

 A new class of fluorescence chloride sensors are derived from a facile citrate-based synthesis platform, enabling low-cost chloride sensing for clinical diagnosis of Cystic Fibrosis through dynamic quenching mechanisms. We demonstrate for the first time a selective multi-halide sensing strategy using a single fluorophore.
A new class of fluorescence chloride sensors are derived from a facile citrate-based synthesis platform, enabling low-cost chloride sensing for clinical diagnosis of Cystic Fibrosis through dynamic quenching mechanisms. We demonstrate for the first time a selective multi-halide sensing strategy using a single fluorophore.
Abstract
Chloride is an essential electrolyte that maintains homeostasis within the body, where abnormal chloride levels in biological fluids may indicate various diseases such as cystic fibrosis. However, current analytical solutions for chloride detection fail to meet the clinical needs of both high performance and low material or labor costs, hindering translation into clinical settings. Here we present a new class of fluorescence chloride sensors derived from a facile citrate-based synthesis platform that utilize dynamic quenching mechanisms. Based on this low-cost platform, we demonstrate for the first time a selective sensing strategy that uses a single fluorophore to detect multiple halides simultaneously, promising both selectivity and automation to improve performance and reduce labor costs. We also demonstrate the clinical utility of citrate-based sensors as a new sweat chloride test method for the diagnosis of cystic fibrosis by performing analytical validation with sweat controls and clinical validation with sweat from individuals with or without cystic fibrosis. Lastly, molecular modeling studies reveal the structural mechanism behind chloride sensing, serving to expand this class of fluorescence sensors with improved chloride sensitivities. Thus citrate-based fluorescent materials may enable low-cost, automated multi-analysis systems for simpler, yet accurate, point-of-care diagnostics that can be readily translated into clinical settings. More broadly, a wide range of medical, industrial, and environmental applications can be achieved with such a facile synthesis platform, demonstrated in our citrate-based biodegradable polymers with intrinsic fluorescence sensing.
Introduction
The transformation of healthcare from curative to predictive medicine relies on the development of efficient point-of-care diagnostic systems, enabling early detection of biological markers for disease.1 Such easy-to-operate sensing systems are essential for the detection of chloride in biological fluids, where abnormal levels in the sweat, urine, serum, and cerebral spinal fluid (CSF) are respective indicators of Cystic Fibrosis (CF), metabolic alkalosis, Addison's disease, and amyotrophic lateral sclerosis (ALS).2 Notably, the sweat chloride test is the gold standard for the diagnosis of CF. Thus early diagnosis of CF, marked by elevated sweat chloride levels, can prevent serious malnutrition and promote long-term growth.3 Yet a wide-scale survey of about 400 clinical labs revealed that sweat chloride for CF diagnosis was determined using manual titration in almost 70% of labs,4 a method based on mercuric nitrate that is time-consuming, labor intensive and prone to technical error.5 Another assessment of 84 hospitals concluded that improper training and inexperience are the greatest factors contributing to the misdiagnosis of CF,6 with false negatives rates reported as high as 12%,7 highlighting the need for automated point-of-care diagnostic tools. However, current chloride analysis techniques fail to meet the clinical needs of both high performance and low material or labor costs, hindering translation into clinical settings.
Material cost is a significant barrier to entry for clinical diagnostics, particularly in the developing world with limited access to sophisticated instrumentation.8 Moreover, sophisticated analytical methods (such as ion exchange chromatography) may yield high sensitivity and selectivity, but these processes cannot be fully automated and are unsuitable for clinical settings due to expensive labor costs and high-maintenance equipment.9–11 To date, fully automated chloride analysis methods include ion-selective electrodes (ISE), coulometry, and colorimetry, yet all three methods suffer from low halide selectivity.9 For example, bromide and iodide respectively produce signals 102 and 103 times greater than chloride in ISE measurements, leading to misdiagnosis:12 positive errors in chloride readings are commonly reported in medical cases of elevated bromide.13,14 Therefore, practical translation of rapid chloride sensing technology to clinical settings relies on significant advances in cost-reduction and automation while maintaining high standards of sensitivity and selectivity.8
In meeting these challenges, fluorescence-based sensors may play a vital role in the transition towards point-of-care; fluorescence offers high sensitivity, minimal background, rapid response kinetics, and low technical complexity for end users. However, existing fluorescence-based chloride sensors such as N-[ethoxycarbonylmethyl]-6-methoxy-quinolinium (MQAE) and other quinolinium-based dyes all suffer from narrow linear range, low fluorescence yield, poor photostability, and high costs, thereby limiting clinical or commercial integration.15 For instance, Wang et al. recently reported a sweat chloride analyzer based on MQAE as the chloride indicator, which showed non-linear response to normal sweat chloride levels (<20 meq. L–1) and significant loss of sensitivity at higher chloride levels.16
Here we identify and isolate a chloride recognition moiety of our previously reported citrate-based biodegradable polymers,17 from which we develop a new class of fluorescence chloride sensors with strong fluorescence properties, and present a rational design for automated and selective chloride sensing. To demonstrate clinical potential, we validated our sensing strategy as an acceptable sweat test method for the diagnosis of CF based on the College of American Pathologists guidelines, and assessed its clinical performance compared to a standard reference method with sweat from individuals with and without cystic fibrosis. Lastly, we performed molecular modeling studies to better understand the mechanism behind chloride sensing, thereby guiding the synthesis of a versatile class of fluorescence sensors with tunable chloride sensitivities that suit the mean and range of various clinical tests.
To address current limitations in chloride sensing, citrate-derived fluorescence sensors represent a new class of halide sensors capable of both selective and automated sensing. Such advances in performance and cost-reduction show promise for reliable point-of-care diagnostic systems to facilitate early detection and timely therapeutic management of cystic fibrosis.
Results and discussion
Synthesis
The new sensors are an extension to our previous work on citrate-based, biodegradable photoluminescent polymers (BPLPs),17 which were later processed into polymeric micelles18 and polylactone co-polymer nanoparticles19 for noninvasive fluorescence bioimaging. To broaden the scope of applications beyond polymeric labeling, we investigated sensing capabilities of our pre-polymers and discovered that its fluorescence is quenched in the presence of chloride ions, strictly under acidic conditions (Fig. 1A, ESI Fig. S1†). Our citrate-based materials retained this chloride sensitivity upon crosslinking into films or immobilizing onto surfaces, suggesting potential as thin-film sensors (ESI Fig. S2†).
Fig. 1. Low-cost synthesis of citrate-based fluorescent sensors. (A) One-pot synthesis scheme of biodegradable photoluminescent polymers (BPLPs), and isolation of CA–cysteine as the chloride recognition element. Chloride sensitivity is observable in a cuvette as drops of NaCl completely quenches the blue fluorescence of CA–cysteine. (B) Facile synthesis scheme of citric acid and various primary amines to expand the family of citrate-based halide sensors with versatile halide sensitivities.

We then processed our polymers into small molecular dyes to conduct direct studies on chloride quenching mechanisms and develop a practical method for chloride sensing.
Accordingly, we identified and isolated the chloride recognition moiety of our polymers (Fig. 1A) based on our previous hypothesis that the strong, intrinsic fluorescence of BPLPs emanated from a conjugated ring formed from citrate and l-cysteine,17 a fluorescent structure later supported by ESI-MS, elemental analysis, 1H and 13C NMR studies by Kasprzyk20 and Hu21 in the characterization of BPLP and BPLP-co-poly(lactide-co-glycolide) respectively. The resulting compound, CA–cysteine, presented match stick-like crystal morphology under SEM (ESI Fig. S3A†) and emitted blue fluorescence (maximal excitation of 365 nm, emission of 445 nm) with a molar absorptivity of 8640 M–1 cm–1. Notably, CA–cysteine exhibited favorable fluorescence properties such as a high quantum yield of 0.81, long lifetime of 10.06 ns, and strong photostability compared to traditional organic dyes, rhodamine B and fluorescein (ESI Fig. S3†). Long lifetimes enhance optimal functioning of LED devices22 while high photostability enables reproducible measurements, particularly for real-time monitoring applications. Chloride sensitivity was similarly pH-dependent (Fig. 2A). We then streamlined the synthesis strategy into a simple one-pot reaction of citric acid and a primary amine (l-cysteine, cysteamine, or methyl serine) in water to establish a facile synthesis platform for a family of citrate-based chloride sensors (Fig. 1B), with chemical structures characterized by HPLC-ESI-MS, FTIR, 1H and 13C NMR studies.23 Hence low-cost and “green” reaction schemes are achieved with this solvent-free synthesis based on simple monomers, enabling in vitro cytocompatibility to our dye as demonstrated in 3T3 cell lines for dosages up to 10.3 mM (ESI Fig. S4†). In comparison, the syntheses of existing chloride indicators are complex and expensive, such as in SPQ which requires the reaction of 6-methoxyquinoline under propanesulton as solvent, resulting in highly prohibitive commercial costs (ESI Table 3†).24
Fig. 2. Fluorescence-based chloride sensing. (A) Chloride quenches CA–cysteine fluorescence strictly under acidic conditions. (B) Stern–Volmer plots linearizing quenching rates over chloride concentration reveal that chloride sensitivity (K SV, or the slope) increases with acidity, R 2 > 0.997. Detection procedures for chloride sensing (shown in schematics) are simplified by standardizing at fixed sulfuric acid concentrations. (C) Comparison of quenching efficiency of common ions, each at 100 mM, in the presence of CA–cysteine at pH 1.3 (except for OH–).

Chloride sensing
In defining the sensing behaviors of CA–cysteine, we first addressed its peculiar chloride recognition that switches on with charge manipulation. To illustrate, hydrochloric acid had twice the quenching power of sulfuric acid or nitric acid (ESI Fig. S5†), although the counter-ions chloride, sulfate, and nitrate by themselves had no effect on fluorescence (ESI Fig. S6†). Fluorescence lifetime studies revealed that although CA–cysteine is generally insensitive to chloride, pH conditions below 2.4 opened accessibility to dynamic quenching mechanisms by both protons and chloride such that chloride sensitivity increased with acidity (ESI Fig. S7, S12† and 2B).
Following, we translated these quenching behaviors into a practical sensing method by linearizing the fluorescence quenching effects of two dynamic quenchers (protons and chloride) according to the modified Stern–Volmer (SV) equation below (derived in ESI Section B†):
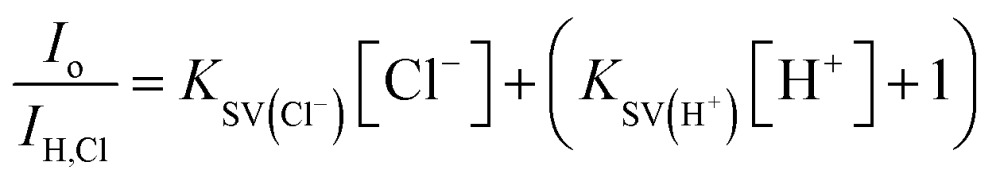 |
1 |
where I o is the unquenched fluorescence intensity of neutral state CA–cysteine, I H,Cl is the quenched fluorescence intensity at the given proton and chloride concentration, and K SV represents the sensitivity of CA–cysteine to protons or chloride. Typically, an SV plot of quenching rate (I o/I H,Cl) versus quencher concentration (such as [Cl–]) produces a slope that is linear for dynamic quenchers. Hence at fixed pH values where the (K SV(H+)[H+] + 1) term becomes a y-intercept constant, we can obtain chloride sensitivity (K SV(Cl–)) from the slope of each standard curve at selected pH levels (Fig. 2B), from which an unknown chloride sample would be analyzed under the same dye strength and acidity.
To simplify detection procedure (and avoid repeated pH measurements), we employed fixed sulfuric acid levels, which is equivalent to fixed pH given that salinity does not significantly affect pH, and neither our dye (at ∼10 μM working concentrations). Based on simple polyprotic acid calculations, 0.16 M sulfuric acid converts to pH 0.77, as listed along Fig. 2B. For clinical applications, we created standard curves at fixed sulfuric acid concentrations of 0.16 M to minimize buffering effects from sweat. We also catalogued K SV(Cl–) under various pH conditions to show that chloride sensitivity increases with acidity, while the reverse showed that proton sensitivity (K SV(H+)) was independent of chloride levels (ESI Fig. S8†).
Lastly, we established the specificity of our sensors by assessing quenching effects of other common acids, salts, bases, and solvents at various pH conditions (Fig. 2C, ESI Fig. S9†). From this systematic investigation, we confirmed that the environment sensitivity of CA–cysteine fluorescence was limited to heavy halides, of which only chloride was deemed clinically relevant for sweat testing since physiological bromide and iodide are typically less than 40 μM and 1.6 μM respectively. Consideration of bromide and iodide in our sensing strategy will be discussed later.
Clinical evaluation
Next, we evaluated the clinical potential of citrate-based sensors as a new sweat test method. The sweat chloride test is the gold standard for the diagnosis for CF, as patients homozygous for the CF gene have sweat chloride concentrations greater than 60 mM.3,7,25 We performed validation of our sensing strategy based on CA–cysteine against sweat controls prepared by a clinical lab, obtaining ranges (ESI Table S3†) well within the evaluation criteria of ±10.0 mM L–1 or 15% according to the external quality assessment set by the College of American Pathologists (CAP) Laboratory Accreditation Program.25
Following, we assessed the clinical performance of CA–cysteine compared to standard mercuric nitrate titration for the determination of sweat chloride from 13 individuals with or without CF. There was excellent correlation and agreement between the two techniques, with an intraclass correlation coefficient (ICC) of 0.984 with a 95% confidence interval of (0.948, 0.995) (Fig. 3A). The Bland & Altman plot showed a mean difference of 2.46 and differences between techniques within 13.3 mM at a 95% limit of agreement, supporting the reliability of our sensing strategy for the diagnosis of CF (Fig. 3B).
Fig. 3. Clinical validation of sweat chloride for the diagnosis of cystic fibrosis. (A and B) Comparison of CA–cysteine-based and standard clinical mercuric nitrate titration methods to determine sweat chloride for the diagnosis of cystic fibrosis based on 13 volunteers, where correlation is measured by the intraclass correlation coefficient (A) and agreement is measured by a Bland & Altman plot (B).

Molecular mechanism
Following these sensing demonstrations, we next sought a molecular mechanism behind such chloride sensing behaviors that change with pH, and the rules that govern quenching interdependencies for heavier halides. Our approach combines fluorescence lifetime studies with computational modeling to decipher the link between charge states of CA–cysteine and its electronic distribution, fluorescence properties, and sensing efficiency. In brief, we propose that each successive protonation step of the 7-carboxyl, 3-carboxyl, and 5-carbonyl of CA–cysteine activates independent halide quenching pathways.
To support this hypothesis, we first performed acid–base titration of CA–cysteine to confirm a diprotic acid with a pK a1 of 2.345 and a pK a2 of 3.41, assigned to the 3- and 7-carboxyl groups respectively (ESI Fig. S11†). We next calculated ground and excited state electronic distributions of these various charge states of CA–cysteine with Hyperchem 8.0 based on the fused, two-ring heterocylic compound numbered in Fig. 4A, with optimized geometry at the ab initio/3-21G level of theory and electronic configurations with ZINDO/S. Generally, molecular orbital (MO) isosurface plots revealed a highly polarizable, conjugated 2-pyridone element as the main spectral transition of CA–cysteine responsible for its bright fluorescence (Fig. 4B and C).
Fig. 4. Sensing mechanisms. (A) Jablonski diagram summarizing the sensing capabilities and mechanisms of CA–cysteine, in which conditions below pH 2.4 leads to excited state protonation of the 5-carbonyl, forming a cationic state that is key to activating halide quenching processes. (B–C) HOMO (B) and LUMO (C) isosurface plots highlight planarity and an aromatic 2-pyridone element responsible for the bright fluorescence of CA–cysteine.

Third, charge density calculations suggested that the electronegative elements nitrogen and carbonyl within this extended aromatic system undergo internal charge transfer (ICT) state upon excitation, favoring the enol resonance structure (ESI Section C†). This strong ICT state enhances the transition dipole moment of CA–cysteine, increasing its absorption and emission intensities as also observed in similar fluorophores.22 More importantly, the ICT state would effectively lower the excited state pK a of the 5-carbonyl by resonance stabilization so that a key cationic state of CA–cysteine would become accessible by excited state protonation even under moderate acidity (Fig. 4A). We speculate that excited state protonation induces greater sp3 character on this carbonyl group, resulting in loss of planarity and rigidity of the conjugated 2-pyridone system since the C-5 carbon has significant atomic orbital (AO) contributions in the excited state (ESI Section C†). This induces out-of-plane vibrations that draw pathways to non-radiative relaxation, a dynamic quenching mechanism commonly reported for heterocyclic aromatics.26–28 Collectively, proton-induced dynamic quenching is primarily attributed to excited state protonation of the 5-carbonyl, which cannot occur until both the 7- and 3-carboxylates are protonated (below pH 3.4 and 2.4 respectively). In support of this model, the SV-plot of fluorescence lifetimes show an abrupt linearity (indicating dynamic quenching) just below pH 2.3 (ESI Fig. S12†).
Halide sensing mechanism
In light of the presented model, we propose that each successive protonation step of the 7-carboxyl, 3-carboxyl, and 5-carbonyl groups in CA–cysteine, at their respective pH regions, lowers electrostatic repulsion to enable halide ion-complex formation in the excited state, activating halide quenching pathways in progressively acidic conditions. Indeed, fluorescence lifetime studies confirm that halide quenching is a dynamic process that only affects excited populations of CA–cysteine (ESI Fig. S7†). We also show that ion-complex formation is an intermediate step in activating halide quenching processes, as described below:
 |
2 |
where eqn (2) is based on the “heavy atom effect”, commonly proposed for halide sensing in quinolinium-based dyes,15 in which partial charge transfer during excited state ionic interactions with halides generate spin–orbit coupling leading to intersystem crossing (ISC) to the triplet state.29,30 As a result, k 1/k –1 depends on the ionization potential of the quencher, i.e. quenching efficiency increases with the mass of halide.31 These enhanced quenching rates of bromide and iodide are also demonstrated in CA–cysteine (Fig. 5D and E), supporting the notion that ionic interaction with halides is indeed a critical step in the quenching process.
Fig. 5. Synthesis scheme of citrate-based sensors, and schematics for automated and selective multi-halide sensing. (A–C) Stern–Volmer plots show the chloride sensitivity of CA–cysteine (A), with sensitivity greatly enhanced with CA–cysteamine (B), and greatly decreased with CA–methyl-serine (C) (R 2 > 0.997, R 2 > 0.999, R 2 > 0.995 respectively). (D and E) Bromide and iodide sensitivity of CA–cysteine were catalogued at fixed sulfuric acid concentrations to obtain halide sensitivity under each acidity, which increase with acidity (R 2 > 0.999 for both). (F) Schematic for automated, multi-halide sensing involves measurement of sample at three pH conditions following standardization to solve a system of three Stern–Volmer equations for chloride, bromide, and iodide concentrations.

Finally, our fluorescence studies show that these protonation steps are linked to significant changes in halide quenching efficiency. Above pH 3.4 where deprotonation of both carboxyl groups (to the dianion) is expected to severely suppress these non-radiative pathways, chloride sensitivity of CA–cysteine is non-existent (K SV = 0.02) while bromide and iodide sensitivities are significantly reduced (K SV = 4.9, 37 respectively) (ESI Fig. S19†). Near pH 2.4, with both carboxylates protonated (to the neutral state), heavier halides experience a jump in sensitivity (K SV = 34 for bromide, 72 for iodide), but accessibility of chloride to the electron-deficient aromatic ring is still restricted (K SV = 1.44) due to the charge separated enolate group (ESI Fig. S19†). Lastly, by increasing acidity below pH 2.4, we promote excited state protonation of the 5-carbonyl (to the cationic state), removing this last electrostatic repulsion (enhancing k 1) and activating halide quenching processes (enhancing k ISC) so that chloride sensitivity is abruptly activated at this pH and continues to improve with increased acidity (up to K SV = 4.8) (Fig. 2B).
In contrast, pH-dependence is not recognized in quinolinium-based halide sensors because of the large spatial separation of electron withdrawing groups (EWGs) from the heterocyclic ring, suggesting that displacing charged substituents may improve halide sensitivity or remove pH-dependence of sensitivity.
Expanding the class of citrate-based sensors
These computational studies also provide an important framework for the development of other citrate-based sensors with improved sensitivities. To illustrate, protonation of the 7-carboxylate lowers LUMO energy levels due to an intrinsic property of the 2-pyridone moiety in which the placement of an additional nodal plane during the HOMO–LUMO transition creates a spatial redistribution of molecular orbitals such that the C-7 carbon has significant AO contributions at the LUMO but minimal contributions at the HOMO (Fig. 4B and C, ESI Fig. S13†). Accordingly, AO isosurfaces showed that C-5 and C-7 carbons are ideal placements for substituents that would affect excited state behaviors, while C-3 and C-8 are ideal for altering ground-state behaviors.
To translate this concept practically, we modified the C-3 substituent by reacting citrate with various primary amines to demonstrate the role of the 3-carboxyl in guiding halide sensing efficiency. We duly synthesized citrate-based sensors structurally similar to CA–cysteine but lacking the 3-carboxyl (CA–cysteamine), or with an additional 3-methyl group (CA–methyl-serine) (Fig. 1B).23 Both compounds exhibited high quantum yields and strong photostability on par with that of CA–cysteine (ESI Table S1†). However, CA–cysteamine enhanced chloride sensitivity 5-fold and bromide sensitivity 2-fold under the same pH conditions as CA–cysteine, while CA–methyl-serine was insensitive to chloride and weakly sensitive to bromide, confirming the notion that charged or bulky substituents at the C-3 position significantly affect excited state kinetics of halide complex formation (Fig. 5B and C, ESI Fig. S14 and 15†). These molecular understandings enable tunable sensing behaviors for the development of a versatile class of citrate-based sensors.
Rational design for multi-halide sensing
Although physiological levels of bromide and iodide are typically low enough to be negligible in chloride measurements, cases of elevated bromide can give positive errors in chloride readings, while bromide determination in the presence of bulk chloride can be highly inaccurate.14 This lack of halide selectivity is also an issue in existing fluorescence-based chloride sensors—yellow fluorescent protein (YFP) derivatives and quinolinium-based dyes15—and has hindered commercial applications of fluorescence quenching-based detection methods. Wolfbeis et al. proposed a solution based on three SV equations to solve for three quencher concentrations,32 but this method requires complex standardization with three different fluorophores, each with distinct halide sensitivities.33
Citrate-based sensors greatly simplify this process due to their unique pH-dependence of halide selectivity, enabling simultaneous detection of multiple halides with a single dye. To the best of our knowledge, this has not been demonstrated in literature. CA–cysteine, for example, acquires the traits of distinct sensors under different pH conditions, yet maintains the advantages of single fluorophore sensing such as predictable degradation and photobleaching rates, enabling reproducibility in serial measurements. This practical advantage greatly simplifies time-dependent corrections in fluorescence sensing to improve reliability and performance.
To justify this multi-halide sensing strategy, we demonstrated that CA–cysteine quenching sensitivities for all halides change with respect to pH (Fig. 2B and 5D and E) in rates and patterns unique to each halide (ESI Fig. S16†), and that halides are independent quenchers amongst themselves (ESI Fig. S17†). Essentially, the repressive effect of each charged substituent (7-carboxyl, 3-carboxyl, and 5-carbonyl) depends on the mass of halide, resulting in pH-dependencies unique to each halide. For example, the charged 7-carboxyl group plays a critical role in suppressing chloride quenching processes, evidenced by a jump in the chloride sensitivity of CA–cysteamine near its pK a of 3.4 (ESI Fig. S18†). However, the effectiveness of hindrance from bulky or charged groups decreases with heavier halides, with bromide and iodide showing a progressively smaller jump near this pH. Further, iodide sensitivity is not affected by protonation of the 5-carbonyl, reaching peak sensitivity under mildly acidic conditions (Fig. 5D), nor by the presence of the 7-methyl on CA–methyl-serine (ESI Fig. S15†). These studies demonstrate that the pattern of pH-dependence in sensitivity is unique to each halide.
Our method for simultaneous sensing of multiple halides is performed by first determining each halide quenching rates (K SV) at three fixed pH conditions to establish standard curves, followed by measuring the unknown sample at the three pH conditions (obtaining I o/I) and solving three SV equations for three unknown concentrations (of chloride, bromide, and iodide) using a matrix solver, all of which can be automated as depicted in Fig. 5F. A simplified proof-of-concept is demonstrated through selective determination of chloride and bromide in sea water using two pH conditions (ESI Section D†).
Next, our design for selective chloride sensing simplifies the above protocol by combining bromide and iodide quenching contributions into a single term, reducing sample measurements to two pH conditions and bypassing individual standardization of bromide and iodide. We provide additional details and a mathematical proof in ESI Section E.†
We can also apply this system towards automated bromide sensing, which has so far been limited by poor halide selectivity because the reactivity, oxidizing potential, and conductivity of bromide are intermediate to that of chloride and iodide.34 For example, bromide and iodide ions generate mercuric complexes and silver halides in colorimetric and coulometric methods respectively, as chloride ions do.9 On the other hand, selective methods lack automation due to the complex chemical or chromatographic separation steps required.35 Thus our selective sensing strategy may significantly improve the accuracy of chloride and bromide readings in fast-paced clinical and industrial settings.
Detection limits
Next, we defined the detection limits of the broader family of citrate-based sensors to identify suitable applications. Since the SV equation is a linear representation of dynamic quenching rates, we defined upper limits based on nonlinearity in the SV plot and determined lower limits from instrumental sensitivity (signal and noise) and halide sensitivity (K SV) values (ESI Section F†). CA–cysteine possesses a particularly wide dynamic range for chloride determination, maintaining linearity in SV plots up to 1400 mM, useful for analyzing urine chloride which fluctuates from 10–280 mM (ESI Fig. S21†). Within the sensor family, CA–cysteamine boasts the lowest detection limits of 373 μM for chloride, 32 μM for bromide, and 20 μM for iodide (ESI Table S4†). The detection limits for bromide may be suitable for analyzing urine or serum bromide, both of which have a baseline of 80 μM.36,37 Elevated serum bromide levels may indicate bromism, which can result from occupational hazards involving fumigants and fire extinguishers as well as administration of drugs such as sedatives and laxatives.38 The development of CA–cysteamine not only enhanced halide detection limits, but also enabled chloride sensing at mildly acidic conditions, with a lower limit of 4 mM at pH 5, as well as bromide and iodide sensing at neutral conditions, with limits of 126 μM and 45 μM at pH 7 respectively, all of which may prove useful in monitoring industrial waste (ESI Table S4†). Furthermore, CA–methyl-serine may provide a chloride-insensitive reference signal incorporated into thin-film sensors for continuous monitoring applications, as depicted in ESI Fig. S22,† since dynamic quenching is a photophysical process that does not degrade the dye.
Conclusions
We have presented a low-cost synthesis platform to establish a repertoire of citrate-based chloride sensors with diverse sensing capabilities to suit the mean and range of various clinical tests. Our computational studies may provide a framework for further optimization of citrate-based sensors with improved chloride sensitivities, conceivably by the placement of electron donating groups near regions of high excited state AO contributions. Particularly, extending chloride sensing to physiological pH may lead to critical developments in organic biosensing, enabling intracellular studies of abnormal transmembrane chloride regulation with minimal cytotoxicity and photobleaching.
We have also performed analytical and clinical validation of CA–cysteine as a highly reliable and convenient sweat test method for the diagnosis of CF, and have demonstrated proof-of-concept for an automated and selective multi-halide analysis system using a single fluorophore. Such advances in performance and cost-reduction meet the clinical needs of a reliable point-of-care diagnostic system for CF. Future clinical sweat studies incorporating selective chloride analysis may improve the diagnostic accuracy of sweat tests by removing bromide interference. Moreover, bromide supplementation studies have shown altered bromide homeostasis presented in the sweat of both CF homozygotes and heterozygotes.39 Thus our novel strategy for multi-halide sensing, combined with discriminant function analysis, may enable the simultaneous measurement of sweat bromide as an additional diagnostic parameter to identify CF carriers and resolve borderline chloride diagnoses, remarkably enriching clinical information obtained from patient sweat. Finally, the intrinsic fluorescence and chloride sensing capability of our citrate-based biodegradable polymers enables great processibility into thin film biosensors to enable a wide range of medical, industrial, and environmental applications.
Experimental
Synthesis of citric acid-based fluorescent materials
Citric acid-based fluorescent dyes are synthesized via a one-pot reaction of citric acid and a primary amine compound dissolved in water or organic solvents. In the case of CA–cysteine synthesis, equimolar ratios of citric acid and l-cysteine were added to a round-bottom flask with just enough distilled water to dissolve both compounds. The reaction took place under 140 °C for 30 minutes, and was terminated by adding 10 mL of DI water in order to dissolve the products. Purification was performed through two cycles of crystallization in DI water and freeze-drying. Citrate-based biodegradable fluorescent polymers were synthesized according to our previous method.17 Briefly, citric acid, 1,8-octanediol, and one of the primary amines (respective molar ratio of 1 : 1 : 0.2) were reacted in a flask at 140 °C under nitrogen for 2 hours, followed by termination of reaction with 1,4-dioxane, precipitation in DI water, and lyophilization.
Fluorescence measurements
Absorbance spectra were recorded on Tecan Infinite M200 Pro UV-vis spectroscopy, and fluorescence spectra were recorded on Horiba FluoroMax-4 fluorospectroscopy at concentrations below 0.1 O.D. (∼10 μM for CA–cysteine) unless otherwise indicated. Quantum yields were measured on the Quantum φ Integrating Sphere accessory to the FluoroMax-4, while lifetime measurements were determined through the Time-Correlation Single Photon Counting (TCSPC) accessory to the FluroMax-4. TCSPC was utilized in conjunction with NanoLED pulsed diode light source with peak excitation wavelength at 352 nm and at 10 000 counts peak preset. Emission was collected at the fluorescence λ max, unless otherwise indicated. For all pH-dependence experiments, sample pH was adjusted with sulfuric acid or sodium hydroxide. For all chloride-dependent quenching experiments, sodium chloride was used unless indicated as magnesium chloride (hexahydrate). Lithium bromide and potassium iodide was used for bromide and iodide-dependent quenching experiments. For halide sensitivities measured at pH 2.6, 3.2, 4.0, and 5.0, a citrate-phosphate buffer made from citric acid and NaH2PO4 at a final concentration of 50 mM was used, while for sensitivities measured at pH 6.0 and 7.0, a phosphate buffer made from Na2HPO4 and NaH2PO4 at a concentration of 50 mM was used. Solution volume and dye concentrations were fixed throughout all experiments.
Lifetime analysis
Fluorescence lifetime decays were fitted with an exponential series according to eqn (3) below:
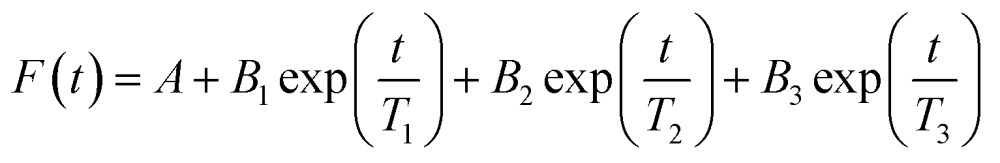 |
3 |
where F(t) is the lifetime decay function with respect to time t, T i is the lifetime value of the emitting species, A is the background offset, and B i is the pre-exponential function of the emitting species. If the lifetime decay is dominated by one emitting species, the equation can be simplified to include only the first two terms. B i measures the relative amplitude of the specified fluorophore, where the fractional contribution of the fluorophore to the lifetime decay, f, is derived from eqn (4):
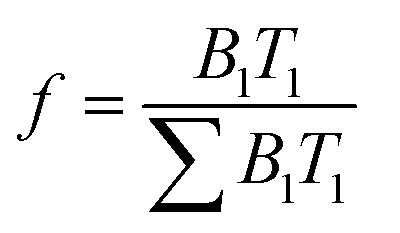 |
4 |
The method of least squares was used to quantify χ 2 based on the decay data and the fitting function, where χ 2 values under 1.2 indicates a good fit, and values above 1.2 indicate need for additional terms in eqn (3).
Molecular modeling
To simulate the electronic effects of CA–cysteine deprotonation, molecular modeling studies were performed with Hyperchem 8.0. The ground state geometry of various protolytic states of CA–cysteine were optimized based on the ab initio method at the 3-21G basis set level of theory, which calculates all molecular integrals under the assumption that atomic orbitals are linear combinations of Gaussian orbitals. The optimization was performed in vacuo using the conjugate gradient Polak–Ribiere algorithm. The theoretical absorption spectra were computed using ZINDO/S, a semi-empirical configuration interaction method commonly used to calculate transition energies.
Validation with sweat controls
To validate the fluorescence-based sweat test method with CA–cysteine, we analyzed three replicates of sweat controls prepared by a clinical sweat test laboratory at the Hershey Medical Center (HMC) in PA. Sweat controls are made from Quantimetrix to simulate human sweat at three levels, where level 1 represents sweat of healthy participants, level 2 represents sweat of participants with cystic fibrosis, and level 3 represent sweat of participants with total lack of reabsorption (chloride levels on par with that of serum), and results are compared with those obtained with a chloridometer by Quantimetrix, reported to be 18–28 mM for level 1, 40.5–61.5 mM for level 2, and 85–127 mM for level 3. The College of American Pathologists (CAP) Laboratory Accreditation Program guidelines require evaluation criteria of ±10.0 mM L–1 or 15% (whichever is greater) for validation.33 Three replicates of each sweat control level and a blank control were prepared by HMC by adding 100 μL of sample to a half piece of gauze in a small bottle, and submerging the gauze with 600 μL of DI water. Diluting the sample with less water gave better accuracy, as only 200 μL of sample is necessary to measure fluorescence with a small-volume cuvette. To measure fluorescence, samples were diluted in concentrated solutions of sulfuric acid to obtain a final molarity of 0.16 M, while final dye concentration was kept consistent, at an optical density below 0.1. The total dilution of sweat control samples in DI water, dye, and acid was 9.1×. Standardization was performed with sodium chloride under the same dye and acid conditions.
Clinical evaluation
Sweat from 8 healthy and 5 CF individuals were collected from each arm at a sweat clinic, where sweat from the right arm was analyzed by mercuric nitrate titration by technicians in clinic and sweat from the left arm was analyzed by our sensing strategy based on CA–cysteine described above. For statistical analysis, the intraclass correlation coefficient (ICC) computed by MedCalc™ and the Bland & Altman (B & A) plot were used to gauge the reliability of measurements. The ICC and B & A plot are typical reliability measures to assess agreement and correlation between two clinical measurement methods. The Bland & Altman plot is based on analysis of differences between two different measurement methods, investigate existence of systematic differences and outliers, where the mean difference is the estimated bias, and 1.96 SD computes the 95% limit of agreement for the comparison of methods.
Conflict of interest
Dr Yang and The Pennsylvania State University have a financial interest in Aleo BME, Inc. These interests have been reviewed by the University's Institutional and Individual Conflict of Interest Committees and are currently being managed by the University.
Acknowledgments
This work was supported in part by National Institutes of Health awards (EB012575, CA182670, HL118498) and National Science Foundation (NSF) award (DMR1313553).
Footnotes
References
- Golubnitschaja O. Croat. Med. J. 2009;50:596. doi: 10.3325/cmj.2009.50.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S., Kimura T., Suenaga K., Wada S., Tsuda K., Kasama S., Takaoka T., Kajiyama K., Takeda M., Yoshikawa H. J. Neurol. Sci. 2009;285:146. doi: 10.1016/j.jns.2009.06.026. [DOI] [PubMed] [Google Scholar]
- Farrell P. M., Kosorok M. R., Rock M. J., Laxova A., Zeng L., Lai H.-C., Hoffman G., Laessig R. H., Splaingard M. L. Pediatrics. 2001;107:1. doi: 10.1542/peds.107.1.1. [DOI] [PubMed] [Google Scholar]
- LeGrys V. A. Arch. Pathol. Lab. Med. 2001;125:1420. doi: 10.5858/2001-125-1420-AOSTPF. [DOI] [PubMed] [Google Scholar]
- Lezana J. L., Vargas M. H., Karam-Bechara J., Aldana R. S., Furuya E. J. Cystic Fibrosis. 2003;2:1. doi: 10.1016/S1569-1993(02)00146-7. [DOI] [PubMed] [Google Scholar]
- Shwachman H., Mohmoodian A. Clin. Chem. 1979;25:158. [PubMed] [Google Scholar]
- LeGrys V. A., Sweat Testing: Sample Collection and Quantitative Analysis, Approved Guideline, National Committee for Clinical Laboratory Standards, 2nd edn, 2000, vol. 20. [Google Scholar]
- Yager P., Domingo G. J., Gerdes J. Annu. Rev. Biomed. Eng. 2008;10:107. doi: 10.1146/annurev.bioeng.10.061807.160524. [DOI] [PubMed] [Google Scholar]
- McClatchey K. D., Clinical laboratory medicine, Lippincott Williams & Wilkins, 2002. [Google Scholar]
- Wise D. L.. Bioinstrumentation and biosensors, CRC Press, 1991. [Google Scholar]
- Lynch A., Diamond D., Leader M. Analyst. 2000;125:2264. doi: 10.1039/b006379g. [DOI] [PubMed] [Google Scholar]
- Bray P., Clark G., Moody G., Thomas J. Clin. Chim. Acta. 1977;80:333. doi: 10.1016/0009-8981(77)90041-9. [DOI] [PubMed] [Google Scholar]
- Blume R. S., MacLowry J. D., Wolff S. M. N. Engl. J. Med. 1968;279:593. doi: 10.1056/NEJM196809122791107. [DOI] [PubMed] [Google Scholar]
- Wenk R., Lustagarten J., Pappas N., Levy R., Jackson R. Am. J. Clin. Pathol. 1976;65:49. doi: 10.1093/ajcp/65.1.49. [DOI] [PubMed] [Google Scholar]
- Geddes C. D. Meas. Sci. Technol. 2001;12:R53. [Google Scholar]
- Wang J., Wu X., Chon C., Gonska T., Li D. Meas. Sci. Technol. 2012;23:025701. [Google Scholar]
- Yang J., Zhang Y., Gautam S., Liu L., Dey J., Chen W., Mason R. P., Serrano C. A., Schug K. A., Tang L. Proc. Natl. Acad. Sci. U. S. A. 2009;106:10086. doi: 10.1073/pnas.0900004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyawali D., Zhou S., Tran R. T., Zhang Y., Liu C., Bai X., Yang J. Adv. Healthcare Mater. 2014;3:182. doi: 10.1002/adhm.201300145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Zhang Y., Liu L., Weng H., Mason R. P., Tang L., Nguyen K. T., Hsieh J. T., Yang J. Adv. Mater. 2014;26:4491. doi: 10.1002/adma.201306070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprzyk W., Bednarz S., Bogdał D. Chem. Commun. 2013;49:6445. doi: 10.1039/c3cc42661k. [DOI] [PubMed] [Google Scholar]
- Hu J., Guo J., Xie Z., Shan D., Gerhard E., Qian G., Yang J. Acta Biomater. 2016;29:307. doi: 10.1016/j.actbio.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz J. R.. Principles of fluorescence spectroscopy, Springer Science & Business Media, 2013. [Google Scholar]
- Yang J.; Kim J. and Xie Z.. US Patent Application PCT/US16/26180, 4 April 2016.
- Verkman A. S. Am. J. Physiol.: Cell Physiol. 1990;259(3):C375–C388. doi: 10.1152/ajpcell.1990.259.3.C375. [DOI] [PubMed] [Google Scholar]
- LeGrys V. A. Pediatr. Pulmonol. 2000;30:476. doi: 10.1002/1099-0496(200012)30:6<476::aid-ppul7>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Shizuka H. Acc. Chem. Res. 1985;18:141. [Google Scholar]
- Kim S., Seo J., Park S. Y. J. Photochem. Photobiol., A. 2007;191:19. [Google Scholar]
- Guzzo A. V., Nelson D. A. Photochem. Photobiol. 1992;55:665. [Google Scholar]
- Hofeldt R., Sahai R., Lin S. J. Chem. Phys. 1970;53:4512. [Google Scholar]
- Chatterjee K., Chakravorti S., Ganguly T., Banerjee S. Il Nuovo Cimento D. 1986;8:541. [Google Scholar]
- Birks J. B., Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970, vol. 704, p. 210s. [Google Scholar]
- Wolfbeis O. S., Urbano E. Anal. Chem. 1983;55:1904. [Google Scholar]
- Zhu C., Bright F. V., Hieftje G. M. Appl. Spectrosc. 1990;44:59. [Google Scholar]
- louden C. e. a. Analyst. 1939;64(756):186. [Google Scholar]
- Farkas L., Lewin M. Anal. Chem. 1947;19:665. [Google Scholar]
- Miller M. E., Kornhauser D. M. Am. J. Dis. Child. 1994;148:266. doi: 10.1001/archpedi.1994.02170030036007. [DOI] [PubMed] [Google Scholar]
- Belkas E., Souliotis A. Analyst. 1966;91:199. [Google Scholar]
- Olszowy H. A., Rossiter J., Hegarty J., Geoghegan P., Haswell-Elkins M. J. Anal. Toxicol. 1998;22:225. doi: 10.1093/jat/22.3.225. [DOI] [PubMed] [Google Scholar]
- Theile H., Gressmann H., Winiecki P. Hum. Genet. 1985;69:277. doi: 10.1007/BF00293041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.