

Abstract
Chemically modified versions of trehalose, or trehalose analogues, have applications in biology, biotechnology, and pharmaceutical science, among other fields. For instance, trehalose analogues bearing detectable tags have been used to detect Mycobacterium tuberculosis and may have applications as tuberculosis diagnostic imaging agents. Hydrolytically stable versions of trehalose are also being pursued due to their potential for use as non-caloric sweeteners and bioprotective agents. Despite the appeal of this class of compounds for various applications, their potential remains unfulfilled due to the lack of a robust route for their production. Here, we report a detailed protocol for the rapid and efficient one-step biocatalytic synthesis of trehalose analogues that bypasses the problems associated with chemical synthesis. By utilizing the thermostable trehalose synthase (TreT) enzyme from Thermoproteus tenax, trehalose analogues can be generated in a single step from glucose analogues and uridine diphosphate glucose in high yield (up to quantitative conversion) in 15-60 min. A simple and rapid non-chromatographic purification protocol, which consists of spin dialysis and ion exchange, can deliver many trehalose analogues of known concentration in aqueous solution in as little as 45 min. In cases where unreacted glucose analogue still remains, chromatographic purification of the trehalose analogue product can be performed. Overall, this method provides a "green" biocatalytic platform for the expedited synthesis and purification of trehalose analogues that is efficient and accessible to non-chemists. To exemplify the applicability of this method, we describe a protocol for the synthesis, all-aqueous purification, and administration of a trehalose-based click chemistry probe to mycobacteria, all of which took less than 1 hour and enabled fluorescence detection of mycobacteria. In the future, we envision that, among other applications, this protocol may be applied to the rapid synthesis of trehalose-based probes for tuberculosis diagnostics. For instance, short-lived radionuclide-modified trehalose analogues (e.g., 18F-modified trehalose) could be used for advanced clinical imaging modalities such as positron emission tomography-computed tomography (PET-CT).
Keywords: Biochemistry, Issue 120, trehalose analogue, enzymatic synthesis, trehalose synthase, TreT, mycobacteria, tuberculosis, click chemistry, fluorescence
Introduction
Trehalose is a symmetrical non-reducing disaccharide consisting of two glucose moieties that are joined by a 1,1-α,α-glycosidic bond (Figure 1A). While trehalose is absent from humans and other mammals, it is found commonly in bacteria, fungi, plants, and invertebrates 1. The primary role of trehalose in most organisms is to protect against environmental stresses, such as desiccation 1. In addition, some human pathogens require trehalose for virulence, including the tuberculosis-causing Mycobacterium tuberculosis, which utilizes trehalose as a mediator of cell envelope biosynthesis and as a building block for the construction of immunomodulatory glycolipids 2.
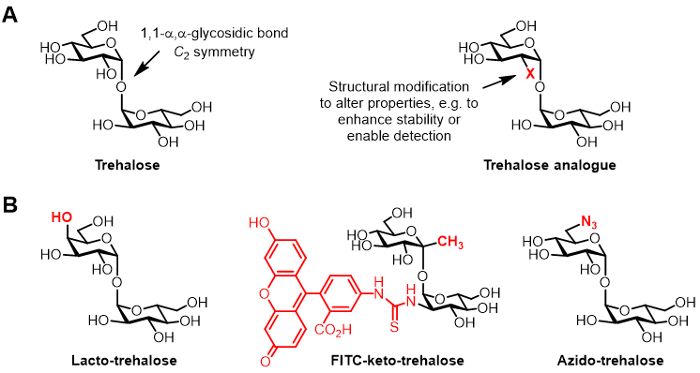 Figure 1:Trehalose and trehalose analogues. (A) Structures of natural trehalose and an unnatural trehalose analogue, where X is a structural modification. (B) Examples of trehalose analogues reported in the literature that have potential applications in biopreservation and bioimaging.
Figure 1:Trehalose and trehalose analogues. (A) Structures of natural trehalose and an unnatural trehalose analogue, where X is a structural modification. (B) Examples of trehalose analogues reported in the literature that have potential applications in biopreservation and bioimaging.
Due to its unique structure and physiological functions, trehalose has drawn significant attention for use in bio(techno)logical and biomedical applications 3. The protective properties of trehalose observed in nature—e.g., its striking ability to help sustain life in "resurrection" plants that have undergone extreme dehydration 4—have spurred its extensive use in biopreservation applications. Trehalose has been used to preserve a wide array of biological samples, such as nucleic acids, proteins, cells, and tissues 3. For instance, trehalose is used as a stabilizing additive in a number of pharmaceuticals that are on the market, including several anti-cancer monoclonal antibodies 3. As well, trehalose is used as a sweetener in the food industry, and it is extensively used for product preservation in both the food and cosmetics industries. The adoption of trehalose for these types of commercial applications was initially limited by the inability to obtain bulk quantities of pure trehalose from natural sources or through synthesis. However, an efficient enzymatic process for the economical production of trehalose from starch has recently been developed, which has spurred its widespread commercial use5.
Chemically modified derivatives of trehalose, referred to herein as trehalose analogues, have gained increasing attention for various applications (generic structure shown in Figure 1A; specific examples of trehalose analogues shown in Figure 1B) 6. For example, lacto-trehalose is a trehalose analogue with one of its glucose units replaced with galactose, thus its 4-position hydroxyl group has an inverted stereochemical configuration. Lacto-trehalose has the same stabilizing properties as trehalose but is resistant to degradation by intestinal enzymes, making it attractive as a non-caloric food additive 6, 7.
Our group's interest in trehalose analogues primarily relates to their value as mycobacteria-specific probes and inhibitors. The Barry and Davis groups developed a fluorescein-conjugated keto-trehalose analogue, named FITC-keto-trehalose, which was shown to metabolically label the cell wall of live M. tuberculosis, enabling its detection by fluorescence microscopy 8. The Bertozzi lab developed smaller azido-trehalose (TreAz) analogues that could metabolically label the cell wall and subsequently be detected using click chemistry and fluorescence analysis 9. These advances point to the possibility of using trehalose-based probes as diagnostic imaging agents for tuberculosis. Trehalose analogues have also been pursued as inhibitors of M. tuberculosis due to their potential to disrupt pathways in the bacterium that are essential for viability and virulence 10,11,12.
So far, the main obstacle to developing trehalose analogues for bio(techno)logical and biomedical applications is the lack of efficient synthetic methods. The two traditional routes to producing trehalose analogues rely on chemical synthesis (Figure 2). One route involves desymmetrization/modification of natural trehalose, while the other involves starting with properly functionalized monosaccharide building blocks and performing chemical glycosylation to forge the 1,1-α,α-glycosidic bond. These approaches, which have recently been discussed in review articles 13, 14, have proven useful for accomplishing multistep synthesis of small quantities of complex trehalose-containing natural products, such as sulfolipid-1 from M. tuberculosis 15. However, both approaches are generally inefficient, time-consuming, inaccessible to non-chemists, and, additionally, are not considered to be environmentally friendly. Thus, for synthesizing certain types of trehalose analogues, these strategies are not ideal.
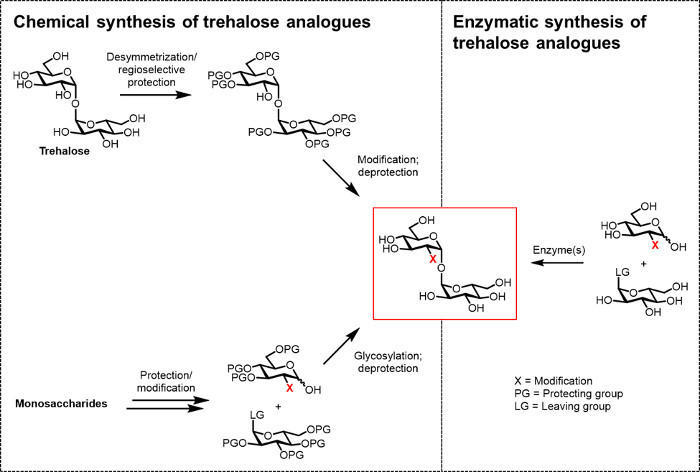 Figure 2:Approaches to trehalose analogue synthesis. Chemical approaches to trehalose analogue synthesis, shown on the left, use multistep procedures that involve difficult protection/deprotection, desymmetrization, and/or glycosylation steps. Enzymatic synthesis, shown on the right, uses enzyme(s) to stereoselectively convert simple, unprotected substrates to trehalose analogues in aqueous solution. The enzymatic protocol reported herein uses a trehalose synthase (TreT) enzyme to convert glucose analogues and UDP-glucose into trehalose analogues in a single step. Please click here to view a larger version of this figure.
Figure 2:Approaches to trehalose analogue synthesis. Chemical approaches to trehalose analogue synthesis, shown on the left, use multistep procedures that involve difficult protection/deprotection, desymmetrization, and/or glycosylation steps. Enzymatic synthesis, shown on the right, uses enzyme(s) to stereoselectively convert simple, unprotected substrates to trehalose analogues in aqueous solution. The enzymatic protocol reported herein uses a trehalose synthase (TreT) enzyme to convert glucose analogues and UDP-glucose into trehalose analogues in a single step. Please click here to view a larger version of this figure.
An efficient biocatalytic route to trehalose analogues would facilitate the production, evaluation, and application of this promising class of molecules. While the commercial enzymatic process for trehalose production5 is not adaptable to synthesizing analogues because it utilizes starch as a substrate, there are other biosynthetic pathways in nature that may be exploited for trehalose analogue synthesis. However, research in this area, which was recently reviewed 6, has been limited. One report used a method inspired by the Escherichia coli trehalose biosynthetic pathway to access a single fluoro-trehalose analogue from the corresponding fluoro-glucose. However, this approach requires a three-enzyme system that has limited efficiency and generality 8. Another approach that has been explored is to use trehalose phosphorylase (TreP) in the reverse direction, which in principle permits the one-step synthesis of trehalose analogues from glucose analogues and glucose-1-phosphate 6, 16, 17. Although this approach may have future promise, both inverting and retaining TrePs currently have drawbacks for analogue synthesis. For example, inverting TrePs have a prohibitively expensive donor molecule (β-D-glucose 1-phosphate) and retaining TrePs have poor enzyme expression yields/stability and limited substrate promiscuity. Significant improvements (e.g., via enzyme engineering) will be needed before TreP-mediated analogue synthesis is practical.
At present, the most practical approach for the enzymatic synthesis of trehalose analogues is to use a trehalose synthase (TreT) enzyme, which converts glucose and uridine diphosphate (UDP)-glucose into trehalose in a single step 6. We recently reported the use of Thermoproteus tenax TreT—a thermostable and unidirectional enzyme 18—to synthesize trehalose analogues from glucose analogues and UDP-glucose (Figure 3) 19. This enzyme only operates in the synthetic direction and avoids the problem of trehalose degradation found in the TreP system. This one-step reaction could be completed in 1 hour, and a broad variety of trehalose analogues were accessed in high yield (up to >99% as determined by high performance liquid chromatography (HPLC)) from readily available glucose analogue substrates (see Table 1 in the Representative Results section).
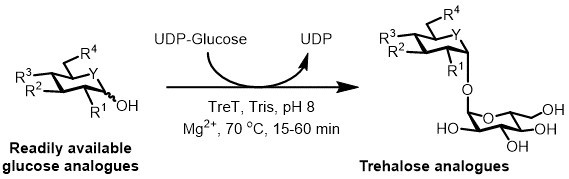 Figure 3:TreT-catalyzed one-step synthesis of trehalose analogues. The TreT enzyme from T. tenax can stereoselectively join readily available glucose analogues and UDP-glucose to form trehalose analogues in one step. R1-R4 = Variable structural modification, for example azido-, fluoro-, deoxy-, thio-, stereochemical, or isotopic label modifications; Y = variable heteroatom, for example oxygen or sulfur, or isotopically labeled heteroatom.
Figure 3:TreT-catalyzed one-step synthesis of trehalose analogues. The TreT enzyme from T. tenax can stereoselectively join readily available glucose analogues and UDP-glucose to form trehalose analogues in one step. R1-R4 = Variable structural modification, for example azido-, fluoro-, deoxy-, thio-, stereochemical, or isotopic label modifications; Y = variable heteroatom, for example oxygen or sulfur, or isotopically labeled heteroatom.
Here, we provide a detailed protocol for the TreT synthesis process, including expression and purification of TreT from E. coli, optimized TreT reaction conditions, and an improved purification method that is carried out entirely in the aqueous phase. This modified protocol enables the expedient and efficient synthesis and purification of diverse trehalose analogues on a semi-preparative scale (10-100 mg). We also demonstrate the use of this protocol for preparing and administering a trehalose-based probe to mycobacteria in less than 1 hour, which enabled the rapid fluorescence detection of mycobacterial cells.
Protocol
1. Expression and Purification of TreT from Top10 E. coli
NOTE: Please contact the authors to request the TreT-expressing E. coli strain (pBAD TreT plasmid, containing the T. tenax tret gene under the control of the AraC protein, transformed into Top10 E. coli 19) and the accompanying material transfer agreement. The following protocol typically gives a protein yield of approximately 4 mg/L.
- Prepare a 3 mL overnight culture of TreT-expressing E. coli.
- Streak Top10 E. coli transformed with pBAD-TreT expression vector on a lysogeny broth (LB) agar plate containing 100 µg/mL ampicillin.
- Incubate the plate at 37 °C for approximately 48 hours.
- Pick a single colony from the plate and inoculate 3 mL of LB liquid medium containing 100 µg/mL ampicillin in a culture tube.
- Place the tube in a shaking incubator at 37 °C x 175 rpm overnight.
- Induce protein expression in TreT-expressing E. coli.
- Add 750 mL Terrific Broth supplemented with 100 μg/mL ampicillin to a 2,800 mL Fernbach culture flask. Transfer 1 mL broth from the flask to a cuvette for later use as a blank.
- Add the 3 mL overnight culture generated in step 1.1.4 to the culture flask, then place the flask in an incubator and shake at 37 °C x 200 rpm. Periodically check the absorbance of the culture at 600 nm versus the blank collected in step 1.2.1.
- Once the absorbance at 600 nm reaches between 0.5-1.0, induce TreT expression by adding 750 µL of 1 M arabinose solution (1 mM final concentration) to the culture. Return the flask to the incubator and shake overnight at 37 °C x 200 rpm.
- Pellet and lyse the TreT-expressing E. coli cells.
- Transfer the culture to a polypropylene bottle and centrifuge for 15 min at 4000 x g at 4 °C.
- Discard the supernatant and re-suspend the pellet in 15 mL of phosphate-buffered saline (PBS).
- Transfer the cell suspension to a 50 mL conical tube and centrifuge for 15 min at 4,000 x g at 4 °C. Discard the supernatant and either proceed to cell lysis (step 1.3.4) or store the pellet indefinitely at -80 °C.
- Dissolve 1 protease inhibitor mini tablet in 20 mL of wash buffer (50 mM NaH2PO4, 500 mM NaCl, 20 mM imidazole, pH 8.0) in a 50 mL conical tube.
- Transfer the protease inhibitor-containing wash buffer to the conical tube containing the pellet. Vortex until the pellet is re-suspended.
- Transfer re-suspended cells to a 100 mL beaker and lyse the cells by sonication (pulse sequence of 45 seconds on, 45 seconds off with a run time of 2 min and 15 seconds at an amplitude of 75 percent).
- Transfer the lysate to a 50 mL metal conical tube and centrifuge for 60 min at 15,000 x g at 4 °C.
- Clarify the lysate by passage through a 0.2-0.45 μm syringe filter into a 50 mL conical tube. NOTE: The typical concentration of lysate obtained is 100 mg/mL.
- Purify TreT from E. coli cell lysate using fast protein liquid chromatography (FPLC).
- Set up the FPLC with a nickel affinity column (5 mL bed volume). Wash the column with 10 mL of deionized water or until the column is clean of any contaminants. Equilibrate the column using 20 mL of wash buffer (50 mM NaH2PO4, 500 mM NaCl, 20 mM imidazole, pH 8.0) at a flow rate of 1 mL/min.
- Load the lysate (20 mL) obtained from step 1.3.8 onto the column and elute untagged proteins with wash buffer at a flow rate of 1 mL/min until the absorbance reaches background levels (typically 80-100 mL of wash buffer are required).
- Elute His-tagged TreT by using a linear gradient of elution buffer (50 mM NaH2PO4, 500 mM NaCl, 250 mM imidazole, pH 8.0) from 1-100% over 60 min at a flow rate of 1 mL/min. Collect 4 mL fractions until TreT has eluted and the absorbance reaches baseline level. NOTE: Typically, 60 mL of elution buffer are required to elute the protein, and the protein elutes in the 60-100% elution buffer range. Approximately 10-15 mL of pure TreT in elution buffer are obtained.
- Determine the concentration of TreT by measuring absorbance at 280 nm against an elution buffer blank.
- Exchange TreT into tris(hydroxymethyl)aminomethane (Tris) buffer by dialysis.
- After preparing the dialysis tubing according to the manufacturer's instructions, prime it by rinsing with deionized water and then Tris buffer (50 mM Tris, 300 mM NaCl, pH 8.0).
- Load the TreT sample into the dialysis tubing using a syringe and blunt needle. Dialyze overnight against 2 L of Tris buffer.
- Determine the concentration of TreT by measuring absorbance at 280 nm against a blank collected from the dialysis washing.
- Transfer the TreT solution to a 50 mL conical tube and proceed to trehalose analogue synthesis (step 2) or store the enzyme at 4 °C. NOTE: TreT is a thermostable protein. The TreT was stored in Tris buffer at 4 °C for several months without observing significant loss of activity.
2. One-step Synthesis of Trehalose Analogues Using TreT Enzyme
NOTE: The protocol below describes a reaction scale based on 4 mL volume, which can deliver approximately 15-30 mg of trehalose analogue depending on reaction efficiency and molecular weight of the product. The reaction components can be scaled to obtain more or less trehalose analogue if desired.
Add glucose analogue (0.080 mmol, mass will depend on the molecular weight), UDP-glucose (0.160 mmol, 97.6 mg), and MgCl2 (0.080 mmol, 16.3 mg) to a 15 mL conical tube. The final concentrations of these components will be 20 mM, 40 mM, and 20 mM, respectively.
Add TreT in Tris buffer (obtained from step 1.5.4) and, if necessary, an appropriate volume of Tris buffer (50 mM Tris, 300 mM NaCl, pH 8.0) to achieve a final enzyme concentration of 300 μg/mL and a final volume of 4 mL. Pipet the mixture up and down gently or invert the tube to dissolve the solids.
Incubate the reaction at 70 °C with shaking at 300 rpm for 1 hour, then place the tube on ice to cool.
3. Purification of Trehalose Analogues from Crude Enzymatic Reaction Mixture
Pre-rinse a centrifugal filter unit (nominal molecular weight limit (NWML) 10 kDa) to remove trace glycerol in the membrane by adding 3 mL of deionized water to the centrifugal filter unit and centrifuging at 3,000 x g until all liquid passes through the filter into the tube (approximately 20 min). Repeat two additional times. Complete this step immediately prior to or during the reaction (step 2.3).
After cooling the enzymatic reaction mixture (obtained from step 2.3), transfer it to the pre-rinsed centrifugal filter unit. Rinse the reaction tube with 1 mL of deionized water and transfer to the centrifugal filter unit. Repeat rinsing of the reaction tube for maximum recovery of product.
Centrifuge the centrifugal filter unit at 3,000 x g until all liquid passes through the filter into the tube (approximately 20 min). Rinse the upper chamber of the centrifugal filter unit with 3 mL of deionized water and centrifuge at 3,000 x g until all liquid passes through the filter into the tube (approximately 20 min). Repeat rinsing for maximum recovery of product.
Discard the upper chamber of the centrifugal filter unit. Add mixed-bed ion-exchange resin (3 g) to the filtrate at the bottom of the tube (typical filtrate volume is 8-15 mL depending on the number of rinses). Stir at room temperature for 1 hour with a magnetic stir bar at a speed sufficient to keep the resin beads suspended in the solution.
Decant the supernatant and filter it to remove the resin. Add 5 mL of deionized water to rinse the remaining resin. Decant the supernatant and filter it, combining it with the product solution from the first decantation. Repeat rinsing of the resin for maximum recovery of product.
Analyze the reaction by thin-layer chromatography (TLC) or HPLC to determine whether complete conversion of the glucose analogue starting material to the trehalose analogue product was achieved. See step 4.1 for TLC analysis and step 4.2 for HPLC analysis.
Remove water by lyophilization or rotary evaporation to give the dried product. If no unreacted glucose analogue was observed during TLC or HPLC analysis, purification by chromatography is unnecessary. Weigh the product to obtain the reaction yield and perform nuclear magnetic resonance (NMR) spectroscopic analysis (step 4.3) to confirm product structure and purity.
- If unreacted glucose analogue was observed during TLC analysis, separate it from the trehalose analogue using a size exclusion column.
- Prepare a 1 x 100 cm column containing deionized water-saturated, extra-fine P2 polyacrylamide bead size exclusion media according to the manufacturer's instructions. NOTE: The size exclusion column can be reused after washing with deionized water.
- Re-dissolve the dried enzymatic reaction product (obtained from step 3.7) in 0.5 mL of deionized water. Apply the product solution to the size exclusion column manually or by using a column flow adapter. Rinse the vial that contained the crude product with another 0.5 mL deionized water, and load it into the size exclusion column.
- Elute the product with deionized water by gravity flow and collect fractions of approximately 2 mL volume.
- Analyze the fractions by TLC (step 4.1). Pool the fractions containing pure trehalose analogue.
- Remove water by lyophilization or rotary evaporation to give the dried product. Weigh the product to obtain the reaction yield and proceed to NMR analysis (see step 4.3).
4. Analysis of Trehalose Analogue Products
- Perform thin-layer chromatography (TLC) analysis of TreT reaction. NOTE: This procedure can also be used to analyze size exclusion column fractions. It may be necessary to concentrate the reaction mixture or column fractions prior to TLC analysis to observe compound staining on the TLC plate.
- Mark lanes on the TLC plate surface with a pencil and apply analyte(s) and relevant standard(s) to the appropriate lanes, including the glucose analogue standard, the trehalose analogue standard (if available), the reaction mixture (or fractions collected from size exclusion column purification), and a co-spot. After applying each sample to the TLC plate, allow the plate to dry. NOTE: For reaction analysis, typically 2 µL of sample is applied to the TLC plate.
- Develop the TLC plate using n-butanol/ethanol/deionized water (5:3:2).
- Dry the developed TLC plate, then dip it in 5% H2SO4 in ethanol (sugar stain) and heat on a hot plate on high setting until sugar-containing spots can be visualized (typically 5 min).
- Perform HPLC analysis of TreT reaction mixtures using any HPLC system capable of separating and detecting carbohydrates. This protocol involves carbohydrate separation using an aminopropyl HPLC column and detection using refractive index.
- Attach aminopropyl column (4.6 x 250 mm) containing a pre-column guard to the HPLC.
- Equilibrate aminopropyl column with 80% acetonitrile in deionized water at a flow rate of 0.4 mL/min.
- Load the solution of reaction product (or standard) onto the aminopropyl column.
- Elute the product (or standard) with 80% acetonitrile in deionized water at a flow rate of 0.4 mL/min and a column temperature of 50 °C. Typically, the run time used is 40 min. NOTE: Both the glucose analogue starting material and the trehalose analogue product can be detected by refractive index, although other methods such as evaporative light scattering detection (ELSD) could be used. Using the conditions described, glucose analogues typically elute between 10-15 min and trehalose analogues elute between 15-25 min.
- NMR analysis of purified trehalose analogues.
- Dissolve purified trehalose analogue in D2O (700 μL) and transfer the solution to an NMR tube.
- Acquire 1H and 13C NMR spectra according to appropriate NMR facility protocols.
5. Application of TreT-synthesized Trehalose Analogues to the Detection of Mycobacteria
- Synthesize, purify, and administer 6-TreAz to M. smegmatis (Msmeg).
- Add 6-azido-6-deoxy glucopyranose (6-GlcAz, 0.020 mmol, 4.1 mg), UDP-glucose (0.040 mmol, 24.4 mg), and MgCl2 (0.020 mmol, 4.1 mg) to a 15 mL conical tube.
- Add TreT in Tris buffer (obtained from step 1.5.4) to achieve a final enzyme concentration of 300 μg/mL and a final volume of 1 mL. Pipet the mixture up and down gently or invert the tube to dissolve the solids.
- Incubate the reaction at 70 °C with shaking for 15 min.
- Dilute the enzymatic reaction mixture with 3 mL of deionized water and transfer it to a pre-washed centrifugal filter unit (NMWL 10 kDa). Centrifuge the filter unit at 3,000 x g until most of the liquid passes through the filter into the tube, approximately 10 min.
- Discard the upper chamber of the centrifugal filter unit. Add mixed-bed ion-exchange resin (0.75 g) to the tube and stir/shake at room temperature for 25 min. Decant the supernatant and filter it to remove the resin. NOTE: Steps 5.1.1-5.1.5 provide an aqueous solution of 6-azido-trehalose (6-TreAz) at approximately 5 mM concentration in less than 1 hour. The 5 mM concentration is estimated based on the quantitative conversion of substrate to product and the dilution that takes places during the purification steps, assuming minimal loss of product during these steps. The solution can be sterile-filtered before addition to a biological sample if desired.
- Add the appropriate volume of 6-TreAz product solution to a log-phase culture of M. smegmatis (Msmeg), typically to achieve a final culture volume of 100-1,000 μL and a final 6-TreAz concentration of ~25 μM. Incubate the cells at 37 °C for the desired amount of time, typically 60 min.
- Perform click chemistry to conjugate a fluorophore to azide-labeled cells. In this protocol, use Cu-catalyzed azide-alkyne cycloaddition (CuAAC) to deliver a fluorophore to cell-surface azides in Msmeg.
- Centrifuge the cells at 3,900 x g for 5 min, and then wash the cells with PBS containing 0.5% bovine serum albumin. Repeat two times.
- Re-suspend the pelleted cells in 4% para-formaldehyde in PBS to fix them. After incubating for 10 min, repeat step 5.2.1 to wash the cells.
- Re-suspend pelleted cells in 138 μL PBS.
- Add 3 μL of a 1 mM stock solution of alkyne-carboxyrhodamine 110 (Alkyne-488) in DMSO.
- Add 3 μL of a freshly prepared 60 mM stock solution of sodium ascorbate in deionized water.
- Add 3 μL of a 6.4 mM stock solution of Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) in tert-butanol/dimethylsulfoxide (DMSO) 4:1.
- Add 3 μL of a 50 mM stock solution of CuSO4 in deionized water.
- Pipet the cell suspension up and down, then incubate in the dark at room temperature for 30 min.
- Repeat step 5.2.1 to wash the cells. Re-suspend the cells in 150 μL PBS.
- Perform cellular fluorescence analysis. In this protocol, use fluorescence microscopy to visualize cellular fluorescence of labeled Msmeg.
- Add 10 μL of bacterial cells suspended in PBS to a microscope slide and lightly spread the liquid into a thin layer using the edge of a coverslip. Allow to air dry in the dark.
- Add 10 μL of mounting medium over the dried sample, then place cover slips over the sample and apply adhesive (e.g., nail polish) to immobilize.
- Image the slides using a fluorescence microscope at 100X magnification.
Representative Results
T. tenax TreT was obtained from E. coli in a yield of approximately 4 mg/L using standard protein expression and purification techniques. A single nickel affinity chromatography step was sufficient to purify TreT from E. coli lysate (a representative FPLC trace is shown in Figure 4). As established in our initial publication on the TreT synthesis process, recombinant T. tenax TreT is capable of converting a broad of variety glucose analogues-many of which are commercially available-to the corresponding trehalose analogues with high efficiency 19. Table 1, which gives HPLC-determined reaction yields for a number of starting glucose analogues using our initially reported protocol, illustrates the scope of TreT's substrate promiscuity and its suitability for synthesis. Diverse structural modifications, including fluoro-, deoxy-, azido-, thio-, and stereochemical modifications at different positions around the sugar ring are well-tolerated by the wild-type enzyme, with the main exception being modifications at the 4-position.
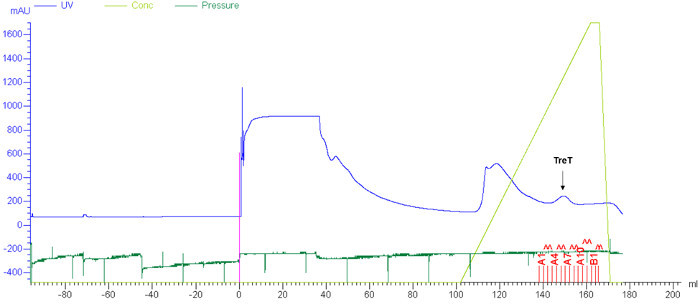 Figure 4:Representative results for TreT purification by FPLC. Purification of recombinant T. tenax TreT from E. coli lysate was carried out using nickel affinity chromatography as described in steps 1.4.1-1.4.4. Blue, ultraviolet (UV) absorbance trace; light green, concentration of elution buffer; dark green, pressure. The peak corresponding to TreT is indicated with an arrow. Please click here to view a larger version of this figure.
Figure 4:Representative results for TreT purification by FPLC. Purification of recombinant T. tenax TreT from E. coli lysate was carried out using nickel affinity chromatography as described in steps 1.4.1-1.4.4. Blue, ultraviolet (UV) absorbance trace; light green, concentration of elution buffer; dark green, pressure. The peak corresponding to TreT is indicated with an arrow. Please click here to view a larger version of this figure.
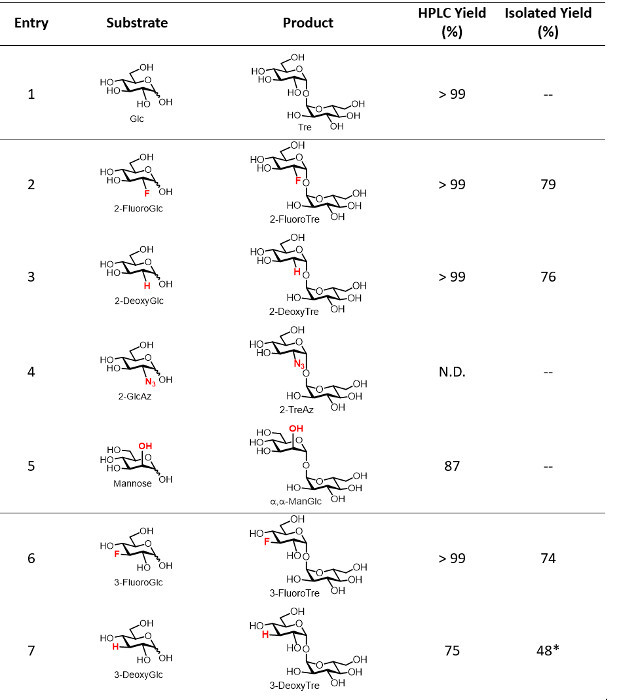 Table 1: Representative yields for the TreT reaction. HPLC-determined and isolated yields of the TreT reaction for several trehalose analogues. N.D., not detected. -- Indicates the reaction was not performed on a semi-preparative scale using the optimized protocol. *Indicates that a size exclusion chromatography step was required. Horizontal dividing lines separate the position of modification on the trehalose sugar ring. Table adapted from reference 19 with permission; updated to include isolated yields from this work. Please click here to download this file.
Table 1: Representative yields for the TreT reaction. HPLC-determined and isolated yields of the TreT reaction for several trehalose analogues. N.D., not detected. -- Indicates the reaction was not performed on a semi-preparative scale using the optimized protocol. *Indicates that a size exclusion chromatography step was required. Horizontal dividing lines separate the position of modification on the trehalose sugar ring. Table adapted from reference 19 with permission; updated to include isolated yields from this work. Please click here to download this file.
Herein, we report optimizations to our initial protocol that improve the overall efficiency and speed of the TreT synthesis process. While the original protocol used 10 mM glucose analogue and 40 mM UDP-glucose, it was determined that comparable conversion could be obtained using 20 mM glucose and 40 mM UDP-glucose, effectively doubling the amount of product generated per reaction and limiting the waste of UDP-glucose, which is relatively expensive. Using equimolar amounts of glucose analogue and UDP-glucose resulted in lower conversions. If desired, reaction times can also be shortened to less than the initially reported 60 min while still retaining comparable conversion for many analogues, which was demonstrated by the quantitative conversion of 6-GlcAz to 6-TreAz in only 15 min at 70 °C (Figures 5 and 6).
The purification protocol reported herein is substantially improved, replacing the original enzyme precipitation/silica gel chromatography method with a non-chromatographic spin dialysis/ion exchange method. The rationale for this modification is that the TreT reaction mixture consists entirely of ionic species except for the neutral trehalose analogue product. Therefore, the mixture can be spin-dialyzed to remove enzyme and then treated with mixed-bed ion exchange resin to remove all of the ionic species, delivering purified trehalose analogue in aqueous solution in as little as 45 min. This all-aqueous purification method avoids environmentally detrimental organic solvents, time-consuming evaporation and filtration steps, and dry-load silica gel chromatography, which is a slow and cumbersome process, especially for non-chemists. For TreT reactions that exhibit quantitative conversion as determined using the described TLC or HPLC analyses, this purification method provides isolated yields of approximately 60-80% at the reported reaction scale, with the product loss likely due to binding of some sugar to the ion exchange resin (see Table 1 for representative isolated yields). In cases where unreacted glucose analogue remains, it can be separated from the desired trehalose analogue product using a polyacrylamide bead-based size exclusion column, eluting with water. If preferred, this could also be accomplished using preparative-scale HPLC with an aminopropyl column. Product purity can be assessed by TLC, HPLC, and/or NMR spectroscopic analysis. Figure 5 shows representative analytical data for 6-TreAz, which was synthesized via the described protocol. The TLC and HPLC data indicated quantitative conversion of 6-GlcAz substrate to 6-TreAz product for this reaction, and the isolated yield after the spin dialysis/ion exchange purification steps was 58%. 1H and 13C NMR spectroscopic analysis confirmed the structure of the 6-TreAz product, most notably including α,α-stereochemistry of the newly formed glycosidic bond, as well as its purity.
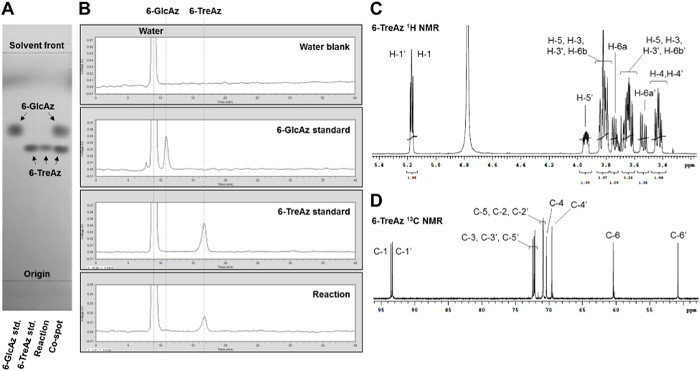 Figure 5: Representative analytical data for the TreT reaction. (A) TLC and (B) HPLC analysis of TreT-catalyzed conversion of 6-GlcAz to 6-TreAz. (C) 1H NMR and (D) 13C NMR spectra of the 6-TreAz product obtained after spin dialysis/ion exchange purification. Please click here to view a larger version of this figure.
Figure 5: Representative analytical data for the TreT reaction. (A) TLC and (B) HPLC analysis of TreT-catalyzed conversion of 6-GlcAz to 6-TreAz. (C) 1H NMR and (D) 13C NMR spectra of the 6-TreAz product obtained after spin dialysis/ion exchange purification. Please click here to view a larger version of this figure.
Given the aforementioned value of trehalose analogues for the specific detection of M. tuberculosis, the TreT process should facilitate the development and use of trehalose-based probes for tuberculosis research and diagnostic applications. To demonstrate this type of application, we used the optimized TreT process to rapidly prepare, purify, and administer 6-TreAz—a trehalose-based click chemistry probe 9—to mycobacteria to enable fluorescence detection (workflow and representative data are shown in Figure 6). Briefly, TreT was used to convert commercial 6-GlcAz to 6-TreAz quantitatively in 15 min, and then the reaction mixture was subjected to the spin dialysis/ion exchange purification method, which took only 45 min (multiple rinse steps were omitted to increase speed). Thus, an aqueous stock solution of pure 6-TreAz of known concentration (~5 mM) was generated in only 1 hour. The 6-TreAz stock was immediately administered to a growing culture of the model bacterium Msmeg (final 6-TreAz concentration ~25 µM) to accomplish azide labeling of the cell surface, which embedded a handle for click chemistry-mediated ligation 20,21 of a fluorescent probe. Subsequently, labeled cells were analyzed by fluorescence microscopy, which showed strong fluorescence for 6-TreAz-treated cells. As expected, control samples that were not treated with 6-TreAz or that were missing a functional trehalose transporter 22, which is required for 6-TreAz uptake and metabolic incorporation 9, showed no fluorescence.
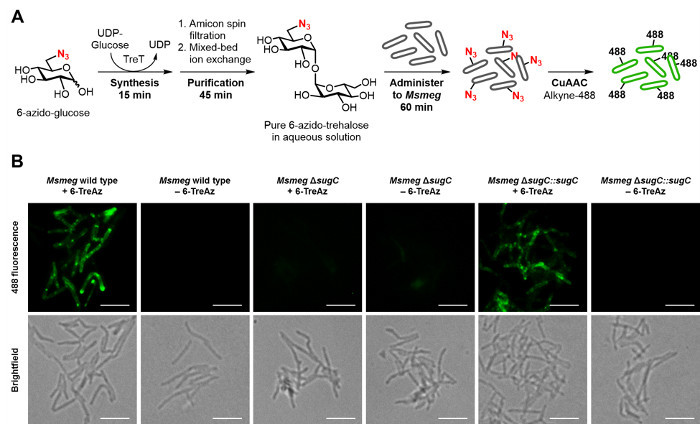 Figure 6: Representative results for the rapid detection of mycobacteria using a TreT-synthesized trehalose analogue. (A) Workflow for the rapid synthesis, purification, and use of 6-TreAz for fluorescence detection of Msmeg. Steps 5.1.1-5.1.6 of the protocol were used to synthesize and purify 6-TreAz (giving a ~5 mM aqueous solution in 1 hour), then administer it to live Msmeg to accomplish metabolic labeling of the cell surface with azides. Next, as described in steps 5.2.1-5.2.9, click chemistry (CuAAC) was performed to react cell-surface azides with an alkyne-modified fluorophore, alkyne-488. (B) Fluorescence imaging of 6-TreAz-treated Msmeg containing a functional trehalose transporter (wild type and ΔsugC::sugC) showed strong fluorescence, while the control samples of Msmeg left untreated or lacking the trehalose transporter (ΔsugC) did not. Figure adapted from reference 19 with permission; updated with workflow and imaging data from the improved TreT protocol. Scale bars, 5 µm. Please click here to view a larger version of this figure.
Figure 6: Representative results for the rapid detection of mycobacteria using a TreT-synthesized trehalose analogue. (A) Workflow for the rapid synthesis, purification, and use of 6-TreAz for fluorescence detection of Msmeg. Steps 5.1.1-5.1.6 of the protocol were used to synthesize and purify 6-TreAz (giving a ~5 mM aqueous solution in 1 hour), then administer it to live Msmeg to accomplish metabolic labeling of the cell surface with azides. Next, as described in steps 5.2.1-5.2.9, click chemistry (CuAAC) was performed to react cell-surface azides with an alkyne-modified fluorophore, alkyne-488. (B) Fluorescence imaging of 6-TreAz-treated Msmeg containing a functional trehalose transporter (wild type and ΔsugC::sugC) showed strong fluorescence, while the control samples of Msmeg left untreated or lacking the trehalose transporter (ΔsugC) did not. Figure adapted from reference 19 with permission; updated with workflow and imaging data from the improved TreT protocol. Scale bars, 5 µm. Please click here to view a larger version of this figure.
Discussion
Trehalose analogues have the potential to impact various fields, from preservation of food and pharmaceuticals to diagnosis and treatment of microbial infections 6. Existing multistep chemical synthesis methods are useful for producing complex trehalose analogues with multiple sites of modification (e.g., naturally occurring complex mycobacterial glycolipids). However, these methods are invariably lengthy and inefficient, even when applied to the synthesis of comparatively simple monosubstituted trehalose analogues 9, 13, 14. Alternative synthetic approaches are needed to rapidly and efficiently produce various types of trehalose analogues that may have value in the aforementioned areas. Biocatalytic approaches that exploit trehalose-synthesizing enzymes from nature hold the most promise. Although several trehalose-synthesizing pathways exist in nature, we consider TreT from T. tenax18 to be the ideal enzyme for trehalose analogue synthesis for several reasons.
TreT from T. tenax is thermostable, allowing reactions to be heated for improved speed and for prevention of microbial contamination in large-scale production settings. TreT from T. tenax is a unidirectional enzyme that is not capable of degrading trehalose. TreT from T. tenax is amenable to expression and purification from E. coli, and its shipment and storage is facile. The TreT reaction is a single step and it involves the simple substrates glucose and UDP-glucose. A large number of structurally/functionally diverse glucose analogues are available from numerous commercial vendors. UDP-glucose is a relatively inexpensive glucose donor, especially in comparison to the TreP donor β-D-glucose 1-phosphate. TreT from T. tenax has high promiscuity for the glucose substrate structure, so various trehalose analogues are accessible. Product purification from the TreT reaction mixture is fast and straightforward.
Herein, we reported a detailed optimized protocol for trehalose analogue synthesis that capitalizes on the above positive attributes of the TreT enzyme. The TreT process can be used to access a variety of pure trehalose analogues on a semi-preparative scale (10-100 mg), though additional scale-up should be possible if desired. Numerous types of analogues were accessed in our prior and present work, including azido-, deoxy-, fluoro-, and thio-trehaloses, as well as trehalose stereoisomers, but other modifications will also be possible (e.g., stable isotope-labeled and radiolabeled trehaloses are of particular interest). The synthesis and purification steps of the TreT process are operationally simple and can easily be carried out by personnel who are not trained in synthetic chemistry. One of the most attractive features of the TreT process is that both the synthesis and purification steps can be executed in a very short period of time, ranging from 1-4 hours depending on the number of wash steps desired (note: if size exclusion column purification is needed to separate the trehalose analogue product from unreacted glucose analogue, this increases the total time to about 2 days, which is still substantially faster than existing methods for trehalose analogue synthesis).
Although the TreT process is the most efficient enzymatic route to accessing trehalose analogues reported yet, it has some drawbacks that provide opportunity for future improvement. First, although UDP-glucose is relatively inexpensive compared to other sugar donors, the efficiency of the TreT process could be further improved by coupling it with an enzymatic synthesis of UDP-glucose from a cheaper source, e.g., α-D-glucose 1-phosphate, or by identifying an inexpensive surrogate for UDP-glucose. A second potential limitation of this method is that the reaction scope is ultimately governed by the substrate tolerance of TreT. While the tolerance of wild-type TreT is quite high (see Table 1), some analogues cannot be synthesized using this method (e.g., 4-position-modified analogues). Therefore, engineered versions of TreT with increased substrate tolerance will be valuable. Third, while many TreT-catalyzed reactions proceed with quantitative conversion, lower conversion necessitates a chromatographic purification step. In this protocol, we focused on using a relatively slow size exclusion process since it is inexpensive, does not require any specialized equipment, and can be performed solely with water elution. Alternatively, HPLC purification employing a preparative-scale aminopropyl column could be used as a faster method for product purification. Despite some of these limitations, the TreT process provides a powerful platform for the rapid and efficient preparation of trehalose analogues, and should expedite their evaluation and application in the bio(techno)logical and biomedical fields. Future improvements to the TreT process, discussed above, will further strengthen research and applications related to trehalose and its derivatives.
Trehalose analogues have potential value in various areas. As mentioned in the introduction, natural trehalose is used for biopreservation and food sweetening applications, so trehalose analogues with properties such as degradation resistance may be attractive. In addition, a variety of stable isotope-labeled and radiolabeled versions of natural trehalose are commercially available for research purposes, and the TreT process can certainly provide fast and efficient access to these molecules. Trehalose analogues are also of interest for targeting pathogens with probes and inhibitors since trehalose is absent from mammals. One application in this area that is of particular interest is the detection of mycobacteria. M. tuberculosis, the causative agent of tuberculosis, which kills 1.5 million people per year, contains unique trehalose-processing pathways that are absent from mammals. For example, the trehalose-recycling transporter and downstream enzymes that incorporate trehalose into outer membrane-resident glycolipids are not present in mammals 22. Targeting this mycobacteria-specific machinery with detectable trehalose analogues represents an attractive approach to in vivo imaging of M. tuberculosis infection in model systems and possibly human patients. Indeed, the above examples of FITC-keto-trehalose and TreAz (Figure 1B) have paved the way for such developments 8,9. To facilitate access of these tools to the research community, we reported herein a protocol for rapidly synthesizing and using 6-TreAz to image mycobacteria in combination with click chemistry.
This protocol can be adapted for the development of other detectable trehalose analogues that may be useful for tuberculosis diagnostic applications. For instance, Barry and Davis first proposed the possible use of 18F-fluoro-trehalose as a positron emission tomography (PET) probe for in vivo imaging of mycobacterial infection 8. The TreT process may be ideal for realizing this goal. TreT is capable of rapidly and quantitatively generating 2-fluoro-trehalose from 2-fluoro-glucose (see Table 1), which is critical because 18F-2-fluoro-glucose is short-lived (18F half-life = 110 min), necessitating extremely fast chemistry. Furthermore, 18F-2-fluoro-glucose is readily available because it is already used in the clinic for PET imaging of tumors. Use of the described TreT process for the development of 18F-fluoro-trehalose and other trehalose analogues for various applications is currently underway in our laboratories.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was funded by a grant from the National Institutes of Health (R15 AI117670) to B.M.S and P.J.W, as well as a Cottrell College Scholar Award from the Research Corporation (20185) to P.J.W. L.M.M. was supported by a Provost's Fellowship from CMU.
References
- Elbein AD, Pan YT, Pastuszak I, Carroll D. New insights on trehalose: a multifunctional molecule. Glycobiology. 2003;13:17–27. doi: 10.1093/glycob/cwg047. [DOI] [PubMed] [Google Scholar]
- Tournu H, Fiori A, Van Dijck P. Relevance of trehalose in pathogenicity: some general rules, yet many exceptions. PLoS Pathog. 2013;9:1003447. doi: 10.1371/journal.ppat.1003447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake S, Wang YJ. Trehalose: Current use and future applications. J. Pharm. Sci. 2011;100:2020–2053. doi: 10.1002/jps.22458. [DOI] [PubMed] [Google Scholar]
- Adams RP, Kendall E, Kartha KK. Comparison of free sugars in growing and desiccated plants of Selaginella lepidophylla. Biochem. Syst. Ecol. 1990;18:107–110. [Google Scholar]
- Kubota M. In: Glycoenzymes. Ohnishi M, editor. Japan Scientific Societies Press; 2000. [Google Scholar]
- Walmagh M, Zhao R, Desmet T. Trehalose analogues: latest insights in properties and biocatalytic production. Int. J. Mol. Sci. 2015;16:13729–13745. doi: 10.3390/ijms160613729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-M, Chang Y-K, Ryu S-I, Moon S-G, Lee S-B. Enzymatic synthesis of a galactose-containing trehalose analogue disaccharide by Pyrococcus horikoshii trehalose-synthesizing glycosyltransferase: Inhibitory effects on several disaccharidase activities. J. Mol. Catal. B: Enzym. 2007;49:98–103. [Google Scholar]
- Backus KM, et al. Uptake of unnatural trehalose analogs as a reporter for Mycobacterium tuberculosis. Nat. Chem. Biol. 2011;7:228–235. doi: 10.1038/nchembio.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts BM, et al. Probing the mycobacterial trehalome with bioorthogonal chemistry. J. Am. Chem. Soc. 2012;134:16123–16126. doi: 10.1021/ja3062419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JD, et al. Synthesis and biological evaluation of trehalose analogs as potential inhibitors of mycobacterial cell wall biosynthesis. Carbohydr. Res. 2002;337:105–120. doi: 10.1016/s0008-6215(01)00288-9. [DOI] [PubMed] [Google Scholar]
- Wang J, et al. Synthesis of trehalose-based compounds and their inhibitory activities against Mycobacterium smegmatis. Bioorg. Med. Chem. 2004;12:6397–6413. doi: 10.1016/j.bmc.2004.09.033. [DOI] [PubMed] [Google Scholar]
- Gobec S, et al. Design, synthesis, biochemical evaluation and antimycobacterial action of phosphonate inhibitors of antigen 85C, a crucial enzyme involved in biosynthesis of the mycobacterial cell wall. Eur. J. Med. Chem. 2007;42:54–63. doi: 10.1016/j.ejmech.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Sarpe VA, Kulkarni SS. Regioselective protection and functionalization of trehalose. Trends in Carbohydr. Res. 2013;5:8–33. [Google Scholar]
- Chaube MA, Kulkarni SS. Stereoselective construction of 1,1-alpha,alpha-glycosidic bonds. Trends in Carbohydr. Res. 2013;4:1–19. [Google Scholar]
- Leigh CD, Bertozzi CR. Synthetic studies toward Mycobacterium tuberculosis sulfolipid-I. J. Org. Chem. 2008;73:1008–1017. doi: 10.1021/jo702032c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaen H, et al. Efficient enzymatic synthesis of disaccharide, alpha-D-galactosyl-alpha-D-glucoside, by trehalose phosphorylase from Thermoanaerobacter brockii. J. Appl. Glycosci. 2001;48:135–137. [Google Scholar]
- Vander Borght J, Soetaert W, Desmet T. Engineering the acceptor specificity of trehalose phosphorylase for the production of trehalose analogs. Biotechnol. Progr. 2012;28:1257–1262. doi: 10.1002/btpr.1609. [DOI] [PubMed] [Google Scholar]
- Kouril T, Zaparty M, Marrero J, Brinkmann H, Siebers B. A novel trehalose synthesizing pathway in the hyperthermophilic Crenarchaeon Thermoproteus tenax: the unidirectional TreT pathway. Arch. Microbiol. 2008;190:355–369. doi: 10.1007/s00203-008-0377-3. [DOI] [PubMed] [Google Scholar]
- Urbanek BL, et al. Chemoenzymatic synthesis of trehalose analogues: rapid access to chemical probes for investigating mycobacteria. ChemBioChem. 2014;15:2066–2070. doi: 10.1002/cbic.201402288. [DOI] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Tornøe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Kalscheuer R, Weinrick B, Veeraraghavan U, Besra GS, Jacobs WR. Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2010;107:21761–21766. doi: 10.1073/pnas.1014642108. [DOI] [PMC free article] [PubMed] [Google Scholar]