation, we tested E1177 and E1172 under extended hybridization periods (Fig. 6). We used probe E146 as a control, since it was a very bright probe of ordinary length (20 nucleotides), unlikely to hybridize more efficiently because of longer incubation. Figure 6 clearly shows that with the conventional 3-h hybridization (i.e., hybridization time used in the generation of the accessibility maps [7, 13] and in the region-specific analyses of this study) both E1172 and E1177 were far from reaching equilibrium. After 3 days of hybridization, the fluorescent response of E1172 was 6.5 times higher than by the end of the 3-h hybridization period. Similarly, the brightness of E1177 increased 2.6-fold when allowed to hybridize for 3 to 4 days. The control probe showed a moderate increase of 28% in brightness when incubated for an extended period of time, indicating that the conventional 3-h hybridization period (35) may be, in general, too short to allow the establishment of equilibrium. Nevertheless, E146 was closer to equilibrium after 3 h than the longer E1172 and E1177 probes. Interestingly, even after reaching equilibrium, E1177 was still brighter than E1172. Thus, the lower brightness of E1172 despite its higher predicted affinity (Fig. 5) is not related to kinetic limitations, a result that undermines the intuitive reasoning that long probes experience transport limitations at the cell wall, and leaves the mechanism unclear.
FIG. 6.

Effect of hybridization time on brightness of probes targeting helices 39, 43, and 45. Data points show averages and standard deviations of duplicate hybridizations. Probe E146 was used as a control.
On the other hand, a more striking and useful outcome of Fig. 6 is that E1177 and E1172, which are dim with 3-h hybridization, reach fluorescence intensities comparable to that of E146 given sufficient time, demonstrating that this seemingly inaccessible site can be an adequate target of FISH probes. The slower hybridization kinetics of E1172 and E1177 are likely due to the secondary rRNA structure (15, 18) or even-higher-order structure of the ribosome at their target site. To evaluate the mechanism more directly, we used formamide, a denaturant not expected to affect permeability, to increase the pace of the hybridization reaction by accelerating the denaturation of the target region (4, 15). As shown in Fig. 7, the fluorescence intensity of E1177 and E1172 after 3 h of hybridization remarkably increased with the addition of formamide, showing clearly that the higher-order structure of the ribosome was impeding hybridization when formamide was not used. For probe E146, formamide had an opposite effect on hybridization efficiency, likely because unfolding of the target site was not a limiting factor and formamide had a more pronounced effect on destabilizing the probe-target hybrid.
FIG. 7.

Effect of formamide concentration on brightness of probes targeting helices 39, 43, and 45. Data points show averages and standard deviations of two independent experiments. Probe E146 was used as a control.
Does the model apply to other targets?
Using other available accessibility maps, we expanded the statistical evaluation of the hybridization model to the 23S rRNA of E. coli (14) and the ssu rRNA of three other organisms representing the three different domains of life (7). In Table 4, we report the correlations of free energy changes with fluorescence intensity for these data sets. The r values were statistically significant except for ΔG°2 when the targets were the 23S rRNA of E. coli and the 16S rRNA of M. sedula. The signs of r values confirmed the expected direction of the correlation with no exception. For each target, ΔG°overall-i had the highest correlation score, with a confidence level above 99.9%. These observations are consistent with the trends obtained for the 16S rRNA of E. coli (Table 3), indicating that the model provides useful information regarding the hybridization efficiency of FISH probes for other targets as well.
TABLE 4.
Correlation coefficients r(X,I) and significance level P(X,I) describing the relation between calculated free energy changes (X) and the reported fluorescent intensity (I) of cy3-labeled probes used by Fuchs et al. (14) and Behrens et al. (7) for the generation of accessibility maps
| Calculated parameter (X) | Target rRNA
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 23S of E. colia
|
16S of Pirellula sp.b
|
16S of M. sedulac
|
18S of S. cerevisiaed
|
|||||
| r(X,I) | P(X,I) | r(X,I) | P(X,I) | r(X,I) | P(X,I) | r(X,I) | P(X,I) | |
| ΔG°1 | −0.44 | 0.000 | −0.41 | 0.000 | −0.34 | 0.001 | −0.31 | 0.001 |
| ΔG°2 | 0.08 | 0.260 | 0.35 | 0.001 | 0.13 | 0.189 | 0.19 | 0.050 |
| ΔG°3-i | 0.23 | 0.002 | 0.24 | 0.023 | 0.35 | 0.000 | 0.44 | 0.000 |
| ΔG°overall-i | −0.54 | 0.000 | −0.60 | 0.000 | −0.51 | 0.000 | −0.66 | 0.000 |
Based on 183 probes reported by Fuchs et al. (14).
Based on 88 probes reported by Behrens et al. (7).
Based on 100 probes reported by Behrens et al. (7). The additional 61 probes of the original study were not used because of an apparent mistake in the internet-reported table.
Based on 112 probes reported by Behrens et al. (7).
DISCUSSION
The affinity model proposed in this study to simulate nucleic acid hybridization during FISH includes the probe-target and the intramolecular interactions that are likely to take place in situ (Fig. 1). The formulation and application of the model takes advantage of recent advances in the understanding of nucleic acid interactions (20, 21, 33) and of available software (39) and data (25, 30) for the calculation of the thermodynamic parameters. Testing and validation of the model was primarily through the use of the accessibility map data sets available in the literature (7, 13, 14) and was complemented with experiments targeting specific regions of the 16S rRNA. The correlation analyses of the effect of probe affinity on hybridization efficiency unequivocally demonstrated that probe affinity was a significant factor defining the brightness of a hybridized probe. However, the overall relationship (Fig. 2a) was also characterized by a large scattering of the data, indicating that predictable probe affinity was not the only component of hybridization efficiency. We were able to determine three potential factors that constrain the predictive ability of the model, namely, the uncertainty in ΔG°3 estimations, the presence of proteins at the target region, and the kinetics of hybridization.
First, since different methods of estimating ΔG°3 yield different results, it is clear that predictive errors in ΔG°3 can cause inaccuracy in the estimation of probe affinity. The potential share of this uncertainty in the scattering of the affinity-brightness data was demonstrated as the improvement from Fig. 2a to c when an optimal combination of ΔG°3 calculations was employed. The four methods used for calculating ΔG°3 differ in the restrictions imposed on mfold (i.e., length of input sequence and conformational constraints based on native folding) to generate the target conformation before (Rf) and after (Ru) hybridization with the probe. Method i relies on mfold-predicted structures; thus, conformational changes involving a global rearrangement of the rRNA folding within a domain are allowed to take place in response to probe binding. At the other extreme, method iv strictly imposes the native rRNA structure models (10) to mfold and, therefore, the resulting ΔG°3 defines the penalty for the disruption of the local structure at the target site when the rRNA cannot readjust. Methods ii and iii lie in between these two extremes. The evaluation of all the methods was necessary, since there are a number of uncertainties, such as whether the native secondary structure of the rRNA is preserved after fixation and during the hybridization at 46°C or if the physical restrictions at the in situ ribosome (e.g., proteins) prevent global reorganization of rRNA. In addition, kinetic barriers in the folding and unfolding of the large rRNA molecule may not allow a global equilibrium to be established (27). Without a detailed mechanistic understanding of these complications, we preferred method i in our calculations, which is the simplest to implement and also gives relatively strong correlations between ΔG°overall and fluorescence intensity (Table 3).
Secondly, with a rough approximation of protein blockage at target sites quantified by ΔASAts, ribosomal proteins were shown to adversely affect the predictive ability of affinity (Fig. 2). Since ΔASAts did not correlate with fluorescence intensity for cy3-labeled probes and the significant correlation found for fluorescein-labeled probes was weak (r = −0.18), the evidence for proteins directly blocking the target sites is not strong. However, we should note that the analysis of spatial blockage was based on a native structure model (32), which may or may not represent the configuration of the fixed ribosome appropriately. Thus, the direct effect of protein coverage may be underestimated in our results. Other potential mechanisms by which proteins can influence hybridization efficiency include the direct reduction of probe affinity via specific intimate contacts with the target site (9) and the prevention of the local or global reorganization of rRNA upon hybridization, both of which might affect the thermodynamics or kinetics of reaction 3 (Fig. 1).
Finally, the key assumption of chemical equilibrium behind the calculation of ΔG°overall was shown to be invalid for two target sites on the 16S rRNA (Fig. 6). We have also seen in recent experiments that equilibrium is not established by the end of the conventional 3-h hybridization period for several other regions of 16S rRNA (data not shown). In particular, the relatively large kinetic barrier affecting probes E1177 and E1172 (Fig. 6) was shown to be imposed by the higher-order structure of the ribosome at their target site (Fig. 7). Therefore, varied levels of progress towards the state of equilibrium at different locations in the 16S rRNA likely contribute to the scattering in Fig. 2a. It is important to note that the effects of these factors are likely interwoven. For example, proteins might be limiting the kinetics of hybridization by impeding global conformational changes in the rRNA. Even though in this case the system can be far from equilibrium, the limitations at the target site could be better captured by switching to a method of calculating ΔG°3 that imposes restrictions in the rearrangement of the secondary rRNA structure.
Despite these and probably other factors contributing to the data scattering in Fig. 2a, we were able to show stronger correlations with local analyses (Fig. 3, 4, and 5), possibly because the only significant variable sensitive to small positional changes in these confined regions of the 16S rRNA was the sequence-dependent predictable affinity. As Fig. 5 shows, only at one of the four regions analyzed were we not able to manipulate fluorescent intensity by adjusting probe affinity. In this region, the limitation turned out to be the severe kinetic inaccessibility of the target site, and the desired level of hybridization efficiency was easily attained with probes E1177 and E1172 by letting the system progress towards equilibrium (Fig. 6 and 7). In Fig. 5, we show the improvement in the signal intensity of E1177 upon using 30% formamide to circumvent the kinetic limitations. Since formamide is routinely used in FISH applications to control probe specificity (2, 28), one can consider the brightness that E1177 reached at the moderate stringency of 30% formamide as the maximum effective value for this probe (fluorescence intensity drops at 40% formamide [data not shown]). In the end, Fig. 5 reveals that the hybridization efficiency at every location targeted could be manipulated successfully by a careful evaluation of probe affinity and equilibrium.
Comprehensive model of hybridization efficiency.
Based on the mechanistic interpretation of FISH, facilitated by the affinity model developed and used in the analysis of experimental data in this study, we formulate here a comprehensive model of the multiple factors affecting hybridization efficiency (Fig. 8).
FIG. 8.

Venn diagram representation of the relation among the factors affecting fluorescence intensity in FISH.
(i) Affinity and accessibility.
For an organism with constant and not limiting ribosome content, the hybridization efficiencies of FISH probes can be defined as mainly a function of two factors, namely, the thermodynamic affinity of the probe to the target site and the accessibility of the target site. Thermodynamic affinity is defined by the predictable free energy change of the overall reaction (ΔG°overall) shown in Fig. 1, and accessibility can be regarded as the ability of the probe to reach the target site, regardless of whether hybridization will occur.
Out of the three values defining probe affinity, ΔG°1 and ΔG°2 (Fig. 8) characterize the stability of the DNA-RNA and DNA-DNA interactions, respectively, and depend only on the probe sequence. The third element of the affinity definition, ΔG°3, describes the thermodynamic penalty to break the secondary RNA-RNA interactions at the target site. Altogether, the three thermodynamic components of affinity can be combined assuming equilibrium to give ΔG°overall, which describes the stability of the DNA/RNA hybrid and, as follows from equation 8, ΔG°overall can be correlated with the brightness of a hybridized probe. When the assumption of equilibrium is not valid, the kinetics of the reactions in Fig. 1 will also affect hybridization efficiency. Because association reactions are much faster than dissociation reactions in nucleic acid hybridizations (33, 36), reaction 1 is unlikely to be rate limiting. Between reactions 2 and 3, the latter is more likely the rate-determining step due to the high self-complementarity and the complex kinetic barriers expected in the large 16S rRNA molecule (27). Nevertheless, kinetic limitations can be easily eliminated or minimized with extended incubation periods (Fig. 6) or with the presence of formamide in the hybridization buffer (Fig. 7).
Since breaking the secondary RNA interactions is critical for the probe to reach the target site, we consider ΔG°3 to be influencing both affinity and accessibility and, therefore, we placed this parameter in the intersection of the affinity and accessibility sets in Fig. 8. Other factors included in the definition of accessibility are the tertiary RNA-RNA interactions and the RNA-protein interactions (Fig. 8). Like proteins, tertiary RNA contacts might influence hybridization by direct noncovalent bonding with the target site, by restricting the reorganization of the rRNA, or by directly blocking the target nucleotides. Thermodynamically, the tertiary interactions at the target region are supposed to be much less stable compared to the secondary stacks (33), and as a first approximation, the burden of breaking these bonds can be neglected in comparison to ΔG°3. Thus, the effect of tertiary RNA structures on hybridization is more likely to be kinetic rather than thermodynamic.
The general concept of accessibility involves the intuitive steric hindrance of the target site within the 3D structure of the ribosome (6, 7, 13). Our idea of target site blockage (ΔASAts) was inspired by this geometric view, but we could not find a strong correlation of ΔASAts with hybridization efficiency, as discussed earlier. An equally interesting question is whether the water-accessible surface area of the target site within the overall 3D structure of the ribosome (ASAts) would correlate better than ΔASAts. Our statistical analysis showed no correlation of ASAts with the brightness of fluorescein-labeled probes (13) and a significant, but weak, correlation (r = −0.17; P = 0.02) for the cy3 data set (7). The negative correlation is counterintuitive, since a larger ASAts should indicate better accessibility and, therefore, despite our quantitative attempts, the effect of the spatial positioning of the target site within the ribosome remains inconclusive. This is consistent with the findings of Behrens et al. (6), who used another computer-generated homology model of the E. coli ssu to qualitatively investigate the relation between fluorescence intensity of cy3-labeled probes (7) and spatial location of target sites and concluded that the 3D model could not explain the variation in brightness. In addition to possible structural changes upon formaldehyde fixation, neither approach considered the tertiary contacts between the rRNAs of the small and large subunits of the ribosome (38), as they are not accounted for in the atomic models used (6, 32). Note that the absence of lsu atomic coordinates was not as big of a concern for our protein analysis, as proteins do not contribute significantly to the interaction of the two subunits (6, 32). In the end, we still cannot validate or rule out intuitive spatial accessibility as a factor of hybridization efficiency.
(ii) Probe size.
The comparison of probes E1177 and E1177+5N showed that increasing probe length reduced hybridization efficiency even when ΔG°overall was high. One possibility is that either the cell wall or the target site or both can be more inaccessible to the longer probes, but since the mechanism is not clear at this time, in Fig. 8 we placed probe size as a factor additional to affinity and accessibility. Hybridization experiments with isolated ribosomes would be necessary to further clarify the potential effect of the cell wall on the hybridization efficiency of long probes.
(iii) Fluorophore-related factors.
The fluorescence efficiency of the fluorophore in its microenvironment is not related to the number of hybrids per cell. Therefore, as shown in Fig. 8, it is the complement of hybridization efficiency in the universal set of signal intensity. The significant differences between the fluorescein (13) and cy3 (7) maps for the 16S rRNA of E. coli indicate that this is an important factor that should not be underestimated. While the current knowledge relates this phenomenon to the specific interactions of the fluorophore with the nucleotides (19, 23, 31), it is possible that similar interactions with proteins can also influence the fluorescence of the dye in FISH experiments. This idea is consistent with our correlation analyses for ΔASAts, which showed that protein coverage significantly affected signal intensity in the fluorescein map but not in the cy3 map (Table 3).
Implications of the mechanistic modeling of probe affinity.
The hybridization efficiency formulations developed for FISH in this study derive much more information from probe and target sequences than the typically used rough approximations based on GC% and probe length (13, 28). This is evident from our statistical analysis of the accessibility map data sets (7, 13, 14), for which the probes were designed to have a Td between 48 and 60°C using equation 1. Since the hybridization temperature was 46°C, the Td restriction was assumed to indicate a stable hybrid for a fully accessible target site. Thus, variations in the hybridization efficiency of probes would reflect the differences in the accessibility of target regions. In our comprehensive model (Fig. 8), this strategy is equivalent to eliminating the probe-specific variables ΔG°1, ΔG°2, and probe size, so that hybridization efficiency is a function of accessibility only. However, our correlation analyses (Tables 3 and 4) showed that ΔG°1, and most of the time also ΔG°2, contributed significantly to the variation of probe brightness in the accessibility maps. Therefore, the design restriction with Td was not sufficient to eliminate probe sequence as a factor of hybridization efficiency. We attribute this primarily to the inability of equation 1 to properly define the stability of DNA-RNA interactions in FISH applications, as this equation was empirically derived from DNA-DNA hybridization experiments under specific wash conditions that do not allow a rigorous evaluation of the thermodynamics of duplex formation (29). Instead of such approximations, we propose the use of ΔG°overall-i as a more accurate predictor of probe affinity, because it not only offers a better utilization of probe sequence information with ΔG°1 and ΔG°2 but also includes a numerical evaluation of target site accessibility with ΔG°3.
In fact, the practical value of the numerical information contained in ΔG°overall-i is comparable to that of the empirical information stored in the accessibility maps. Behrens et al. (7) showed that the brightness of 77 probes targeting homologous sites on the 16S rRNAs of E. coli and Pirellula sp. correlated with a significant r value of 0.47. The correlation between ΔG°overall-i and fluorescence intensity was as strong with E. coli (Table 3; r = −0.47 for cy3-labeled probes) and even stronger with Pirellula sp. (Table 4; r = −0.60). Furthermore, ΔG°overall-i can be calculated for any target site, thus overcoming the predictive limitations of empirical maps on rRNA regions where the brightness of probes is sensitive to small positional shifts. This implies that for a given organism, thermodynamic probe affinity is better at predicting the hybridization efficiency of FISH probes than the accessibility data set of the closest organism that has been mapped (e.g., M. sedula map for archaeal organisms). However, since numerical predictions are more meaningful if there is a reference data set connecting affinity to actual fluorescence intensity (e.g., Fig. 2a), the benefits of the model are maximized when used in combination with existing hybridization efficiency data sets. This is illustrated in Fig. 5, which shows that we could take advantage of our numerical approach to increase hybridization efficiency beyond satisfactory levels by increasing the magnitude of ΔG°overall-i provided that kinetic barriers were circumvented, if there were any.
On the other hand, enhanced hybridization efficiency is a necessary but not sufficient condition for a FISH probe to be useful. The design generally requires the optimization of signal intensity and specificity. Since increasing the affinity of a probe by elongating it may cause nonspecific binding to organisms with few mismatches, it is important to know the threshold of ΔG°overall-i which would ensure efficient hybridization (as defined by the designer) with the target organism while preventing an excessive increase in affinity for nontarget organisms. Thus, we specifically recommend a ΔG°overall-i of ca. −13.0 kcal/mol to increase the possibility of designing a working probe. This threshold value is where equation 9 reaches the upper plateau in Fig. 4 at the typical probe concentration of 250 nM (ca. 1.5 ng/μl). Significantly lower magnitudes of ΔG°overall-i will likely result in dim probes (ΔG°overall-i > −10 kcal/mol) or probes within the transition zone described in Fig. 3 and 4 and, therefore, hybridization efficiency could be very susceptible to other factors or uncertainties. It is noteworthy that setting ΔG°overall-i to −13.0 kcal/mol is analogous to keeping Td slightly higher than the hybridization temperature, as was done with the design of probes for the generation of the accessibility maps. When all the accessibility map data sets are considered, the percentage of probes that are class 3 or brighter (i.e., moderate to high brightness) is between 40 and 63%, indicating that nearly half of the probes did not hybridize efficiently with the common constraint of 48°C < Td < 60°C. However, when only the probes with ΔG°overall-i of <−13.0 kcal/mol are considered, the percentage of class 3 or brighter probes increases to 68 to 94%, demonstrating that the probability of designing a useful probe increases with this constraint. The effectiveness of probe design should be expected to be even higher if equilibrium is established, since this study also revealed that some sites identified as inaccessible in the maps could be strongly influenced by the kinetics of the hybridization reaction. Indeed, recent studies in our lab have shown that when the threshold of −13.0 kcal/mol is used as a design criterion and the hybridization is allowed to reach equilibrium, over 90% of the probes yield moderate to high brightness (Yilmaz et al., unpublished data). In conclusion, in addition to providing a mechanistic approach to predict hybridization efficiency in FISH via ΔG°overall-i, our analysis revealed that currently published accessibility maps are conservative predictors of accessibility, since sites reported as inaccessible can be effectively targeted by increasing probe affinity and/or allowing equilibrium to be reached. Besides the immediate implications discussed above, new mechanistic approaches based on the affinity model should lead to the design of new experiments for the continuous improvement of our understanding of the mechanisms that govern in situ hybridizations.
Acknowledgments
This research was supported by National Science Foundation grants BES-9875642 and BES-0302618.
We thank Diana M. Downs for providing us with the E. coli K-12 pure culture. We are indebted to Julie Zilles for experimental recommendations and helpful discussions. The technical assistance of Hatice Eser Okten is gratefully acknowledged.
REFERENCES
- 1.Amann, R., B. M. Fuchs, and S. Behrens. 2001. The identification of microorganisms by fluorescence in situ hybridisation. Curr. Opin. Biotechnol. 12:231-236. [DOI] [PubMed] [Google Scholar]
- 2.Amann, R., W. Ludwig, and K.-H. Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amann, R. I., B. J. Binder, R. J. Olson, S. W. Chisholm, R. Devereux, and D. A. Stahl. 1990. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56:1919-1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amann, R. I., S. J., R. Devereux, R. Key, and D. A. Stahl. 1992. Molecular and microscopic identification of sulfate-reducing bacteria in multispecies biofilms. Appl. Environ. Microbiol. 58:614-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ban, N., P. Nissen, J. Hansen, P.-B. Moore, and T. A. Steitz. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289:905-920. [DOI] [PubMed] [Google Scholar]
- 6.Behrens, S., B. M. Fuchs, L. Mueller, and R. Amann. 2003. Is the in situ accessibility of the 16S rRNA of Escherichia coli for Cy3-labeled oligonucleotide probes predicted by a three-dimensional structure model of the 30S ribosomal subunit? Appl. Environ. Microbiol. 69:4935-4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behrens, S., C. Rühland, J. Inácio, H. Huber, A. Fonseca, I. Spencer-Martins, B. Fuchs, and R. Amann. 2003. In situ accessibility of small-subunit rRNA of members of the domains Bacteria, Archaea, and Eucarya to Cy3-labeled oligonucleotide probes. Appl. Environ. Microbiol. 69:1748-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biegala, I. C., F. Not, D. Vaulot, and N. Simon. 2003. Quantitative assessment of picoeukaryotes in the natural environment by using taxon-specific oligonucleotide probes in association with tyramide signal amplification-fluorescence in situ hybridization and flow cytometry. Appl. Environ. Microbiol. 69:5519-5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brodersen, D. E., W. M. Clemons, A. P. Carter, B. T. Wimberly, and V. Ramakrishnan. 2002. Crystal structure of the 30S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16S RNA. J. Mol. Biol. 316:725-768. [DOI] [PubMed] [Google Scholar]
- 10.Cannone, J. J., S. Subramanian, M. N. Schnare, J. R. Collett, L. M. D'Souza, Y. Du, B. Feng, N. Lin, L. V. Madabusi, K. M. Muller, N. Pande, Z. Shang, N. Yu, and R. R. Gutell. 2002. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed]
- 11.DeLong, E., G. Wickham, and N. Pace. 1989. Phylogenetic stains: ribosomal RNA-based probes for the identification of single cells. Science 243:1360-1363. [DOI] [PubMed] [Google Scholar]
- 12.Frischer, M. E., P. J. Floriani, and S. A. Nierzwicki-Bauer. 1996. Differential sensitivity of 16S rRNA targeted oligonucleotide probes used for fluorescence in situ hybridization is a result of ribosomal higher order structure. Can. J. Microbiol. 42:1061-1071. [DOI] [PubMed] [Google Scholar]
- 13.Fuchs, B., G. Wallner, W. Beisker, I. Schwippl, W. Ludwig, and R. Amann. 1998. Flow cytometric analysis of the in situ accessibility of Escherichia coli 16S rRNA for fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 64:4973-4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuchs, B. M., K. Syutsubo, W. Ludwig, and R. Amann. 2001. In situ accessibility of Escherichia coli 23S rRNA to fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 67:961-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gamper, H. B., G. D. Cimino, and J. E. Hearst. 1987. Solution hybridization of crosslinkable DNA oligonucleotides to bacteriophage M13 DNA. J. Mol. Biol. 197:349-362. [DOI] [PubMed] [Google Scholar]
- 16.Hubbard, S. J., and J. M. Thornton. 1993. NACCESS. University College, London, England.
- 17.Kirk, R. E. 1984. Elementary statistics, 2nd ed. Brooks/Cole Publishing Company, Monterey, Calif.
- 18.Lima, W. F., B. P. Monia, D. J. Ecker, and S. M. Freier. 1992. Implications of RNA structure on antisense oligonucleotide hybridization kinetics. Biochemistry 31:12055-12061. [DOI] [PubMed] [Google Scholar]
- 19.Marras, S. A. E., F. R. Kramer, and S. Tyagi. 2002. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 30:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathews, D., M. Burkard, S. Freier, J. Wyatt, and D. Turner. 1999. Predicting oligonucleotide affinity to nucleic acid targets. RNA 5:1458-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathews, D., J. Sabina, M. Zuker, and D. Turner. 1999. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 288:911-940. [DOI] [PubMed] [Google Scholar]
- 22.Moter, A., and U. B. Gobel. 2000. Fluorescence in situ hybridization (FISH) for direct visualization of microorganisms. J. Microbiol. Methods 41:85-112. [DOI] [PubMed] [Google Scholar]
- 23.Nazarenko, I., R. Pires, B. Lowe, M. Obaidy, and A. Rashtchian. 2002. Effect of primary and secondary structure of oligodeoxyribonucleotides on the fluorescent properties of conjugated dyes. Nucleic Acids Res. 30:2089-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roller, C. M., M. Wagner, R. Amann, W. Ludwig, and K.-H. Schleifer. 1994. In situ probing of gram-positive bacteria with high DNA G+C content using 23S rRNA-targeted oligonucleotides. Microbiology 140:2849-2858. [DOI] [PubMed] [Google Scholar]
- 25.SantaLucia, J., Jr. 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 95:1460-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schonhuber, W., B. M. Fuchs, S. Juretschko, and R. Amann. 1997. Improved sensitivity of whole-cell hybridization by the combination of horseradish peroxidase-labeled oligonucleotides and tyramide signal amplification. Appl. Environ. Microbiol. 63:3268-3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sohail, M., S. Akhtar, and E. M. Southern. 1999. The folding of large RNAs studied by hybridization to arrays of complementary oligonucleotides. RNA 5:646-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stahl, D., and R. Amann. 1991. Development and application of nucleic acid probes, p. 205-248. In E. Stackebrandt and M. Goodfellow (ed.), Nucleic acid techniques in bacterial systematics. John Wiley & Sons Ltd., West Sussex, United Kingdom.
- 29.Suggs, S. V., T. Hirose, T. Miyake, E. H. Kawashima, and M. J. Johnson. 1981. Use of synthetic oligodeoxyribonucleotides for the isolation of specific cloned DNA sequences, p. 683-693. In D. Brown and C. F. Fox (ed.), Developmental biology using purified genes. Academic Press, Inc., New York, N.Y.
- 30.Sugimoto, N., S. Nakano, M. Katoh, A. Matsumura, H. Nakamuta, T. Ohmichi, M. Yoneyama, and M. Sasaki. 1995. Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry 34:11211-11216. [DOI] [PubMed] [Google Scholar]
- 31.Torimura, M., S. Kurata, K. Yamada, T. Yokomaku, Y. Kamagata, T. Kanagawa, and R. Kurane. 2001. Fluorescence-quenching phenomenon by photoinduced electron transfer between a fluorescent dye and a nucleotide base. Anal. Sci. 17:155-160. [DOI] [PubMed] [Google Scholar]
- 32.Tung, C., S. Joseph, and K. Y. Sanbonmatsu. 2002. All-atom homology model of the Escherichia coli 30S ribosomal subunit. Nat. Struct. Biol. 9:750-755. [DOI] [PubMed] [Google Scholar]
- 33.Turner, D. 2000. Conformational changes, p. 259-334. In V. A. Bloomfield, D. M. Crothers, and I. J. Tinoco (ed.), Nucleic acids: structures, properties, and functions. University Science Books, Sausalito, Calif.
- 34.Wagner, M., M. Horn, and H. Daims. 2003. Fluorescent in situ hybridisation for the identification characterization of prokaryotes. Curr. Opin. Microbiol. 6:302-309. [DOI] [PubMed] [Google Scholar]
- 35.Wallner, G., R. Amann, and W. Beisker. 1993. Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry 14:136-143. [DOI] [PubMed] [Google Scholar]
- 36.Wetmur, J. G. 1991. DNA probes: applications of the principles of nucleic acid hybridization. Crit. Rev. Biochem. Mol. Biol. 26:227-259. [DOI] [PubMed] [Google Scholar]
- 37.Wimberly, B.-T., D.-E. Brodersen, V.-M.-.J. Clemons, R. Morgan-Warren, C. von Rhein, T. Hartsch, and V. Ramakrishnan. 2000. Structure of the 30S ribosomal subunit. Nature 407:327-339. [DOI] [PubMed] [Google Scholar]
- 38.Yusupov, M. M., G. Z. Yusupova, A. Baucom, K. Lieberman, T. N. Earnest, J. H. Cate, and H. F. Noller. 2001. Crystal structure of the ribosome at 5.5 Å resolution. Science 292:883-896. [DOI] [PubMed] [Google Scholar]
- 39.Zuker, M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]