

Abstract
The hypoxia-inducible factor 1α (HIF-1α) and its microRNA target, miR-210, are candidate tumor-drivers of metabolic reprogramming in cancer. Neuroendocrine neoplasms such as paragangliomas (PGLs) are particularly appealing for understanding the cancer metabolic adjustments because of their associations with deregulations of metabolic enzymes, such as succinate dehydrogenase (SDH), and the von Hippel Lindau (VHL) gene involved in HIF-1α stabilization. However, the role of miR-210 in the pathogenesis of SDH-related tumors remains an unmet challenge. Herein is described an in vivo genetic analysis of the role of VHL, HIF1A and SDH on miR-210 by using knockout murine models, siRNA gene silencing, and analyses of human tumors. HIF-1α knockout abolished hypoxia-induced miR-210 expression in vivo but did not alter its constitutive expression in paraganglia. Normoxic miR-210 levels substantially increased by complete, but not partial, VHL silencing in paraganglia of knockout VHL-mice and by over-expression of p76del-mutated pVHL. Similarly, VHL-mutated PGLs, not those with decreased VHL-gene/mRNA dosage, over-expressed miR-210 and accumulate HIF-1α in most tumor cells. Ablation of SDH activity in SDHD-null cell lines or reduction of the SDHD or SDHB protein levels elicited by siRNA-induced gene silencing did not induce miR-210 whereas the presence of SDH mutations in PGLs and tumor-derived cell lines was associated with mild increase of miR-210 and the presence of a heterogeneous, HIF-1α-positive and HIF-1α-negative, tumor cell population. Thus, activation of HIF-1α is likely an early event in VHL-defective PGLs directly linked to VHL mutations, but it is a late event favored but not directly triggered by SDHx mutations. This combined analysis provides insights into the mechanisms of HIF-1α/miR-210 regulation in normal and tumor tissues potentially useful for understanding the pathogenesis of cancer and other diseases sharing similar underpinnings.
Keywords: succinate dehydrogenase, hypoxia inducible factor, von hippel lindau, paragangliomas, miR-210
INTRODUCTION
A defining hallmark of cancer is metabolic reprograming. Most of cancer cells use glycolysis-based metabolism for proliferation and reprogramming of the tricarboxylic acid cycle and mitochondrial oxidative phosphorylation to adapt to challenging conditions [1]. Aside this general cancer feature, there is a wealth of evidences proving that altered metabolic enzymes and their metabolites are also oncogenic and not simply a product of tumor proliferation [2]. This is particularly relevant in pheochromocytomas and paragangliomas (PCC/PGLs) which frequently develop in patients with germline mutations of genes encoding the succinate dehydrogenase mitochondrial complex II (SDH). These associations provide a useful tumor model to unravel the molecular connections between mitochondrial activity and cancer. Importantly, besides PCC/PGLs, SDH inactivation is increasingly observed in other neuroendocrine malignancies as well as in gastrointestinal neoplasms and renal cell carcinomas [3], and, thus, it is expected that it will acquire a more general clinical relevance in cancer pathogenesis.
PGLs are rare neuroendocrine tumors of the autonomous nervous system. Although the majority of PGLs occur sporadically, about 30-45% of these tumors are hereditary due to germline mutations in the succinate dehydrogenase SDHB, SDHC, SDHD, SDHA, and SDHAF2 genes (hereafter SDHx genes), among other predisposing genes [4]. Mutations in any of the SDHx genes have been associated to succinate accumulation. In vitro assays revealed that excess of intracellular succinate may lead to increased transcriptional activity of hypoxia-inducible factors (HIF) [5, 6] and up-regulation of pro-tumorigenic HIF target genes, such as carbonic anhydrase 9 (CA9) and hypoxia-inducible factor prolyl hydroxylase 3 (EGLN3), irrespective of oxygen levels (a phenomenon defined as ‘pseudohypoxia’). PGLs may also associate with germline mutations in the von Hippel-Lindau (VHL) tumor suppressor [7]. Inherited mutations in the VHL gene cause the VHL disease, an autosomal-dominant neoplastic disease that is associated with various tumour types, including clear cell renal cell carcinomas, haemangioblastomas, pancreatic neuroendocrine tumours and PCC/PGLs [8]. VHL type 1 disease is characterized by development of clear cell renal cell carcinomas and hemangioblastomas, but not PCCs, and is associated with gross deletions in VHL. Type 2 disease is predominantly associated with VHL missense mutations and the development of PCCs (type 2C) or PCCs and hemangioblastomas (type 2A) or PCCs, hemangioblastomas and RCCs (type 2B). Somatic VHL mutations (i.e. mutations in tumor but not germline DNA) have been also described in about 9% of PGLs [9–11]. Intriguingly, the two types of VHL somatic mutations (type 1 and type 2) have been identified in parasympathetic PGLs [11] and are thus thought to be involved in the development of these tumors via a molecular mechanism not completely understood. VHL functions as the substrate recognition component of an E3-ubiquitin ligase, which targets HIF for proteasomal degradation under normoxic conditions [12]. Thus, HIF represent the molecule where SDH and VHL dysfunctions converge in PGLs. Nonetheless, the role of HIF in SDH-related tumorigenesis remains controversial. Some reported transcription profiles have shown that SDHx-mutated PGLs (SDHx-PGLs) showed enrichment in genes associated with the HIF-pathway and accumulate HIF-1α [13, 14] whereas others did not find such an association thus arguing against the pseudohypoxia hypothesis in SDHx-PGLs [11, 15, 16]. In contrast to SDHx-PGLs, the canonical HIF signaling pathway has been more clearly detected in most tumors that carry VHL mutations [11].
Studies in VHL-related PGLs (VHL-PGLs) revealed that the HIF-pathway involves, among other cues, the HIF-1α-microRNA target, miR-210 [11, 16]. Importantly, VHL-PGLs, in addition to accumulate HIF-1α, express low levels of SDHB likely as a result of miR-210 up-regulation which also represses expression of other genes essential for mitochondrial metabolism [17]. This suggests that miR-210 is a potential oncomiR in VHL-deficient tumors which impact on mitochondria metabolism [16]. Thus, understanding of the in vivo mechanisms of miR-210 expression should increase our knowledge on cancer pathogenesis and allow the identification of cancer-specific vulnerabilities that could be exploited therapeutically. However, the role of miR-210 in the pathogenesis of SDH-related tumors remains an unmet challenge. These premises encouraged us to carry out an in-depth analysis of the role of HIF-1α, SDH, and VHL on miR-210 expression by using knockout murine models and analyses of human tumor material.
RESULTS
In vivo roles of VHL and HIF-1α on the expression of miR-210
We first addressed whether the expression of miR-210 in the paraganglionic system is regulated by VHL and/or HIF-1α activity in vivo. To this end, we analyzed miR-210 levels in tissues isolated from mice with either VHL or HIF1A conditionally deleted in the neural crest derived cells. We present here data obtained from adrenal medulla. Figure 1A shows a significant 13-fold increase of miR-210 levels in the adrenal gland of VHL-null mice in comparison with wild type mice. However, miR-210 levels did not change in heterozygous tissues obtained from VHL +/- thus indicating that a single functional copy of VHL produce enough functional VHL. In contrast to VHL, absence of HIF-1α did not alter the constitutive expression of miR-210. As shown in Figure 1B, miR-210 levels in the adrenal gland of normoxic knockout HIF1A −/− or HIF1A +/- mice were similar to those of wild type HIF1A +/+. Similarly, mRNA levels of other HIF-1α-targets, EGLN3 and CA9, did not decrease in the absence or partial loss of HIF-1α. To further analyze the role of HIF-1α on miR-210 expression, wild type HIF1A +/+ and knockout HIF1A −/− mice were exposed to hypoxia (10% O2) for 30 days prior to organ isolation to allow for HIF-1α accumulation in tissues. Under these conditions, the functional inactivation of HIF-1α significantly reduced hypoxic expression of miR-210 as well as EGLN3 in the adrenal medulla (Figure 1C).
Figure 1. In vitro and in vivo analysis of the role of VHL and HIF-1α on miR-210 expression.
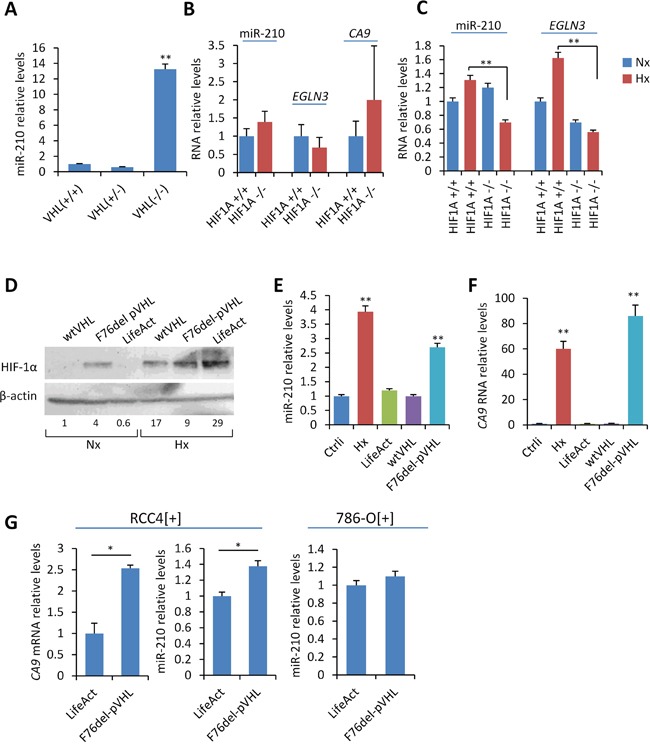
A, B, C. miR-210, CA9 and EGLN3 RNA levels were quantified by RT-qPCR in adrenal gland obtained from the indicated mice genotypes. For analysis of HIF1A knockouts, HIF1A +/+ and HIF1A −/− mice were exposed to hypoxia (10% O2) for 30 days prior to isolation of adrenal glands (panel C, hypoxia) or left under atmospheric O2 conditions (panel B, normoxia). D. Western blot showing HIF-1α protein levels upon ectopic expression of F76del VHL or wild type VHL in SCC40 cells incubated under normoxic (Nx: 21% O2) or hypoxic (Hx: 1% O2) conditions for 24 hours. Cells transiently transfected with LifeAct plasmid were used as controls (mock). β-actin, total protein-loading control. Fold change of HIF-1α protein levels upon normalization to total protein expression is shown below the Western blot image. E, F, G. miR-210 and CA9 RNA relative levels in SCC40 (E, F) and RCC4[+] or 786-O[+] cells (G) treated as indicated in (D). CA9 mRNA levels were undetectable in 786-O[+] cells even when incubated under hypoxic conditions. * P < 0.05 ** indicates P < 0.01.
Given that most tumors carrying VHL inactivating mutations are not necessarily accompanied by loss of heterozygosity, we also analyzed the impact of a VHL cancer-associated mutation, F76del VHL, on miR-210 expression by using SCC40 cells which, endogenously, express wild type VHL [18, 19]. F76del mutant was selected because represents the VHL gene alteration (c.227_229delTCT) associated with a wide spectrum of cancers including PGLs and no-neuroendocrine neoplasms related to VHL disease. We first used a squamous cell carcinoma cell line, SCC40, which has been well-characterized at the genetic and functional level with regard to HIF signaling [18, 19]. SCC40 cells were transfected with a VHL-expressing construct encoding for F76del-VHL. As controls, we used a VHL-expressing construct containing the wild type gene (wtVHL) or a LifeAct construct expressing an actin binding peptide (mock). Figure 1D shows that HIF-1α protein was not detected in mock-transfected cells incubated under normoxic conditions. By contrast, HIF-1α accumulated under normoxic conditions in cells transfected with F76del-VHL with further increase upon hypoxic treatment (1% O2, 24 h). As expected, normoxic accumulation of HIF-1α was not observed by ectopic expression of wild type-VHL. Consistent with our previously reported findings in PGL tissues carrying mutant F76del-pVHL [11], a ~ 3- and ~ 90-fold increase of miR-210 and CA9 RNA levels, respectively, was detected upon ectopic expression of F76del-VHL under normoxic conditions as compared with wild type VHL or mock-transfected cells (Figure 1E and 1F). Because SCC40 cells endogenously express wild type VHL, these data show that a mutant non-functional F76del-VHL may abolish ubiquitination of HIF-1α by wild type VHL thus exerting a dominant negative effect that lead to the activation of the HIF-1/miR-210 pathway.
The above results were replicated in RCC4 renal carcinoma cells expressing wild type VHL (RCC4[+]). In the absence of a human cell model suitable for in vitro analysis of PGLs, cancer cells derived from clear cell renal cell carcinomas are valuable tools for studies of parasympathetic PGLs because of the genetic similarities between both types of tumors [11]. As shown in Figure 1G, a significant increase of miR-210 and CA9 was detected upon expression of the F76del-VHL mutant in RCC4[+] cells. This result was not replicated in 786-O[+] cells that express wild type VHL and the HIF-2α rather than HIF-1α subunit thus suggesting that HIF-1α is the predominant HIF-α subunit involved in the pseudohypoxic up-regulation of miR-210.
Roles of SDHD and SDHB on the expression of miR-210
To determine whether SDH loss of function exert any effect on miR-210 expression, we used previously reported established cell lines (MEF and BMK) obtained from a tamoxifen-inducible SdhD knockout mutant mouse [20]. Homozygous SdhD −/−, but not heterozygous SdhD +/-, MEF and BMK cell lines are known to partially recapitulate the HIF pathway in comparison to wild type homozygous SdhD +/+ cell lines. By contrast, tissues (such as adrenal gland) derived from conditional homozygous SdhD −/− mice do not over-express HIF-1α nor its targets [20] and therefore they were not expected to over-express miR-210. Analysis of miR-210 in MEF and BMK cells revealed that ablation of the two (homozygous SdhD −/−) or one (heterozygous SdhD +/-) SdhD alleles failed to induce miR-210 over-expression in BMK-cells and even caused a reduction of miR-210 expression levels in MEF cells (Figure 2A). These data contrasted with the reported link between SDHD deficiency and HIF-1α accumulation what prompted us to use another cell-system to clarify the role of decreased mitochondrial complex II activity on miR-210 expression. We first used thenoyltrifluoroacetate (TTFA) which binds to the ubiquinone docking sites and abrogates the electron flow at the SDH complex. Figure 2B shows that inhibition of the succinate:ubiquinone oxidoreductase activity by 0.5 mM TTFA did not induce miR-210 over-expression in VHL-deficient (786-O[−]) or VHL-proficient (786-O[+]) cells. On the contrary, TTFA significantly reduced miR-210 levels in VHL-deficient cells. 786-O cells are known to mainly express HIF-2α subunit (although minimal levels of HIF-1α protein have been detected under hypoxic conditions [11]). Thus, we also tested the effect of TTFA on SCC40 cells which express HIF-1α subunit. As shown in Figure 2C, TTFA treatment did not induce miR-210 expression but slightly, although significantly, reduced it in SCC40 cells. Decreased mRNA levels were also detected for EGLN3, another HIF-target gene, in SCC40 cells treated with TTFA and in 786-O[-] cells (Figure 2D).
Figure 2. Analysis of the role of SDHD on miR-210 expression.
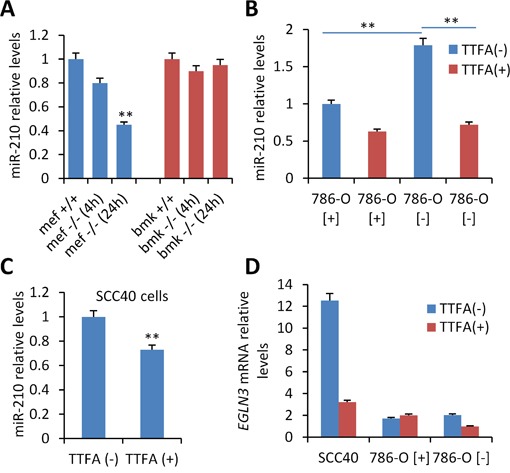
A. Mouse embryonic fibroblasts (MEFs) and baby mouse kidney (BMK) cells obtained from SDHD-ESR −/− mice and their homozygous SDHD flox/+ (+/+) and heterozygous SDHD flox/− (+/−) littermates were cultured in medium supplemented with 4-hydroxy-tamoxifen for 24 hours. B, C, D. VHL-proficient (786-O[+]) and VHL-deficient (786-O[-]) (B, D) or SCC40 cells (C, D) were incubated with 0.5 mM TTFA (TTFA(+)) or solvent (TTFA(-)) for 16 hours prior to miR-210 (B, C) or EGLN3 (G) quantification by RT-qPCR. Values are expressed as mean ratios ± SD **, P <0.01, unpaired t test.
The role of the SDH complex on miR-210 expression was further explored in cultured cell lines by using siRNAs targeting the SDHD or SDHB genes. For this analysis, we used cells expressing wild type VHL such as 786-O[+] cells, which mainly express HIF-2α subunit, and RCC4[+] and SCC40 cells, which mainly express HIF-1α subunit. Endogenous SDHD and SDHB mRNA levels were found to be 40% to 90% lower in cells transfected with either of the two siRNAs (siSDHB or siSDHD) as compared to control siRNA (siCtrl)-transfected cells (data not shown). However, increased miR-210 levels were not found in any of the transfected cells (Figure 3), thus suggesting that full SDH function is not required for HIF-α-independent miR-210 expression. The same experimental strategy was then applied but using cells that were incubated under hypoxic conditions (1% O2 for 48 h) after siRNA transfection to allow for HIF-α-dependent miR-210 over-expression (Figure 3). As expected, increased levels of miR-210 were detected in hypoxic versus normoxic conditions. However, siSDHD or siSDHB did not further increase the hypoxic effect on miR-210 levels. Analysis of CA9 and EGLN3 mRNA levels also revealed that other HIF-α-target genes were not induced by decreased SDHD or SDHB expression either in normoxia or in hypoxia. Only a subtle inductive effect was found for CA9 in RCC4 [+] cells (CA9 gene could not be analyzed in 786-O[+] cells because its mRNA was not detected neither in normoxia nor in hypoxia). In agreement with those data, immunocytochemical analysis of HIF-1α revealed nuclear accumulation of this protein in siSDHB- or siSDHD-transfected cells incubated under hypoxic conditions but not under normoxic conditions (Figure 4).
Figure 3. Absence of miR-210 up-regulation by siRNA-mediated silencing of SDHB or SDHD.

miR-210, CA9 and EGLN3 RNA levels were analyzed in the indicated cells following treatment with SDHD (siSDHD), SDHB (siSDHB) or control (siCtrl) siRNAs. 24 hours after transfection, cell were incubated under normoxic (Nx) or hypoxic (Hx, 1% O2 for 25 hours) conditions. Values are expressed as mean ratios ± SD.
Figure 4. Absence of HIF-1α protein accumulation in nuclei of cells treated with SDHB or SDHD-siRNAs.
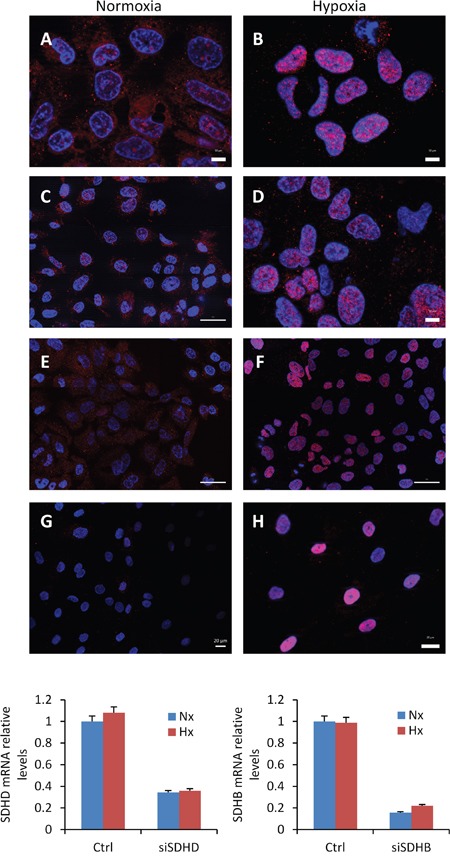
Immunocytochemical analysis of HIF-1α in SCC40 cells treated with Ctrl- (A, B), SDHB- (C, D), or SDHD- (E, F) -siRNAs and exposed to normoxia (A, C, E) or hypoxia (1% O2 for 24 h) (B, D, F). (G, H) Immunocytochemical analysis of HIF-1α in tumor associated fibroblasts derived from human PGL carrying SDHD mutation (SDHD-TAFs) and incubated under normoxic G. or hypoxic (1% O2 for 24 h) H. conditions; note the higher intensity of HIF-1α immunostaining (pink) in hypoxic SDHD-TAF in comparison with siSDHB- or siSDHD-treated SCC40 cells. Quantification of the SDHB or SDHD mRNA levels shows significant reduction of the mRNA levels after siRNA treatments.
The above data shows that HIF-1α accumulation is not triggered, at least in a significant and detectable level, by absence of SDHD activity or decreased SDHB or SDHD protein levels. We next tested whether HIF-1α accumulation could be associated with the presence of SDHx mutations instead of absence/decreased protein levels. All of SDHx genes are tumor suppressors, showing loss of heterozygosity, the loss of the normal allele in the tumors, in conjunction with the germline mutation. To analyze the impact of SDHx cancer-associated homozygous mutations on the expression of HIF-1α and miR-210, we used two PGL-derived cell lines, one composed of tumor cells which had been obtained from a tympanic PGL with homozygous SDHC exon 2 deletion (SDHC-PGL) [16] and other composed of tumor-associated fibroblasts which were derived from a SDHD-mutated (c.88_314+247del, p.His30Asn fs*11) jugular PGL (SDHD-TAFs). Thus, SDHC-PGL cells lack wild type SDHC protein whereas SDHD-TAFs express mutated and wild type SDHD gene (data not shown). Analysis of HIF-1α protein expression revealed that the SDHC-mutated cell line contains mixed populations of tumor cells, i.e. 72% were negative for HIF-1α protein expression but about 28% of cells accumulate nuclear HIF-1α even under normoxic conditions (Figure 5A). No HIF-1α accumulation was observed, however, in SDHD-TAFs in normoxia but they did accumulate higher HIF-1α levels, under hypoxic conditions, than the siSDHB or siSDHD-treated SCC40 cells thus suggesting that they are primed for a more intense HIF-1α-related cellular response (Figure 4). Figure 5B shows that both types of cells, SDHC-PGLs and SDHD-TAFs, have slightly but significant higher levels of miR-210 expression in normoxia than human paraganglia (normal carotid body) or non-tumoral fibroblasts, respectively. Analysis of EGLN3 mRNA showed ~ 3-fold and ~ 18-fold higher levels in SDHD-TAF and SDHC-PGLs than in normal fibroblasts or carotid body, respectively (Figure 5C). Thus, in contrast to the data obtained in the SDHD knock-out mouse model or in the cell-based assays, cells carrying SDHC or SDHD mutations express higher levels of HIF-1α protein and its targets, miR-210, CA9 and EGLN3 than their normal counterparts. Nevertheless, the effect of mutations on HIF activation, if anything, seems to be rather subtle as compared with the stronger effect exerted by hypoxia (Figure 5B).
Figure 5. Mild heterogeneous activation of HIF-1α/miR-210 signaling axis in PGL-derived cells carrying SDHx mutations.
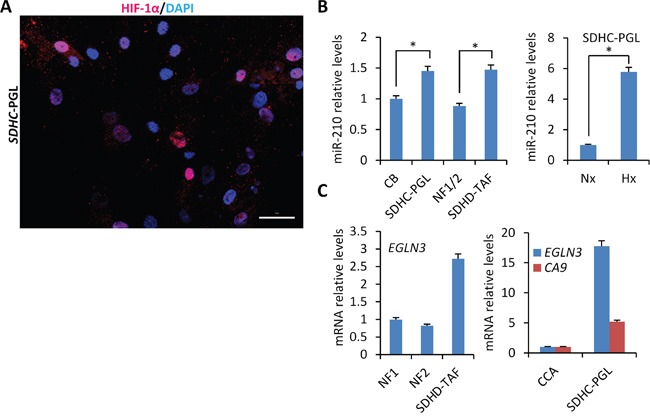
A. Immunocytochemical analysis of HIF-1α in cells derived from human PGL carrying SDHC exon 2 deletion (SDHC-PGL). Note the heterogeneity of the SDHC-PGL cell line which is composed of HIF-1α-positive and -negative cells. B and C. miR-210 (B), EGLN3 and CA9 (C) mRNA levels were analyzed by RT-qPCR in SDHC-PGL or SDHD-TAFs and compared with those of human normal carotid body (CB) or normal fibroblasts (NF), respectively. Two NF cell lines (NF1 and NF2) were used in this analysis. NF1/2 refers to the miR-210 data (mean ± SD value) obtained in NF1 and NF2 cells. CA9 mRNA levels were undetectable in NF and TAF cells. Values are expressed as mean ratios ± SD.
Collectively, the data indicates that the constitutive expression of miR-210 requires a functional wild type VHL protein but it is not influenced by partial VHL losses. HIF-1α, in turn, is involved in the hypoxic-induction of miR-210 but do not participate in its constitutive expression. Deleterious VHL gene mutations seems to have dominant negative effects on wild type VHL protein and as such activate the HIF-1α pathway. Finally, loss of SDHD or decreased SDHB or SDHD expression levels does not induce the HIF-1α/miR-210 pathway. Instead, mutations of SDHx genes may exert subtle inductive effects on the HIF-1α/miR-210 pathway.
Pseudo-hypoxic fate of VHL- and SDHx-related PGLs
The above data cannot be directly extrapolated to the biology of tumor tissues inasmuch as they all represent cell-based models that do not necessarily recapitulate cell pathophysiology of PGLs. Thus, we next sought to determine whether the genetic-phenotypic correlations identified in the murine and cellular models were also observed in tumor tissues. To this end, we used a cohort of PGLs of parasympathetic origin with known VHL and SDH gene mutations. Germline mutations of SDHD or SDHB, and somatic heterozygous mutations of VHL were found in 11, 7, and 2 patients, respectively, while 25 patients did not carry SDHx and VHL somatic/germline mutations. VHL expression data was unknown for most of those tumors. Thus, we first performed a comprehensive analysis of the VHL gene by examining VHL DNA copy number and mRNA levels. Because, unexpectedly, we had previously identified somatic VHL gene mutations in parasympathetic PGLs [11], analysis of VHL gene dosage was also performed even though chromosomal losses on the 3p have not been previously described as frequent events in PCCs/PGLs [21].
VHL DNA copy number was analyzed by MLPA assay. We identified 6 samples exhibiting decreased copy number of exon 1 of VHL (hereafter VHLdel-PGLs) (Table 1, Figure 6A) in tumor genomic DNA. This was significantly observed in PGLs lacking SDHx mutations (SDHxwt) (6/27, ~22%) but not in SDHx mutant (SDHxm)-PGLs (0/18) (P=0.02) and was not found in peripheral blood lymphocytes from any of these patients. These genetic defects were confirmed by an optimized quantitative PCR method in all tumors (data not shown), and by CGH microarray analysis in one tumor (Figure 6B). We next measured transcriptional levels of the VHL gene by RT-qPCR (n=36) to compare tumor samples with and without VHLdel. As shown in Figure 6C, low VHL transcript levels were encountered in VHLdel-PGLs as compared with PGLs lacking VHL or SDHx gene alterations (SDHwt/VHLwt) or tumors with SDHx mutations. Thus, 9 out of the 32 (28%) non-SDHx mutant PGLs included in this study carry VHL gene alterations (3 with VHL mutations -hereafter VHLm- and 6 with VHLdel). This places this tumor suppressor among the most frequently altered genes described thus far in parasympathetic PGLs. Table 1 shows the association between the clinicopathological characteristics of the patients and the presence of VHLdel. No association was found with age, location, functionality, multiplicity, or malignancy of tumors.
Table 1. Clinicopathological features of patients and association with VHLdel.
| n | del VHL | wtVHL* | P | |
|---|---|---|---|---|
| Age at diagnosis | ||||
| ≤40 years | 23 | 3 | 20 | 0.697 |
| >40 years | 31 | 3 | 28 | |
| UN§ | 3 | 0 | 3 | |
| Location | ||||
| jugulotimpanic | 28 | 4 | 24 | 0.693 |
| vagal | 8 | 0 | 8 | |
| carotid body | 19 | 2 | 17 | |
| abdominal | 1 | 0 | 1 | |
| Functional | ||||
| no | 53 | 4 | 49 | 0.472 |
| yes | 4 | 0 | 4 | |
| Single/Multiple | ||||
| single | 41 | 6 | 35 | 0.478 |
| multiple | 16 | 0 | 16 | |
| Benign/malignant | ||||
| benign | 55 | 5 | 50 | 0.067 |
| malignant | 2 | 1 | 1 | |
| SDHx mutation# | ||||
| none | 32 | 6 | 26 | 0.155 |
| SDHB | 8 | 0 | 8 | |
| SDHD | 16 | 0 | 16 | |
| SDHC | 1 | 0 | 1 |
wild type VHL;
unknown;
germline mutations
Figure 6. VHL exon 1 deletion in PGLs.
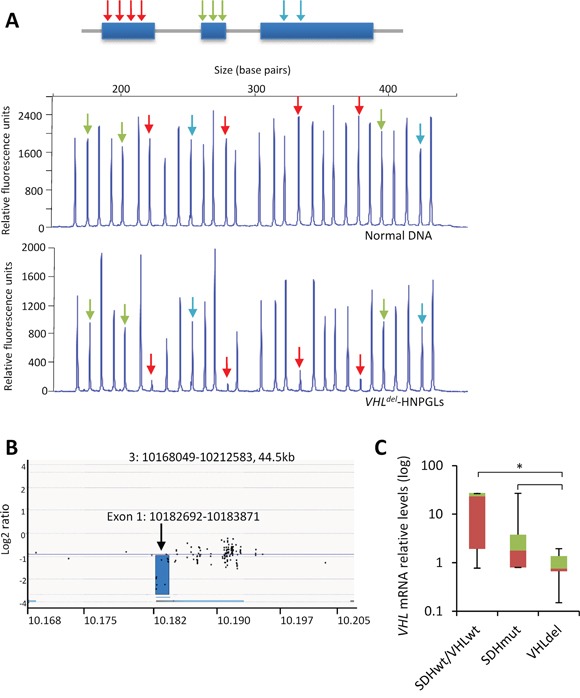
A. Upper panel, schematic representation of the structure of VHL. Arrows indicate exon location of the probe oligonucleotides included in the MLPA test. Middle and lower panels, capillary gel electrophoresis images depicting the MLPA assay for VHL in a normal control DNA and a tumor DNA. Red arrows indicate deleted exons. B. Array CGH screening of genomic DNA in a PGL sample with VHL exon 1 deletion as detected by MLPA assay. Profile is displayed as normalized log2 signal intensity ratios of each spot on the array to the genomic position at chromosome 3. C. RT-qPCR was performed for VHL in the indicated tumor types. Values are expressed as mean ratios ± SD. * p < 0.05 paired Student's t test.
We next analyzed the relative HIF-1α and miR-210 levels in tumors with specific genetic backgrounds: SDHxwt/VHLwt, VHLm, VHLdel, and SDHxm (SDHB or SDHD)m. miR-210 quantifications revealed that, as previously described, VHLm-PGLs expressed higher levels of miR-210 than SDHxm- or SDHxwt/VHLwt-PGLs (Figure 7A). However, VHLdel-PGLs expressed low levels of miR-210 which were similar to those detected in SDHxwt/VHLwt-PGLs. In addition, SDHxm-PGLs expressed slightly higher, statistically significant, levels of miR-210 compared to SDHxwt/VHLwt-PGLs (p=0.045).
Figure 7. HIF-1α/miR-210 expression in SDHx- and VHL-mutant PGLs.
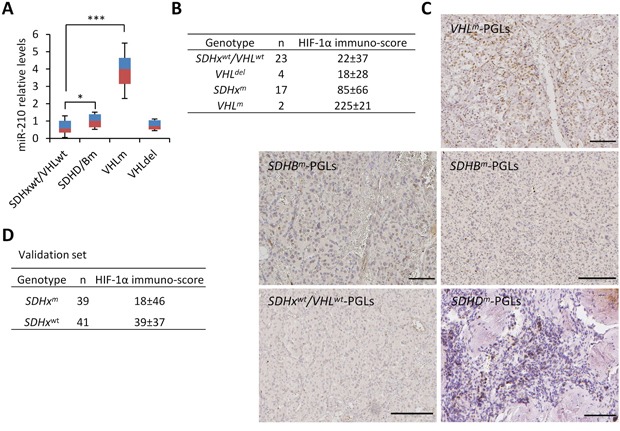
A. RT-qPCR was performed for miR-210 in the indicated tumor types. Values are expressed as mean ratios ± SD * p < 0.05, *** p < 0.001, paired Student's t test. B and D. Immuno-scores of HIF-1α in the tumor samples from patients with the indicated genotypes. C. Representative images of HIF-1α immunostainings in PGL samples from patients with VHLm, SDHBm or SDHDm or from a patient lacking SDH and VHL mutations (SDHwt/VHLwt).
Then, we analyzed HIF-1α protein expression in PGL tumor tissues (n=46) by immunohistochemistry. Given that many of the samples showed non-homogeneous distribution of positive nuclei and distinct intensity of immunostaining, both, the percentage of positively stained cells and the relative intensity of immunostaining, were considered for HIF-1α quantification. Some of the tumors had been chemoembolized prior to surgery, but we did not find correlation between the percentage of positively immunostained cells and presurgical chemoembolization thus ruling out a “iatrogenic” effect of embolization not related to tumor biology. Overall, HIF-1α immuno-scores ranged from 0 to 240 with a mean value of 52 and median value of 17. Comparison of the immuno-scores in tumors revealed a gradient in the levels of HIF-1α protein spanning from very low (SDHxwt/VHLwt- and VHLdel-PGLs) to low (SDHxm-PGLs) and high (VHLm-PGLs) expression levels (Figure 7B). The difference of immunostaining in VHLm-PGLs and SDHxm-PGLs lies in that the first has a high percentage of positive nuclei homogeneously distributed in the whole tumor section whereas the last takes on a patched appearance apparently not related to the distance to blood vessels (see representative immunohistochemical images in Figure 7C). In line with the miR-210 expression data, differences in HIF-1α immunostainings were statistically significant between VHLdel-PGLs and VHLm- PGLs (p<0.0001) or SDHx-PGLs (p=0.03), but not between VHLdel-PGLs and SDHxwt/VHLwt-PGLs. Differences were also statistically significant between SDHxm-PGLs and SDHxwt/VHLwt- (p=0.044).
Next, HIF-1α protein expression and SDHx genetic analysis was extended to a total of 112 parasympathetic PGLs (41 SDHxwt/VHLwt-PGLs and 39 SDHxm-PGLs all of them lacking VHL gene mutations) for validation. In addition to genetic analysis, the presence of SDHx mutations was analyzed by SDHB immunohistochemistry in 87 of the tumors given that negative immunostaining has been identified as a biomarker of SDHx mutations [22]. Indeed, our data showed that SDHB immunohistochemical negativity was significantly associated with the presence of mutations in any of the SDHx genes (p<0.0001). Comparison of the HIF-1α protein levels in tumors harboring or lacking SDHx mutations confirmed previously reported data revealing that SDHx-PGLs have a significantly higher percentage of HIF-1α positive cells than tumors lacking SDHx mutations (Figure 7D, p=0.008). No differences in HIF-1α protein levels were found, however, between SDHD- and SDHB-mutated PGLs/PCCs (p=0.999). Importantly, none of the SDHx-mutated tumors showed the very high percentage of positive nuclei that had been observed in VHL-PGL.
DISCUSSION
In this report we have used cellular- and knockout murine-models and neuroendocrine PGLs to determine whether the HIF-1α and VHL proteins, implicated in the miR-210-mediated hypoxia/pseudohypoxia response in vitro, and the mitochondrial SDH complex, involved in HIF-1α stabilization, are essential for miR-210 expression in vivo. We demonstrated that ablation of VHL or HIF1A genes in neural crest-derived cells, from which the paraganglia originates, increases the basal levels of miR-210 (VHL knockout) or blunt the inducing effect of hypoxia (HIF1A knockout) on miR-210 indicating that these proteins are essential for the in vivo expression of miR-210 under hypoxic/pseudohypoxic conditions in paraganglia. By contrast, the mitochondrial SDH complex activity, related with tumorigenesis and supposedly involved in HIF-1α regulation [5], does not appear to have a direct effect on HIF-1α-dependent miR-210 over-expression but it favors it via a mechanism which has not been clearly defined as yet. Importantly, the data obtained in the murine/cellular models were corroborated by the use of human tumor materials containing known alterations of the VHL and SDHx genes. PGLs were chosen as tumor model because of their associations with mutations in those genes. The genotype-phenotype association studies revealed that the HIF-1α/miR-210 pathway is strongly activated in VHLm-PGLs, weakly activated in SDHxm-PGLs, and not activated in VHLdel- and SDHxwt/VHLwt-PGLs. A functional link between defects of the VHL gene and activation of the miR-210 signaling pathway has also been demonstrated in other tumors such as PCCs, ccRCC and ovarian cancer [11, 23, 24]. However, the reported connections between HIF-1α and SDH in human tumors are more scarce and controversial.
The relative contribution of HIF-2α isoform in the development of PGL and miR-210 expression has not been explored in this report. As previously reported, nuclear HIF-2α protein expression is not detected in PGLs, irrespective of whether they carry SDHx or VHL mutations [11]. Therefore, although several lines of evidence suggest a predominant role for HIF-1α in miR-210 expression [11], the relative contribution of HIF-2α isoform in PGL development remains to be fully determined.
Role of SDH on HIF-1α/miR-210 expression
Several studies have shown that the HIF-1α gene expression pathway is activated in SDHxm-tumors whereas others did not find such an association [11, 14, 16, 25]. Only a few of those studies have focused on correlation analysis between miR-210 expression and SDHx-mutations showing that overexpression of miR-210 was associated with SDHx mutations of PCCs, PGLs, and gastrointestinal stromal tumours [26, 27]. In the present report, we addressed the functional role of the SDH mitochondrial complex II components on the hypoxic/pseudohypoxic miR-210 up-regulation. By using RNA interference as well as conditional knock-out of SDHD via the Cre-Lox system, we demonstrate that absence of SDHD does not induce HIF-1α dependent miR-210 up-regulation. Similar data were obtained in cells with siRNA-induced SDHB deficiency. We also show that this effect was mimicked by TTFA, a SDH inhibitor that blocks the mitochondrial electron flow by binding to the ubiquinone docking sites at the SDH complex [28]. Conversely, PGL-derived cells carrying SDHC or SDHB mutations do show mild activation of HIF-1α and miR-210. These data agree with those observed in PGLs, i.e. HIF-1α/miR-210 levels were found higher in SDHxm-PGLs as compared to SDHxwt-PGLs although lower than those detected in VHLm-PGLs. Given that the data obtained in murine and cellular models support that SDH loss of function do not directly activate the HIF-1α pathway, that HIF-1α nuclear accumulation is only observed in about 38% of tumor cells in SDHxm-PGLs, and that these tumors show mild miR-210 over-expression, we postulate that HIF-1α up-regulation in these tumors is a late event that it is favored by SDHx mutations but is triggered by still undefined biochemical or genetic event/s. Mitochondrial complex II activity catalyzes the oxidation of the metabolite succinate, which is involved in HIF-1α hydroxylation, to fumarate. The electrons generated in this reaction are then channeled, in a not completely understood pathway, to ubiquinone, which is reduced to ubiquinol by the succinate:ubiquinone oxidoreductase activity of the complex. Electrons provided by succinate oxidation may leak out from complex II and lead to the formation of sub-lethal levels of ROS. Although existing literature includes many contradictory findings, mitochondrial production of ROS is involved in regulating hypoxia-induced HIF-1α protein stabilization [29, 30]. Thus, other possibility is that SDHx mutations favor an intracellular microenvironment rich in succinate and/or ROS that only when they reach a threshold level may trigger HIF-1α protein accumulation. This provides an explanation for the published contradictory data regarding the link of SDH dysfunction and HIF-1α. Further biochemical studies will be required to unravel the specific mechanisms involved in regulation of the HIF-1α/miR-210 axis by pathogenic mutations on SDHx genes.
Role of VHL on HIF-1α/miR-210 expression
Another major finding of this study is the identification of recurrent deletions of exon 1 of the VHL gene (19% of tumors) in PGLs developed in patients lacking germline SDHx mutations. Previous studies revealed that losses of VHL are rare in PGLs [31–33] but not in renal cell cancer or VHL-related PCCs. Thus, this is the first description of somatic VHL gene exon deletions in parasympathetic PGLs. Nevertheless, the phenotypic consequences of VHL gene alterations seem to be heterogeneous depending on the type of defect. As stated above, we definitively demonstrated that PGLs carrying VHLm are grossly defective with respect to HIF regulation and induce a strong activation of miR-210. However, we also discovered that partial loss of VHL in tumors is not accompanied by HIF-1α activation or miR-210 over-expression. In accord with these findings, miR-210 over-expression was not detected in the adrenal medulla from heterozygous VHL +/- mice. Absence of correlations between phenotypes induced by loss of gene expression versus gene mutation has been demonstrated in zebrafish, mouse and Arabidopsis [34–37]. In this last setting, Rossi et al, provided an explanation for those observations [38]. They found that deleterious mutations, but not gene knockdown, trigger compensatory responses that preclude observation of phenotypes similar to those induced by gene knockdown. Thus, we cannot rule out the possibility that loss of VHL, in contrast to VHL mutations, activates compensatory networks to cushion the deficit. It is also possible that VHL genetic defects may represent “passenger” events not relevant to carcinogenesis. Alternatively, VHL partial deficiency may activate HIF-independent pathways which may be relevant to its tumour suppressor function [39, 40]. Ultimately, it can also be speculated that VHL-reduced expression versus VHL-gene mutations may elicit different effects on VHL stability and/or protein-protein interactions and lead to a distinct non canonical HIF-1α-related biochemical outputs [41]. Prior to this study, microarray gene expression studies had been performed in PGLs including a few of the VHLdel-PGLs identified in this study [11]. Re-analysis of the microarray data has revealed a constrained list of HIF-1α-related deregulations in VHLdel-PGLs which was limited only to genes involved in angiogenesis and extracellular matrix remodeling, molecular processes heavily related to cellular proliferation and differentiation. These data suggest that the HIF-1α protein levels reached under these circumstances, if any, are not sufficient for induction of miR-210 expression but they are possibly enough to induce other HIF-1α-related genes. If confirmed with broader studies, the observed genotype-phenotype associations would argue in favor of VHL as a driver gene in sporadic PGLs. Further genetic and cell-based assays are required to fully understand the functional significance of the observed genotype-phenotype associations.
We also provide experimental evidences supporting that a mutant VHL protein, F76del, acts as a dominant-negative inhibitor of the wild-type VHL by inducing the pro-tumorigenic HIF/miR-210 pathway even in the presence of wild type VHL. This study was of interest because many VHL mutations involved in human cancer, especially those found in PGLs, are not always accompanied by loss of the wild type allele. Thus, the question remained whether mutant VHLs are also defective for HIF-1α regulation and able to up-regulate miR-210 in the presence of wild type VHL. We selected the F76del mutant VHL to address this issue because this mutation has been associated with distinct types of cancer such as renal cell carcinomas, PCCs and PGLs [42]. Previous association studies had shown that mutations of VHL in tumors correlate with miR-210 over-expression [11, 23, 26, 27]. In addition, a disturbance of the coupling between HIF-1α and mutants VHL, including F76del, has been experimentally confirmed by other authors using a series of in vitro assays [43]. However, most of those studies were performed in VHL-deficient cells in which the VHL gene expression was restored by transfection of the mutated VHL gene [44]. Thus, other than association studies, direct evidences for a role of F76del on miR-210 expression was lacking and it was not known whether loss of the wild type allele was required for the activation of the pro-tumorigenic HIF pathway by the mutant VHL.
In summary, the analyses of different pseudo-hypoxic biomarkers in different settings are fully consistent and strongly suggest that the HIF-1α-related expression profile may be finely adjusted by SDH and VHL. VHL mutations are associated with a strong HIF-1α-dependent miR-210 over-expression and accumulation of HIF-1α in most tumor cells whereas SDHx mutations are associated with mild increase of miR-210 and the presence of a heterogeneous, HIF-1α-positive and HIF-1α-negative, tumor cell population. Thus, accumulation of HIF-1α protein seems to be an early event in the tumorigenic pathway led by VHL mutations but a deferred episode in tumors harboring SDHx mutations. The question remains as to whether the distinct pseudo-hypoxic landscapes identified in PGLs translate into different clinical phenotypes as, for example, the multiplicity of tumors, the risk of developing PGLs of sympathetic or parasympathetic origin, or the different tendency to develop metastasis [45]. Certainly, the data obtained in the present study will acquire a more general clinical relevance in cancer pathogenesis and will pave the way for the understanding of the effect of genetic events on pseudo-hypoxic signaling and its role in the pathogenesis of tumors, a knowledge that will allow the use of optimal and individualized therapeutic targeting of tumors harboring these genetic changes.
MATERIALS AND METHODS
Tumor specimens
Tumor samples were obtained from 57 patients with parasympathetic PGLs, diagnosed and treated between 1996 and 2012 in the Hospital Universitario Central de Asturias and Hospital Gregorio Marañón (Table 1). For validations, independent set of tumors from 119 patients was used. For RNA/DNA-based studies, fragments containing more than 70% tumor cells were obtained from the core of the tumor. Tumor specimens were snap-frozen at time of surgical resection and stored at −80 °C in RNAlater (Ambion) until processed. All but two tumors had benign clinical behavior. One of the patients with malignant PGL developed regional lymph node metastasis and died six years after diagnosis. Informed consent was obtained from all patients. The study was approved by the Ethical Committee of the Hospital Central of Asturias and the methods were carried out in accordance with the approved guidelines.
Cell culture
SCC40 cell line, derived from larynx squamous cell carcinoma, was kindly provided by Dr. R. Grenman (Department of Otolaryngology, University Central Hospital, Turkey, Finland). SCC40, 786-O and RCC4 cells were grown in DMEM supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 200 μg/mL streptomycin, 2mmol/L L-glutamine, 20 mmol/L HEPES (pH 7.3), and 100 μmol/L nonessential amino acids. Cell line authentication was performed by array CGH [19]. Growth curve analysis was performed regularly, and no phenotype changes were observed through the duration of this study. To avoid cross-contamination and phenotype changes, these cell lines have not been maintained in long-term cultures. Mouse embryonic fibroblasts (MEFs) and baby mouse kidney (BMK) cells were obtained from SDHD-ESR mice (SdhD−/−) and their homozygous SdhDflox/+ (SdhD+/+) and heterozygous SdhDflox/− (SdhD+/−) littermates and cultured in medium supplemented with 4-hydroxy-tamoxifen for 4 or 24 hours as previously described [20].
Genetic testing
Genomic DNA was isolated using the QIAmp DNA Mini kit (Qiagen, Inc., Chatsworth, CA) and subsequently treated with RNase A (1unit/mL) at 37°C for 5 minutes. Mutation analysis was performed by direct sequencing as previously described [16]. VHL gene was analyzed for the presence of large deletions in tumor DNA using the Multiplex Ligation-dependent Probe Amplification (MLPA) (SALSA MLPA P016, MRC Holland, The Netherlands) method as recommended by the manufacturer. Analysis of the amounts of the MLPA PCR products per gene was done on an ABI PRISM 3100 Genetic Analyzer platform (Applied Biosystems) according to the manufacturer's instructions. Each gene peak area was divided by the sum of the control peak areas for that sample. These relative areas were then compared with the corresponding average relative area obtained from three normal mucosa samples, thus creating a ratio (case/normal). The mean ratio obtained from at least two independent MLPA reactions for each sample was considered the final result. For a normal mucosa, this ratio was expected to be 1. DNA copy numbers for VHL gene were confirmed by real time PCR and Array Comparative Genomic Hybridization (array CGH).
Real time PCR was performed following as previously described [46]. Relative DNA quantity was determined using the delta-Ct formula. For normalization of the relative quantities, the VHL copy numbers were divided by the geometric mean of the reference GPR15 copy number.
Screening of genome-wide copy number variants was carried out by array-based CGH using the OncoNIM® Familial Cancer platform, a 60 k Agilent based custom array-CGH (Nimgenetics; Madrid, Spain). This custom array covers the whole genome with a median spatial resolution of 1 probe per 150 kb, with high density coverage in 20 genes related to familial cancer (100 bp median spatial resolutions for these genes). Hybridizations were performed according to the manufacturer's protocols. A commercially available male DNA sample (Promega, Madison, WI, USA) was used as reference DNA. Microarray data were extracted and visualized using the Feature Extraction Software v10.7 and Agilent Genomic Workbench v.5.0 (Agilent Technologies, Santa Clara, CA) using ADAM-2 (windown 0.5Mb, A=10) as aberration detection statistic. Genomic build NCBI37 (Hg19) was used for delineating the genomic coordinates of the detected CNVs.
RT-qPCR
Total RNA was isolated with the mirVana™ miRNA Isolation Kit (Ambion) according to the manufacturer's instructions. cDNA synthesis and real-time PCR were performed as previously described [18]. Briefly, real-time PCR reactions were performed by using SYBR Green PCR Master mix (Applied Biosystems) and the thermocycler conditions recommended by the manufacturer. Each sample was analyzed for cyclophilin A mRNA to normalize for RNA input amounts and to perform relative quantification. Primers were designed using the computer program Primer Express (Applied Biosystems). Primers were as follows: VHL, forward 5’-CCCAGGTCATCTTCTGCAAT-3’- and reverse, 5’-ACATTTGGGTGGTCTTCCAG-3’; cyclophilin A, forward, 5’-CATCTGCACTGCCAAGACTGA-3’ and reverse, 5’-TTGCCAAACACCACATGCTT-3’; CA9, forward, 5’- CGCTGAGGAAGGCTCAGAGA-3’ and reverse 5’- GGCAGGAGTGCAGATATGTCC-3’; EGLN3 5’-CCGGCTGGGCAAATACTATG-3’ and reverse 5’- GATAGCAAGCCACCATTGCC-3’. Melting curve analysis showed a single sharp peak with the expected Tm for all samples. mRNA relative quantities were obtained using the 2ΔΔCt method. Individual TaqMan assay (Applied Biosystems) was used to analyze the expression of the mature human miR-210. 10 ng of total RNA was used in the RT reaction and the transcribed cDNA was then used for subsequent PCR amplification using TaqMan 2X Universal PCR Master Mix, No AmpErase UNG (Applied Biosystems) as described by the manufacturer. RNU44 expression was assayed for normalization. All reactions were performed in triplicate, and relative miRNA expression was normalized against endogenous controls using the comparative delta-delta CT method.
VHL and HIF-1α knockout mice
In order to delete HIF1A or VHL production specifically in neural crest derived cells, we used conditional knockout mice carrying floxed alelles of HIF1A [47] or VHL [48] genes crossed with transgenic mice carrying a tamoxifen-inducible CRE recombinase whose expression is driven by the promoter of the intermediate neurofilament Nestin (Nes-CreERT2 mice) [49]. The resulting strains undergo deletion of HIF1A or VHL specifically in neural crest-derived tissues expressing nestin protein and only after tamoxifen treatment. We injected 14 mg/kg of tamoxifen to the 8-weeks old mice in 3 doses in a week. HIF1A −/− or control mice HIF1A +/+ were put in a hypoxic chamber at 10% O2 for 1 month. Animals were sacrificed and adrenal medulla was dissected. All experiments were performed in accordance with institutional guidelines and were approved by the animal ethics committee of the University of Oviedo.
Site-direct mutagenesis
Site-direct mutagenesis was performed in pcDNA3.1 HA-VHL construct, kindly provided by Holger Moch (University Hospital Zurich, Institute of Surgical Pathology) using the GeneArt Site-Directed Mutagenesis System (Life Technologies) in order to introduce F76del mutation in the VHL cDNA. Gene amplification and bacterial transformation followed the protocol described by the manufacturer and the mutated DNA sequences were screened by nucleotide sequencing. Primers used to generate the mutations were: 5’-CCTCCCAGGTCAGCAATCGCAGTCCGC-3’ and 5’-GCGGACTGCGATTGCATATGACCTGGGAGG-3’.
siRNA treatment
siRNA duplex oligonucleotides were purchased from Dharmacon Research (Lafayette, CO, USA). siCONTROL Nontargeting pool (Dharmacon) were used as control siRNA. SCC40 cells were transfected with 35 pmol/ml siRNAs using Lipofectamine 2000. mRNA analysis analyses revealed a substantial inhibition of SDHB or SDHD expression 48–72 h after transfection. The transfected cells were used for subsequent experiments within that interval of time.
Western blot analyses
Protein extracts were obtained from SCC40 cells at 80–90% confluence as previously described [18]. Equal amounts of proteins were fractionated on SDS–PAGE and transferred to PVDF membranes. Membranes were probed with anti-HIF-1α, (Becton Dickinson Transduction Laboratories, Erembodegem, Belgium), or anti-β-actin (Sigma-Aldrich, St Louis, MO, USA) antibodies at 1:1000 dilutions. Bound antibodies were detected using enhanced chemiluminescence reagent (Life Technologies) according to the protocol of the manufacturer. Densitometry of Western blot analyses was conducted with ImageJ analysis software.
Immunostainings
HIF-1α expression was evaluated by immunohistochemistry in tissue sections from a retrospective cohort of 46 surgically resected PGLs as previously described [16]. Following published recommendations [50], both, the percentage of immunostained cells and the intensity of staining were used to quantify HIF-1α protein expression. Staining intensity was scored from 0 to 3+. Data were reviewed independently by 3 investigators. For immunofluorescence analysis, Alexa555-conjugated anti-mouse IgG was used as secondary antibody and cells were pictured using Zeiss AxioObserver Z1 microscope with AxioCam MRM and ApoTome 2 (Carl Zeiss, Germany).
Acknowledgments
We thank Eva Allonca for excellent technical assistance.
Footnotes
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
GRANT SUPPORT
This work was supported by Instituto de Salud Carlos III-Fondo de Investigación Sanitaria [FIS PI11/929]; Red Temática de Investigación Cooperativa en Cáncer [RD12/0036/0015] Instituto de Salud Carlos III (ISCIII), Spanish Ministry of Economy and Competitiveness & European Regional Development Fund (ERDF); the European Regional Development's funds (FEDER) (CIBERONC) and Obra Social CajAstur-Instituto Universitario de Oncología del Principado de Asturias.
REFERENCES
- 1.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–73. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 2.Morin A, Letouzé E, Gimenez-Roqueplo A-P, Favier J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int J cancer. 2014;135:2237–48. doi: 10.1002/ijc.29080. [DOI] [PubMed] [Google Scholar]
- 3.Evenepoel L, Papathomas TG, Krol N, Korpershoek E, de Krijger RR, Persu A, Dinjens WNM. Toward an improved definition of the genetic and tumor spectrum associated with SDH germ-line mutations. Genet Med. 2015;17:610–20. doi: 10.1038/gim.2014.162. [DOI] [PubMed] [Google Scholar]
- 4.Gimenez-Roqueplo A-P, Tischler AS. Pheochromocytoma and Paraganglioma: progress on all fronts. Endocr Pathol. 2012;23:1–3. doi: 10.1007/s12022-011-9190-7. [DOI] [PubMed] [Google Scholar]
- 5.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 6.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–42. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King KS, Pacak K. Familial pheochromocytomas and paragangliomas. Mol Cell Endocrinol. 2014;386:92–100. doi: 10.1016/j.mce.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011;19:617–23. doi: 10.1038/ejhg.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaal J, van Nederveen FH, Erlic Z, Korpershoek E, Oldenburg R, Boedeker CC, Kontny U, Neumann HP, Dinjens WNM, de Krijger RR. Parasympathetic paragangliomas are part of the Von Hippel-Lindau syndrome. J Clin Endocrinol Metab. 2009;94:4367–71. doi: 10.1210/jc.2009-1479. [DOI] [PubMed] [Google Scholar]
- 10.Burnichon N, Vescovo L, Amar L, Libé R, de Reynies A, Venisse A, Jouanno E, Laurendeau I, Parfait B, Bertherat J, Plouin P-F, Jeunemaitre X, Favier J, et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet. 2011;20:3974–85. doi: 10.1093/hmg/ddr324. [DOI] [PubMed] [Google Scholar]
- 11.Merlo A, de Quirós SB, de Santa-María IS, Pitiot AS, Balbín M, Astudillo A, Scola B, Arístegui M, Quer M, Suarez C, Chiara M-D. Identification of somatic VHL gene mutations in sporadic head and neck paragangliomas in association with activation of the HIF-1α/miR-210 signaling pathway. J Clin Endocrinol Metab. 2013;98:E1661–6. doi: 10.1210/jc.2013-1636. [DOI] [PubMed] [Google Scholar]
- 12.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 13.Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–9. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 14.Pollard PJ, El-Bahrawy M, Poulsom R, Elia G, Killick P, Kelly G, Hunt T, Jeffery R, Seedhar P, Barwell J, Latif F, Gleeson MJ, Hodgson S V, et al. Expression of HIF-1alpha, HIF-2alpha (EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J Clin Endocrinol Metab. 2006;91:4593–8. doi: 10.1210/jc.2006-0920. [DOI] [PubMed] [Google Scholar]
- 15.Blank A, Schmitt AM, Korpershoek E, van Nederveen F, Rudolph T, Weber N, Strebel RT, de Krijger R, Komminoth P, Perren A. SDHB loss predicts malignancy in pheochromocytomas/sympathethic paragangliomas, but not through hypoxia signalling. Endocr Relat Cancer. 2010;17:919–28. doi: 10.1677/ERC-09-0316. [DOI] [PubMed] [Google Scholar]
- 16.Merlo A, de Quiros SB, Secades P, Zambrano I, Balbín M, Astudillo A, Scola B, Arístegui M, Suarez C, Chiara M-D. Identification of a signaling axis HIF-1α/microRNA-210/ISCU independent of SDH mutation that defines a subgroup of head and neck paragangliomas. J Clin Endocrinol Metab. 2012;97:E2194–200. doi: 10.1210/jc.2012-2410. [DOI] [PubMed] [Google Scholar]
- 17.Ivan M, Huang X. miR-210: fine-tuning the hypoxic response. Adv Exp Med Biol. 2014;772:205–27. doi: 10.1007/978-1-4614-5915-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Secades P, de Santa-María IS, Merlo A, Suarez C CM. In vitro study of normoxic epidermal growth factor receptor-induced hypoxia-inducible factor-1-alpha, vascular endothelial growth factor, and BNIP3 expression in head and neck squamous cell carcinoma cell lines: Implications for anti-epidermal growth fact. Head Neck. 2015;37:1150–62. doi: 10.1002/hed.23733. [DOI] [PubMed] [Google Scholar]
- 19.Secades P, Rodrigo JP, Hermsen M, Alvarez C, Suarez C, Chiara M-D. Increase in gene dosage is a mechanism of HIF-1alpha constitutive expression in head and neck squamous cell carcinomas. Genes Chromosomes Cancer. 2009;48:441–54. doi: 10.1002/gcc.20652. [DOI] [PubMed] [Google Scholar]
- 20.Millán-Uclés A, Díaz-Castro B, García-Flores P, Báez A, Pérez-Simón JA, López-Barneo J, Piruat JI. A conditional mouse mutant in the tumor suppressor SdhD gene unveils a link between p21(WAF1/Cip1) induction and mitochondrial dysfunction. PLoS One. 2014;9:e85528. doi: 10.1371/journal.pone.0085528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castro-Vega LJ, Letouzé E, Burnichon N, Buffet A, Disderot P-H, Khalifa E, Loriot C, Elarouci N, Morin A, Menara M, Lepoutre-Lussey C, Badoual C, Sibony M, et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6044. doi: 10.1038/ncomms7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, Sleddens HF, Derkx P, Rivière J, Dannenberg H, Petri BJ, Komminoth P, Pacak K, Hop WC, Pollard PJ, Mannelli M, Bayley JP, Perren A, Niemann S, Verhofstad AA, de Bruïne AP, de KRD. Maher. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10:764–71. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCormick RI, Blick C, Ragoussis J, Schoedel J, Mole DR, Young AC, Selby PJ, Banks RE, Harris AL. miR-210 is a target of hypoxia-inducible factors 1 and 2 in renal cancer, regulates ISCU and correlates with good prognosis. Br J Cancer. 2013;108:1133–42. doi: 10.1038/bjc.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu T, Zhao L, Chen W, Li Z, Hou H, Ding L, Li X. Inactivation of von Hippel-Lindau increases ovarian cancer cell aggressiveness through the HIF1α/miR-210/VMP1 signaling pathway. Int J Mol Med. 2014;33:1236–42. doi: 10.3892/ijmm.2014.1661. [DOI] [PubMed] [Google Scholar]
- 25.López-Jiménez E, Gómez-López G, Leandro-García LJ, Muñoz I, Schiavi F, Montero-Conde C, de Cubas AA, Ramires R, Landa I, Leskelä S, Maliszewska A, Inglada-Pérez L, de la Vega L, et al. Research resource: Transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol Endocrinol. 2010;24:2382–91. doi: 10.1210/me.2010-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsang VHM, Dwight T, Benn DE, Meyer-Rochow GY, Gill AJ, Sywak M, Sidhu S, Veivers D, Sue CM, Robinson BG, Clifton-Bligh RJ, Parker NR. Overexpression of miR-210 is associated with SDH-related pheochromocytomas, paragangliomas, and gastrointestinal stromal tumours. Endocr Relat Cancer. 2014;21:415–26. doi: 10.1530/ERC-13-0519. [DOI] [PubMed] [Google Scholar]
- 27.de Cubas AA, Leandro-García LJ, Schiavi F, Mancikova V, Comino-Méndez I, Inglada-Pérez L, Perez-Martinez M, Ibarz N, Ximénez-Embún P, López-Jiménez E, Maliszewska A, Letón R, Gómez Graña A, et al. Integrative analysis of miRNA and mRNA expression profiles in pheochromocytoma and paraganglioma identifies genotype-specific markers and potentially regulated pathways. Endocr Relat Cancer. 2013;20:477–93. doi: 10.1530/ERC-12-0183. [DOI] [PubMed] [Google Scholar]
- 28.Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, Rao Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121:1043–57. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 29.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–20. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martínez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, Dufour E, Spelbrink JN, Weinberg SE, et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol Cell. 2016;61:199–209. doi: 10.1016/j.molcel.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bender BU, Gutsche M, Gläsker S, Müller B, Kirste G, Eng C, Neumann HP. Differential genetic alterations in von Hippel-Lindau syndrome-associated and sporadic pheochromocytomas. J Clin Endocrinol Metab. 2000;85:4568–74. doi: 10.1210/jcem.85.12.7015. [DOI] [PubMed] [Google Scholar]
- 32.Friedrich CA. Genotype-phenotype correlation in von Hippel-Lindau syndrome. Hum Mol Genet. 2001;10:763–7. doi: 10.1093/hmg/10.7.763. [DOI] [PubMed] [Google Scholar]
- 33.Brauch H, Weirich G, Brieger J, Glavac D, Rödl H, Eichinger M, Feurer M, Weidt E, Puranakanitstha C, Neuhaus C, Pomer S, Brenner W, Schirmacher P, et al. VHL alterations in human clear cell renal cell carcinoma: association with advanced tumor stage and a novel hot spot mutation. Cancer Res. 2000;60:1942–8. [PubMed] [Google Scholar]
- 34.Gao Y, Zhang Y, Zhang D, Dai X, Estelle M, Zhao Y. Auxin binding protein 1 (ABP1) is not required for either auxin signaling or Arabidopsis development. Proc Natl Acad Sci U S A. 2015;112:2275–80. doi: 10.1073/pnas.1500365112. [cited 2016Oct 28] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Souza AT, Dai X, Spencer AG, Reppen T, Menzie A, Roesch PL, He Y, Caguyong MJ, Bloomer S, Herweijer H, Wolff JA, Hagstrom JE, Lewis DL, et al. Transcriptional and phenotypic comparisons of Ppara knockout and siRNA knockdown mice. Nucleic Acids Res. 2006;34:4486–94. doi: 10.1093/nar/gkl609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Law SHW, Sargent TD. The serine-threonine protein kinase PAK4 is dispensable in zebrafish: identification of a morpholino-generated pseudophenotype. PLoS One. 2014;9:e100268. doi: 10.1371/journal.pone.0100268. [cited 201628] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kok FO, Shin M, Ni C-W, Gupta A, Grosse AS, van Impel A, Kirchmaier BC, Peterson-Maduro J, Kourkoulis G, Male I, DeSantis DF, Sheppard-Tindell S, Ebarasi L, et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev Cell. 2015;32:97–108. doi: 10.1016/j.devcel.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M, Stainier DYR. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524:230–3. doi: 10.1038/nature14580. [DOI] [PubMed] [Google Scholar]
- 39.Kanno H, Saljooque F, Yamamoto I, Hattori S, Yao M, Shuin T, HS U. Role of the von Hippel-Lindau tumor suppressor protein during neuronal differentiation. Cancer Res. 2000;60:2820–4. [PubMed] [Google Scholar]
- 40.Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 2015;15:55–64. doi: 10.1038/nrc3844. [DOI] [PubMed] [Google Scholar]
- 41.Leisz S, Schulz K, Erb S, Oefner P, Dettmer K, Mougiakakos D, Wang E, Marincola FM, Stehle F, Seliger B. Distinct von Hippel-Lindau gene and hypoxia-regulated alterations in gene and protein expression patterns of renal cell carcinoma and their effects on metabolism. Oncotarget. 2015;6:11395–406. doi: 10.18632/oncotarget.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Limaverde-Sousa G, de A Barreto E, Ferreira CG, Casali-da-Rocha JC. Simulation of the mutation F76del on the von Hippel-Lindau tumor suppressor protein: mechanism of the disease and implications for drug development. Proteins. 2013;81:349–63. doi: 10.1002/prot.24191. [DOI] [PubMed] [Google Scholar]
- 43.Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, Ratcliffe PJ, Maher ER. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet. 2001;10:1029–38. doi: 10.1093/hmg/10.10.1029. [DOI] [PubMed] [Google Scholar]
- 44.Rathmell WK, Hickey MM, Bezman NA, Chmielecki CA, Carraway NC, Simon MC. In vitro and in vivo models analyzing von Hippel-Lindau disease-specific mutations. Cancer Res. 2004;64:8595–603. doi: 10.1158/0008-5472.CAN-04-1430. [DOI] [PubMed] [Google Scholar]
- 45.Costa MHS, Ortiga-Carvalho TM, Violante AD, Vaisman M. Pheochromocytomas and Paragangliomas: Clinical and Genetic Approaches. Front Endocrinol (Lausanne) 2015;6:126. doi: 10.3389/fendo.2015.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoebeeck J, van der Luijt R, Poppe B, De Smet E, Yigit N, Claes K, Zewald R, de Jong G-J, De Paepe A, Speleman F, Vandesompele J. Rapid detection of VHL exon deletions using real-time quantitative PCR. Lab Invest. 2005;85:24–33. doi: 10.1038/labinvest.3700209. [DOI] [PubMed] [Google Scholar]
- 47.Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–57. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A. 2001;98:1583–8. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balordi F, Fishell G. Mosaic removal of hedgehog signaling in the adult SVZ reveals that the residual wild-type stem cells have a limited capacity for self-renewal. J Neurosci. 2007;27:14248–59. doi: 10.1523/JNEUROSCI.4531-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gong L, Zhang W, Zhou J, Lu J, Xiong H, Shi X, Chen J. Prognostic value of HIFs expression in head and neck cancer: a systematic review. PLoS One. 2013;8:e75094. doi: 10.1371/journal.pone.0075094. [DOI] [PMC free article] [PubMed] [Google Scholar]