

Abstract
The production of a stable cDNA copy of an unstable RNA molecule by reverse transcription is a widely used and essential technology for many important applications, such as the construction of gene libraries, production of DNA probes, and analysis of gene expression by reverse transcriptase PCR (RT-PCR). However, the synthesis of full-length cDNAs is frequently inefficient, because the RT commonly used often produces truncated cDNAs. Synthesizing cDNA at higher temperatures, on the other hand, can provide a number of improvements. These include increasing the length of cDNA product, greater accuracy, and greater specificity during reverse transcription. Thus, an RT that remains stable and active at hot temperatures may produce better-quality cDNAs and improve the yield of full-length cDNAs. Described here is the discovery of a gene, designated trt, from the genome of the thermophilic bacterium Bacillus (Geobacillus) stearothermophilus strain 10. The gene codes for an open reading frame (ORF) similar to the ORFs encoded by group II introns found in bacteria. The gene was cloned and overexpressed in Escherichia coli, and its protein product was partially purified. Like the host organism, the Trt protein is a heat-stable protein with RT activity and can reverse transcribe RNA at temperatures as high as 75°C.
Heat-stable DNA polymerases are an essential and widely used tool in molecular biology research. Such applications as cloning DNA via the PCR, determining the sequence of DNA, and generating random mutations in DNA all use these thermostable DNA polymerases. Of particular utility is the process of reverse transcriptase PCR (RT-PCR), where an RNA molecule is converted to a more stable cDNA copy by reverse transcription, followed by amplification of the cDNA via PCR (9). This is a powerful technique used to detect and analyze RNA molecules from many types of cells.
In addition to a thermostable DNA polymerase, RT-PCR also usually requires a dedicated enzyme like an RT, whose RNA-dependent, DNA polymerase activity allows RNA to be reverse transcribed into cDNA. cDNA synthesis is most often done using RTs derived from animal viruses at temperatures below about 50°C. However, there are a number of advantages to synthesizing cDNA at hot temperatures (above 50°C). For example, higher temperatures will melt the secondary structures that can form in the RNA template and thus increase the length of cDNA product synthesized by the RT (7, 10). In addition, higher temperatures also increase the specificity and accuracy of DNA synthesis during reverse transcription (8, 11, 14). Thus, an RT that remains stable and active at high temperatures is especially useful for cDNA synthesis in RT-PCRs, as well as other applications.
A search of the nearly completed genome sequence of the eubacterial, thermophile Bacillus stearothermophilus (now designated Geobacillus stearothermophilus) (19) indicated the presence of an open reading frame (ORF) that we have designated as trt for thermostable intron RT. This ORF potentially codes for a 420-amino-acid protein with strong similarity to the protein ORFs encoded by group II introns found in bacteria and yeast mitochondria (12, 15, 38). Group II introns are self-splicing RNAs that interrupt genes found in the genomes of mitochondria and chloroplasts and are also common in bacteria. The autocatalytic RNAs splice to produce a characteristic lariat structure similar to the excision of nuclear introns by the eukaryotic spliceosome (12, 15). Some group II introns are also retrotransposons that can “reverse splice” and insert back into a specific DNA site within an allele that lacks the corresponding intron in a process called homing (4). On rare occasions, group II introns can also insert into nonhomologous sites in DNA in a process called retrotransposition (3). The ability of some group II introns to transpose to new locations is mediated by a multifunctional protein encoded by the intron. This protein contains a maturase activity that helps the intron to splice, an RT activity that converts the spliced intron RNA into a cDNA copy and, in some cases, a DNA endonuclease which helps the intron insert into a new site in DNA (15, 38).
It is surmised that the trt ORF from G. stearothermophilus may code for a protein with RT activity that is also heat stable, since this ORF is present in the genome of a thermophilic bacterium and is closely similar to ORFs encoded by group II introns (32). Such properties could make this DNA polymerase a useful tool for synthesizing cDNA at hot temperatures in important applications like RT-PCR. As a first step to possibly developing the Trt protein into a useful tool, this paper describes the cloning, overexpression, and purification of Trt in Escherichia coli. In addition, this protein is shown to have RT activity and to retain its activity after exposure to temperatures as high as 75°C.
MATERIALS AND METHODS
Bacterial strains and plasmids.
G. stearothermophilus strain 10 was a kind gift from Bruce Roe, University of Oklahoma, and was used for cloning the trt gene (26). Cultures were grown in trypticase soy agar plates at 55°C. The plasmid pUC18 was used for routine subcloning of DNA fragments (36). The plasmid pET28a and the E. coli strain BL21(DE3) were from Novagen (Madison, Wis.) and were used for heterologous expression of the Trt protein in E. coli.
Cloning the trt gene.
The following primers were used to amplify the trt gene from the genome of G. stearothermophilus strain 10: Bst755, 5′-AGACAACATATGCGGCAAGACCTGAATCTCAT-3′ (the underlined sequence contains an NdeI restriction site for cloning into the pET28a expression vector); Bst1396, 5′-AATGGATCCGCTGGCGAACATCCTTCTC-3′ (the underlined sequence contains a BamHI restriction site); Bst2015, 5′-ATTACTGCAGAGCGGTCCAGTAGGTTTTG-3′ (the underlined sequence contains a PstI restriction site); Bst2198, 5′-ACTCAAGCTTGAGAAGGGCTTGACGTTCATG-3′ (the underlined sequence contains a HindIII restriction site for cloning into the pET28a expression vector). Amplification of the trt gene was done in two stages (see Fig. 2, below) using a single colony of G. stearothermophilus as a source of template. A single colony from an overnight plate culture was suspended in 10 μl of water. One microliter of this cell suspension was added to a 50-μl PCR mix containing 1.5 mM MgCl2, Taq polymerase buffer (Promega, Madison, Wis.), a 0.2 μM concentration of each deoxynucleoside triphosphate (dNTP), a 0.5 μM concentration of each primer, and 2 U of Taq polymerase. The reaction was amplified under the following conditions: one cycle at 95°C for 2 min and 30 cycles at 95°C for 1 min, 50°C for 2 min, and 72°C for 2 min. Amplified DNAs were purified by gel electrophoresis, digested with the appropriate restriction endonuclease, and ligated into either pUC18 or directly into the pET28a expression vector.
FIG. 2.

DNA sequence of the trt ORF from G. stearothermophilus. The DNA sequence shown is from plasmid p12B43, containing the trt intron cloned from G. stearothermophilus strain 10 and the incomplete genomic DNA sequence of G. stearothermophilus strain 10 (B. Roe, NSF-EPSCOR Program, University of Oklahoma). This DNA codes for a 420-amino-acid ORF containing the Trt protein (GenBank accession number AY672067; protein_id number AAT72329). The location of DNA sequences used to design the primers Bst755, Bst1396, Bst2015, and Bst2198 are designated with arrows. These primers were used to amplify the trt ORF for cloning.
DNA and protein sequence analyses.
PCR primers were designed based on the DNA sequence recovered from a BLAST search (2) of the unfinished genome sequence of G. stearothermophilus from the “BLAST with bacterial genomes” web page at the National Center for Biotechnology Information (www.ncbi.nlm.nih-gov/BLAST) (see also Fig. 1, below). The program Primer3 (available at the website http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) was used to aid in design of primers. The DNA sequences of plasmid pTrt#16 and other constructions were determined with the BigDye terminator cycle sequencing using an ABI 327 automated DNA sequencer at the sequencing service (Molecular Biology Core Facility) provided by the Quillen College of Medicine at East Tennessee State University. Multiple amino acid sequence alignments of the trt ORF with other known intron ORFs were done using Clustal W (30) (available at the BCM search launcher website [http://searchlauncher.bc.tmc.edu/]).
FIG. 1.

Comparison of Trt with group II intron-encoded ORFs by multiple amino acid sequence alignment. The ORF designated B.st. codes for the Trt protein and was cloned from the genome of G. stearothermophilus strain 10 (26). The amino acid sequence of Trt is compared with the amino acid sequence from three related proteins encoded by group II introns from bacteria. These sequences include an ORF from B. halodurans (B.ha.; accession no. AB031210) (18), an RT-maturase protein from Clostridium acetobutylicum (C.ac.; accession no. NC003030) (20), and a group II intron protein from P. alcaligenes (P.al.; accession no. U77945) (37). As observed in all RTs, the Trt sequence contains most of the highly conserved amino acids that fall into seven distinct domains (underlined sequences labeled I to VII). These seven conserved domains correspond to important structural regions shared by all RTs (35). In addition, the Trt sequence also contains most of the highly conserved amino acids contained in a region of the protein designated X. This domain is associated with the maturase function of group II intron-encoded proteins. The most conserved amino acids found in the X region of bacterial group II intron proteins (according to Zimmerly et al. [38]) are shown above the alignment in italics (boxed amino acids).
Protein analysis.
Soluble and insoluble protein fractions were compared by resuspending cells (from a 50-ml induced culture) in a 1/10 volume of buffer (50 mM Tris-HCl [pH 8.0], 2 mM EDTA) containing lysozyme (100 μg/ml) plus 1% Triton X-100. After incubation at 30°C for 15 min and sonication, the cell extract was centrifuged at 12,000 × g for 15 min. The supernatant was mixed with an equal volume of sodium dodecyl sulfate (SDS) sample buffer, and this served as the soluble protein fraction for protein gels. The pellet of cell debris was mixed with SDS sample buffer, and this served as the insoluble protein fraction for protein gels.
Heterologous expression and purification of Trt.
Overexpression of the Trt protein in E. coli was done using the T7 RNA polymerase system and the pET28a expression vector (Novagen, Madison, Wis.) (27). Since the upstream primer (Bst755) used to amplify the trt gene was engineered with an NdeI restriction site, this allowed the amplified DNA containing the trt ORF to be ligated into the NdeI (plus a HindIII site for the downstream primer) restriction site of the expression vector. This produced an in-frame fusion between the polyhistidine tag element in the pET28a vector and the trt ORF. The Trt fusion protein was overexpressed in E. coli strain BL21 lysogenic for λDE3 as follows. Cells of E. coli strain BL21(DE3) transformed with the plasmid pTrt#16 (containing the trt fusion construction) were induced by addition of isopropyl-β-d-thiogalactopyranoside (IPTG; 1 mM). After 3 h of induction, cells from a 100-ml culture were harvested and resuspended in binding buffer (1×) for nickel ion column purification (Novagen). A cell extract containing the Trt fusion protein was prepared by incubating the cell suspension in fresh lysozyme (1 mg/ml), followed by three cycles of quick-freeze-thaw (10 min at −80°C followed by 10 min at 37°C), followed by sonication. Centrifugation (15,000 × g) and filtration (0.45-μm-pore-size filter) produced a cleared, crude protein preparation. The cleared extract was then loaded onto a premade nickel ion column (His-bind column; Novagen), and purified fractions were collected as per the manufacturer's instructions (Novagen). Column fractions containing the purified Trt fusion protein were pooled, dialyzed into buffer A (50 mM Tris [pH 7.5], 1 mM EDTA, 1 mM dithiothreitol [DTT], and 10% glycerol) using either a Microcon 30 membrane concentrator (Amicon, Beverly, Mass.) or dialysis tubing, and then used to assay for RT activity.
Assay for RT activity.
A product-enhanced RT assay known as the PERT assay was used to detect RT activity and was essentially as described previously (21, 24, 33). The PCR amplification reaction mix was assembled first in the bottom of a 0.2-ml tube and contained MgCl2-free PCR buffer (1×; 50 mM KCl, 10 mM Tris-HCl [pH 9.0], 0.1% Triton X-100), 1 μM (each) BMV-PCR1 primer (5′-CGTGGTTGACACGCAGACCTCTTAC-3′) and BMV-PCR2 primer (5′-TCAACACTGTACGGCACCCGCATTC-3′), 0.8 mM (each) dNTP, and Taq polymerase (Promega). The RT reaction mix was then assembled on top after sealing the lower PCR mix with a layer of wax using an Ampliwax pellet (PCR-Gem 50; Applied Biosystems, Roche). The RT reaction mix contained RT buffer (50 mM Tris [pH 8.3], 75 mM KCl, and 10 mM DTT), 2.5 mM MgCl2, 0.17% NP-40, 10 U of RNasin (Promega), 0.8 mM (each) dNTP, 0.02 μM RT primer (5′-GGTCTCTTTTAGAGATTTACAGTG-3′), 100 ng of brome mosaic virus (BMV) RNA (Promega), and a source of RT. The source of RT added to the reaction mixture was either the purified Trt fusion protein described above or commercially prepared Moloney murine leukemia virus (MMLV) RT (2 U). The reaction tube was then placed in a thermocycler under the following conditions: 1 cycle at 37°C for 1 h (reverse transcription reaction); 1 cycle at 94°C for 1 min; 30 cycles at 94°C for 15 s, 56°C for 15 s, and 72°C for 15 s (amplification step); and finally 72°C for 5 min. Amplified DNA was detected by electrophoresis of the reaction mix on a 5% polyacrylamide gel followed by staining with ethidium bromide.
In addition to the PERT assay, a second more quantitative assay was used to measure the effect of increasing temperature on the RT activity of the Trt protein. This assay involves the incorporation of radiolabeled nucleotide during cDNA synthesis and was done essentially as described by Moran et al. (17). Briefly, 5 μl of the purified Trt protein (pooled column fractions; about 0.5 μg) (see Fig. 3B, lanes 9 to 11) was added to a 15-μl reaction mixture containing 1 μg of poly(rA)-oligo(dT)18 as template-primer, 10 μCi of [α-32P]dTTP (3,000 Ci/mmol; Amersham), 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, and 10 mM DTT. After incubating the reaction mixture at 37°C for 30 min, 10 μl of the reaction mixture was spotted onto a DE81 paper disk (Whatman) and allowed to air dry. The disks were then washed four times, 5 min per wash, in 2× SSC (0.3 M NaCl, 0.03 M sodium citrate) and then air dried. The amount of radioactivity retained on the disks was measured by Cerenkov counting in a scintillation counter.
FIG. 3.

Overexpression of the Trt fusion protein in E. coli. (A) E. coli cells (BL21) containing the plasmid pTrt#16 were induced with IPTG to overexpress the Trt fusion protein via the T7 promoter system. Total protein extracts prepared from induced and noninduced cells were analyzed by electrophoresis on an SDS-polyacrylamide protein gel (stained with Coomassie blue). A prominent protein band migrating at about 48 kDa (arrow) is apparent in induced cells (lane 2) but absent from noninduced cells (lane 1) and also absent from control cells containing just the plasmid vector alone (data not shown). In addition, much of the overexpressed protein band appears in the insoluble cell fraction (lane 4 [soluble cell fraction] versus lane 3 [insoluble cell fraction]). (B) Affinity column purification of the Trt fusion protein. The soluble fraction from a cell extract expressing the Trt fusion protein was loaded onto a nickel ion affinity column. A single-step elution of the polyhistidine-tagged fusion protein yielded a partially purified fraction containing the 48-kDa protein band (arrow). Column fractions 1 to 7 were analyzed by electrophoresis on an SDS-polyacrylamide protein gel and stained with Coomassie blue (lanes 5 to 11, respectively). The RT activity of each column fraction was stabilized by dialysis into buffer A. Lane 3 contains the soluble cell fraction that was loaded onto the column. Lane 4 contains the column flowthrough. Lanes 1 and 2 contain total cell extract from uninduced and induced cells, respectively. Lane S contains a protein standard with the size (in kilodaltons) of each known protein indicated on the left.
Heat treatment of Trt.
The column-purified Trt fusion protein (as described above) was added (15 μl) to a microcentrifuge tube and heated in a water bath at either 65 or 75°C for 15 min. After heat treatment, the tube was centrifuged and 7.5 μl of the cleared supernatant was added directly to the PERT assay reaction. MMLV RT diluted in 1× RT buffer (5 U) was treated in a similar fashion.
To run the PERT assay at different temperatures, the reverse transcription reaction mixture was incubated in a separate reaction tube preheated at either 37, 55, 75, or 85°C. After 1 h of incubation, RNase A was added to stop the reaction and the reaction mix was immediately added to the top of the PCR mix and processed as described above.
Nucleotide sequence accession number.
The DNA sequence of the trt ORF has been deposited in GenBank under accession number AY672067. The translated Trt protein sequence is deposited under the protein_id number AAT72329.
RESULTS
Discovery of the trt gene.
Retroelements are genetic elements that code for an RT and employ the process of reverse transcription in some stage of their replication or mobility. A large variety of these retroelements are found in nearly every type of eukaryotic organism. They include some RNA viruses, DNA viruses, transposons, introns, and even mitochondrial plasmids (6). Bacteria also contain genetic elements encoding an RT that fall into two basic types. One type is the group II introns, found in a variety of bacteria, that contain an ORF that codes for a multifunctional protein with RT activity (15, 38). The other type of retroelement is called a retron, which is responsible for the production of an unusual satellite DNA called msDNA (13). The amino acid sequences from 10 different retron RTs were compared with the amino acid sequences from five different bacterial group II intron ORFs by multiple sequence alignment (Clustal W alignment) (data not shown). This multiple sequence alignment was used to generate a consensus amino acid sequence for RTs found in bacteria. The bacterial consensus sequence was then used as the query sequence in a BLAST search of both the GenBank database as well as the bacterial genomes database that contains both completed as well as unfinished bacterial species. These searches revealed several, not previously described, ORFs with similarity to the consensus sequence. This included a 420-amino-acid ORF from the unfinished genome sequence of G. stearothermophilus. The amino acid sequence of this G. stearothermophilus ORF was further analyzed by comparison with other RTs and found to be strongly similar to group II intron ORFs from both bacteria and mitochondria (Fig. 1 and data not shown). The amino acid sequences of RTs are generally highly variable. However, multiple amino acid sequence alignments clearly demonstrate the presence of a few highly conserved amino acids shared among all RTs. These conserved amino acids fall into seven domains (designated I to VII) (Fig. 1) that correspond to conserved secondary structures within the folded RT protein (25, 35). A short distance beyond domain VII is an additional conserved region, designated domain X, that is found only in group II intron-encoded proteins. Domain X is associated with the maturase function found in group II intron ORFs (38). Although domain X is not well conserved among bacterial group II intron proteins, the G. stearothermophilus ORF appears to contain most of the conserved amino acids of domain X shared among bacterial group II introns (Fig. 1). Thus, the G. stearothermophilus ORF appears to have most of the highly conserved amino acids present in both the RT region and the maturase region (or domain X) of group II intron proteins (Fig. 1). For this reason the G. stearothermophilus ORF is clearly not a retron-type RT and was thus designated trt, for thermostable intron RT. Some proteins encoded by group II introns also contain a third, zinc finger domain that imparts an endonuclease activity on this multifunctional protein (38). However, based on sequence comparisons, this endonuclease domain appears to be absent from the trt ORF (data not shown).
Cloning the trt gene.
Several oligonucleotide primers were designed to amplify the trt ORF. However, only two pairs of primers worked successfully to specifically amplify the trt gene from the chromosome of G. stearothermophilus. The first primer pair, designated Bst755 and Bst2015, was used to specifically amplify most, but not all, of the trt ORF via a simple PCR protocol (Fig. 2). A second primer pair, designated Bst1396 and Bst2198, was used to amplify a region that includes the last 16 amino acids at the C terminus of the trt ORF (Fig. 2).
Cloning of the trt gene into a plasmid vector for expression in E. coli was done in two stages. First, a 1.26-kbp amplified DNA product produced by the first primer pair (Bst755 and Bst2015) was ligated into the expression plasmid pET28a (see Materials and Methods). This produced an in-frame fusion between the polyhistidine tag element found in the expression plasmid and the trt ORF. DNA sequence determination confirmed that the predicted fusion protein construction began with an ATG start codon (just downstream of the T7 promoter system) followed by six histidine codons, a thrombin cleavage site, and 15 additional amino acids from the beginning of the amplified DNA insert, before finally reaching the first amino acid of the trt ORF. To capture the remaining 16 amino acids at the C terminus of Trt, a naturally occurring EcoRI site (Fig. 2) was used to splice the 3′ end of the PCR product produced by the second primer pair (Bst1396 and Bst2198) to the first amplified DNA to yield an expression plasmid, pTrt#16, containing the entire predicted ORF.
Overexpression and purification of Trt.
The plasmid pTrt#16 was used to overexpress the Trt protein in E. coli by induction of the T7 promoter system with IPTG (see Materials and Methods). Only cell extracts from IPTG-induced cultures containing pTrt#16 showed a prominent protein band. The overexpressed protein migrated at about 48 kDa in size after electrophoresis in a polyacrylamide protein gel (Fig. 3A, lane 2). This is close to the size expected (52 kDa) for the predicted fusion construction of Trt in plasmid pTrt#16. In addition, Western blot analysis confirmed the presence of a polyhistidine tag in the 48-kDa protein band with a specific (antipolyhistidine) antibody probe (data not shown). Much of the overexpressed fusion protein appears to fractionate into the insoluble cell debris after high-speed centrifugation of the cell extract (Fig. 3A, lane 3). However, some of the 48-kDa fusion protein also appears in the soluble cell fraction (Fig. 3A, lane 4) and may produce detectable RT activity. For this reason, the 48-kDa fusion protein was purified from the soluble cell fraction by nickel ion affinity chromatography. A single-step elution of the polyhistidine-tagged fusion protein yielded a partially purified fraction containing predominately a 48-kDa protein band (Fig. 3B, lanes 6 to 11). Each eluted column fraction was dialyzed and concentrated into a new buffer (buffer A) to stabilize the purified protein (see Materials and Methods). The polyhistidine tag at the N terminus of the purified Trt protein was not removed, because this short extension of the protein was not expected to affect the RT activity of the fusion protein. This is based on the expression of mammalian viral RTs and other recombinant eukaryotic RTs in E. coli using similar technology (34).
Assay for RT activity.
A highly sensitive assay known as the PERT assay was used to detect RT activity (21, 24, 33). The assay requires the reverse transcription of a BMV RNA template to produce a cDNA; a small region of this cDNA is then further amplified by PCR. The presence of a 168-bp PCR product in a DNA gel, following electrophoresis, indicates the presence of RT activity. The assay was run on both crude cell extracts as well as purified column fractions. Essentially, no RT activity was detected in any of the crude cell extracts tested. In addition, no RT activity was detected from the purified fractions eluted from the nickel ion affinity column used to purify the fusion protein (Fig. 4A, lane 1, and data not shown). However, when the eluted column fractions were dialyzed and concentrated into buffer A, RT activity was detected in some of the column fractions (Fig. 4B, lanes 1 to 3). The most likely explanation for this is the presence of potent inhibitors of RT activity that must be removed from both the crude cell extract and the relatively harsh buffer conditions used to elute the fusion protein from the affinity column (29). For example, when commercially prepared MMLV RT was added to the PERT assay mixture, as expected a 168-bp DNA was produced, indicating RT activity (Fig. 4A, lane 4). However, if MMLV RT was mixed with column fraction 7, no RT activity was detected (data not shown).
FIG. 4.
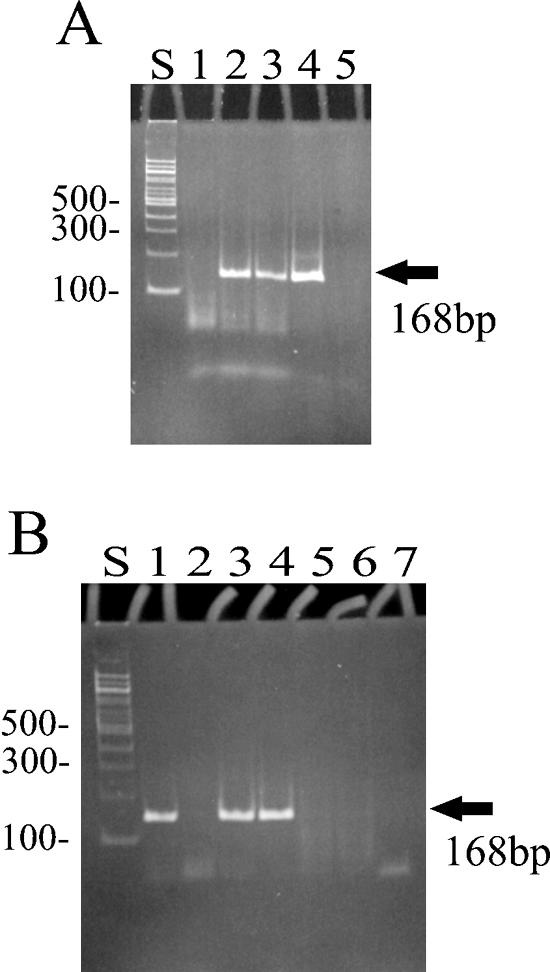
Detection of RT activity in a purified preparation of the Trt fusion protein. Purified preparations of the Trt protein were added to the PERT assay mixture as a source of RT. Various control reactions were also run in the PERT assay. The production of a 168-bp amplified DNA indicates RT activity. All reactions were run at 37°C. (A) Lane S, 100-bp molecular weight marker; lane 1, affinity-purified Trt protein, with column fraction 2 added as a source of RT; lane 2, column fraction 2 after dialysis in buffer A; lane 3, the same as lane 2 but supplemented with MMLV RT; lane 4, MMLV RT in RT buffer (serving as a positive control); lane 5, MMLV RT but with one of the PCR primers absent from the reaction mix (a negative control). (B) Lane S, 100-bp molecular weight markers; lanes 1 to 3, column fractions 2 to 4, respectively, after dialysis in buffer A; lane 4, MMLV RT in RT buffer (positive control); lane 5, MMLV RT plus RNase A added to the reaction mix; lane 6, water added as an exogenous source of RT; lane 7, column fraction 2 (dialyzed in buffer A) plus RNase A. The lack of a positive reaction in lane 2 is thought to be due to the presence of inhibitors copurifying with Trt in this column fraction.
Various control reactions were run to eliminate false-positive reactions (alternative mechanisms) that could also produce the 168-bp DNA product. For example, if one of the PCR primers was omitted from the PERT assay, no DNA product was produced (Fig. 4A, lane 4 [with both primers] versus lane 5 [with only one primer]). This indicated that the 168-bp DNA product is the result of specific amplification of the reverse-transcribed cDNA and not by some other mechanism. In another control reaction, RNase was added to the fraction to be tested for RT activity. For example, when RNase was mixed with dialyzed column fraction number 2, no DNA product was observed (Fig. 4B, lane 1 [no RNase added] versus lane 7 [with RNase added]). This indicates that the 168-bp DNA is amplified from a cDNA that is reversed transcribed from the BMV RNA template present in the assay mixture and is not due to the amplification of some contaminating DNA carried over from previous assay reactions. A final control reaction contained only water as the source of RT to be tested for RT activity (Fig. 4B, lane 6). As expected, no DNA product was produced. This indicated that only an exogenous source of RT added to the assay reaction mixture and not the Taq polymerase present in the assay mixture was responsible for cDNA formation (and thus the amplification of the 168-bp DNA).
Heat-stable RT activity.
The column-purified Trt fusion protein was subjected to either no heat treatment (Fig. 5A, lane 2), heated at 65°C for 15 min (Fig. 5A, lane 3), or heated at 75°C for 15 min (Fig. 5A, lane 4). After heat treatment, the purified fraction was tested for RT activity in the PERT assay. The same experiment was repeated with commercially prepared MMLV RT. The purified Trt protein appeared to retain its RT activity even after exposure to temperatures of 75°C (Fig. 5A, lane 4). In contrast, no RT activity was detected after heat treatment (at both 65 and 75°C) of the mesophilic MMLV RT (Fig. 5A, lanes 6 and 7).
FIG. 5.
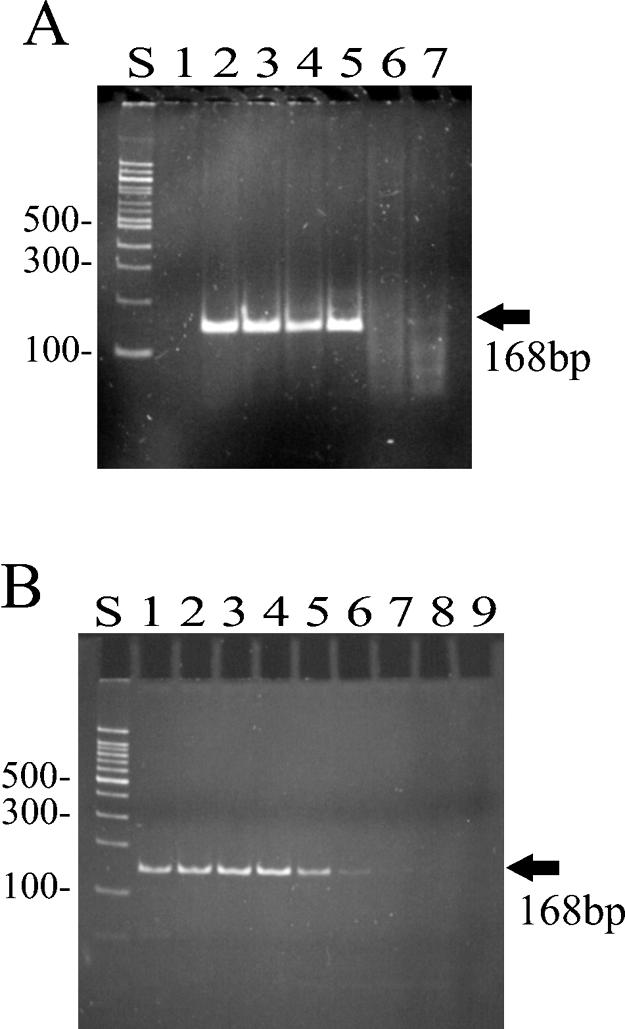
Detection of heat-stable RT activity. The PERT assay was used to detect RT activity of the purified Trt protein under various temperature conditions. (A) Column-purified fractions of Trt (dialyzed in buffer A) were pooled and preincubated at the indicated temperatures for 15 min and then added to the PERT assay mixture. Lane S, 100-bp molecular weight marker; lane 1, MMLV RT with one PCR primer missing (serving as a negative control); lane 2, purified Trt protein with no heat treatment; lane 3, purified Trt protein heated to 65°C; lane 4, purified Trt protein heated to 75°C; lane 5, commercial preparation of MMLV RT with no heat treatment; lane 6, MMLV RT heated to 65°C; lane 7, MMLV RT heated to 75°C. (B) The PERT assay itself was run at four different temperatures with either the purified Trt protein or MMLV RT added as a source of RT. Lane S, 100-bp molecular weight marker; lanes 1 to 4, PERT assay mixture containing the purified Trt protein incubated at 37, 55, 75, and 85°C, respectively; lanes 5 to 8, PERT assay mixture containing MMLV RT incubated at 37, 55, 75, and 85°C, respectively; lane 9, PERT assay reaction mixture with no source of RT added (serving as a negative control).
In a second experiment, the purified Trt protein was tested for RT activity by running the PERT assay at four different temperatures: 37, 55, 75, and 85°C (Fig. 5B). Again, the purified Trt protein was not affected in its ability to synthesize cDNA by reverse transcription, even at the highest temperature tested (Fig. 5B, lane 1 versus lane 4). By contrast, cDNA production appeared to be greatly reduced or absent at temperatures above 55°C for the mesophilic mammalian RT (Fig. 5B, lane 5 versus lane 7).
PERT is a highly sensitive, all-or-nothing assay to detect RT activity. However, this assay is not a quantitative assay for activity and thus provided limited information about the direct effect of temperature on the RT activity of the Trt protein. Therefore, a more quantitative assay, based on the incorporation of a radiolabeled nucleotide into cDNA, was used to measure the RT activity of the Trt protein at various temperatures.
Pooled column fractions (Fig. 3B, lanes 9 to 11) of the purified Trt protein were assayed for RT activity at various temperatures, and the level of activity was then compared with that of MMLV RT (Fig. 6). As expected, the RT activity of MMLV RT (a mesophilic enzyme) declined dramatically at temperatures above 45°C (Fig. 6). In contrast, the RT activity of the Trt protein remained uniformly high at all temperatures, up to and including 65°C. In fact, the RT activity of Trt appeared to increase slightly as the temperature increased from 37°C, reaching its highest activity at 65°C. Only at the highest temperature of 75°C did the activity of Trt decrease significantly (Fig. 6).
FIG. 6.

RT activity of Trt at different incubation temperatures. RT assay mixtures were incubated for 30 min at various temperatures with poly(rA)-oligo(dT)18 as the template-primer. RT activity was measured as the amount of incorporation of [α-32P]dTTP into high-molecular-weight cDNA retained on DEAE paper and is expressed as radioactive counts per minute. Values plotted on the graph are means of the RT assays run in triplicate for each temperature. White bars are for assay mixtures containing MMLV RT, and black bars are for assay mixtures containing the purified Trt protein as a source of RT.
DISCUSSION
The discovery of the trt gene reported here is, to the best of our knowledge, the first description of an RT from a thermophilic bacterium, and it is most likely encoded by a group II intron element. Indeed, the DNA sequence just downstream of the stop codon of the trt ORF was analyzed for conserved intron structures. Group II intron RNAs are self-splicing, autocatalytic RNAs that fold into a complex series of secondary stem-loop structures designated domains I through VI (16, 22). Similarity in the nucleotide sequence between different intron RNAs is usually poor; however, the secondary structural domain V, which plays an important role in splicing, is generally the most conserved in its primary nucleotide sequence (22, 31).
Sequence information just downstream of the trt ORF was analyzed to determine if the conserved domain V could form in a folded intron RNA structure. A postulated folded structure for part of the Trt intron RNA is shown in Fig. 7 and shows the stem-loop structures IV, V, and VI. Because the amino acid sequence of the trt ORF is most similar to the intron ORF from Bacillus halodurans (28) (Fig. 1), this intron RNA, whose folded structure has been determined (31), was used as a guide to produce the folded structure for the Trt intron. Like the intron from B. halodurans, the trt RNA contains an atypical domain V with a shorter stem-loop structure (30 nucleotides rather than the typical 34). In addition, the conserved sequence at the base of the stem in domain V is CCGC rather than the more typical RAGC (Fig. 7). These unusual structural features appear to place the Trt intron in what is known as the bacterial class C introns, which also include group II introns from B. halodurans, Streptococcus pneumoniae, Pseudomonas alcaligenes, and Pseudomonas putida (15, 31). The class C introns are apparently the most atypical in their RNA structure among bacterial group II introns, and it remains to be determined if the Trt intron is functional in splicing and transposition.
FIG. 7.

The proposed secondary structure for part of the trt intron RNA is shown. The postulated RNA ribozyme for the trt intron forms a series of folded stem-loop structures. The stem-loop designated (domain) IV contains a large loop region of 1,424 nucleotides and contains the ORF encoding the Trt protein. Stem-loop V contains the conserved sequence CCGC (boxed sequence) shared by most bacterial group II introns. Stem-loop VI contains the so-called bulging A nucleotide (circled A), where intron splicing will initiate to form the branched linkage of the intron lariat product.
Based on Southern hybridization experiments with the trt gene serving as a probe, there appear to be many additional copies of the gene, or at least genes very similar to trt, in the genome of G. stearothermophilus strain 10 (data not shown). Bacterial introns are commonly found inserted into DNA transposon elements, and this could explain the multiple copy number. However, an analysis of group II intron insertion sites in bacterial genomes (5) indicates that class C-type introns, such as the intron from B. halodurans, appear to insert just after the stem-loop structure of rho-independent transcription terminators. Thus, because insertion is linked to a potential stem-loop structure, the insertion sites for these introns are quite variable and show little sequence conservation (5). It will be interesting to determine the insertion sites for the Trt intron and if similar insertion events have occurred in the G. stearothermophilus genome.
The trt gene was cloned and overexpressed in E. coli, and the protein product was partially purified and, like its host organism, shown to be a thermostable protein that can carry out reverse transcription at high temperatures. Overexpression of the Trt fusion protein in E. coli resulted in a significant amount of the protein being separated into insoluble material during purification. Because RTs from other group II introns are known to be more stable when in a complex with their intron RNA, this could explain why much of the Trt protein is insoluble (23, 39). The plasmid pTrt#16 used to overexpress the trt gene in E. coli does not contain the entire intron element. Thus, overexpression of cloned DNA containing the entire intron element could increase the stability and activity of the Trt protein. This is because overexpression of the Trt protein would occur in tandem with expression of its intron RNA and allow the protein to form a more stable ribonucleoprotein complex.
As demonstrated here, the Trt protein can carry out reverse transcription at very hot temperatures (cDNA synthesis was detectable at 85°C [Fig. 5]) and is a novel type of RT that may be exploitable for applications where synthesis of cDNA at high temperatures is preferable. Many enzymes from thermophilic bacteria show an increase in their enzymatic activity as temperatures increase from 37 to 55 or 65°C (32). For the Trt protein, only a slight increase in RT activity was observed between 37 and 65°C. At 75°C the RT activity of the Trt protein dramatically declined (Fig. 6).
Regardless of whether the Trt protein proves to be of practical use, the discovery of this novel gene presents some interesting questions for further investigation. For example, what is the basis for the enhanced thermostability of this enzyme? Can determination of the structural features that provide heat stability to this protein guide the engineering of changes in commonly used mesophilic RTs to make them more heat stable? Is the trt ORF part of a functional group II intron? Can the postulated intron RNA self-splice at high temperatures? That is, can this intron RNA form a thermostable ribozyme that can cleave and splice intron RNA at high temperatures? Recently, a bacterial group II intron from Azotobacter (a mesophile) was described to have unusually high thermostability and required heat (more than 50°C) to activate splicing (1). Some understanding of these questions should provide important insights into this novel intron.
Acknowledgments
We gratefully acknowledge the close consultation and expert advice of Masayori Inouye on this project.
This work was generously supported by a grant from Takara Bio Inc. (to B.C.L.).
REFERENCES
- 1.Adamidi, C., O. Fedorova, and A. M. Pyle. 2003. A group II intron inserted into a bacterial heat-shock operon shows autocatalytic activity and unusual thermostability. Biochemistry 42:3409-3418. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic logical alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 3.Cousineau, B., S. Lawrence, D. Smith, and M. Belfort. 2000. Retrotransposition of a bacterial group II intron. Nature 404:1018-1021. [DOI] [PubMed] [Google Scholar]
- 4.Cousineau, B., D. Smith, S. Lawrence-Cavanagh, J. E. Mueller, J. Yang, D. Mills, D. A. Nanias, G. M. Dunny, A. M. Lambowitz, and M. Belfort. 1998. Retrohoming of a bacterial group II intron: mobility via complete reverse splicing, independent of homologous DNA recombination. Cell 94:451-462. [DOI] [PubMed] [Google Scholar]
- 5.Dai, L., and S. Zimmerly. 2002. Compilation and analysis of group II intron insertions in bacterial genomes: evidence for retroelement behavior. Nucleic Acids Res. 30:1091-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eickbush, T. H. 1994. Origin and evolutionary relationships of retroelements, p. 127-157. In S. S. Morse (ed.), The evolutionary biology of viruses. Raven Press, New York, N.Y.
- 7.Fu, G. K., and L. L. Stuve. 2003. Improved method for the construction of full-length enriched cDNA libraries. BioTechniques 34:954-957. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs, B., K. Zhang, M. G. Rock, M. E. Bolander, and G. Sarkar. 1999. High temperature cDNA synthesis by AMV reverse transcriptase improves the specificity of PCR. Mol. Biotechnol. 12:237-240. [DOI] [PubMed] [Google Scholar]
- 9.Hamilton, S. C., J. W. Farchaus, and M. C. Davis. 2001. DNA polymerases as engines for biotechnology. BioTechniques 31:370-383. [DOI] [PubMed] [Google Scholar]
- 10.Hawkins, P. R., P. Jin, and G. K. Fu. 2003. Full-length cDNA synthesis for long-distance RT-PCR of large mRNA transcripts. BioTechniques 34:768-773. [DOI] [PubMed] [Google Scholar]
- 11.Kim, B., J. C. Ayran, S. G. Sangar, E. T. Adman, S. M. Fuller, N. H. Tran, and J. Harrigan. 1999. New human immunodeficiency virus, type 1 reverse transcriptase (HIV-1) mutants with increased fidelity of DNA synthesis. J. Biol. Chem. 274:27666-27673. [DOI] [PubMed] [Google Scholar]
- 12.Lambowitz, A. M., and M. Belfort. 1993. Introns as mobile genetic elements. Annu. Rev. Biochem. 62:587-622. [DOI] [PubMed] [Google Scholar]
- 13.Lampson, B. C., M. Inouye, and S. Inouye. 2001. The msDNAs of bacteria. Prog. Nucleic Acid Res. Mol. Biol. 67:65-91. [DOI] [PubMed] [Google Scholar]
- 14.Malboeuf, C. M., S. J. Isaacs, N. H. Tran, and B. Kim. 2001. Thermal effects on reverse transcription: improvement of accuracy and processivity in cDNA synthesis. BioTechniques 30:1074-1084. [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Abarca, F., and N. Toro. 2000. Group II introns in the bacterial world. Mol. Microbiol. 38:917-926. [DOI] [PubMed] [Google Scholar]
- 16.Michel, F., K. Umesono, and H. Ozeki. 1989. Comparative and functional anatomy of group II catalytic introns—a review. Gene 82:5-30. [DOI] [PubMed] [Google Scholar]
- 17.Moran, J. V., S. Zimmerly, R. Eskes, J. C. Kennell, A. M. Lambowitz, R. A. Butwo, and P. S. Perlman. 1995. Mobile group II introns of yeast mitrochondrial DNA are novel site-specific retroelements. Mol. Cell. Biol. 15:2828-2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakasone, K., N. Masui, Y. Takaki, R. Sasaki, G. Maeno, T. Sakiyama, C. Hirama, F. Fuji, and H. Takami. 2000. Characterization and comparative study of the rrn operons of alkaliphilic Bacillus halodurans C-125. Extremophiles 4:209-214. [DOI] [PubMed] [Google Scholar]
- 19.Nazina, T. N., T. P. Tourova, A. B. Poltaraus, E. V. Novikova, A. A. Grigoryan, A. E. Ivanova, A. M. Lysenko, V. V. Petrunyaka, G. A. Osipov, S. S. Belyaev, and M. V. Ivanov. 2001. Taxonomic study of aerobic thermophilic bacilli and descriptions of Geobacillus subterraneus gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from petroleum reservoirs and transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermoglucosidasius and Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, G. thermocatenulatus, G. thermoleovorans, G. kaustophilus, G. thermoglucosidasius and G. thermodenitrificans. Int. J. Syst. Evol. Microbiol. 51:433-466. [DOI] [PubMed] [Google Scholar]
- 20.Nolling, J., G. Breton, M. V. Omelchenko, K. S. Markarva, Q. Zeng, R. Gibson, H. M. Lee, J. Dubois, D. Qiu, J. Hitti, Y. I. Wolf, R. L. Tatusova, F. Sabathe, L. Doucette-Stamm, P. Soncaille, M. J. Daly, G. N. Bennett, E. V. Koonin, and D. R. Smith. 2001. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J. Bacteriol. 183:4823-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pyra, H., J. Boni, and J. Schupbach. 1994. Ultrasensitive retrovirus detection by a reverse transcriptase assay based on product enhancement. Proc. Natl. Acad. Sci. USA 91:1544-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qin, P. Z., and A. M. Pyle. 1998. The architectural organization and mechanistic function of group II intron structural elements. Curr. Opin. Struct. Biol. 8:301-308. [DOI] [PubMed] [Google Scholar]
- 23.Saldanha, R., B. Chen, H. Wank, M. Matsuura, J. Edwards, and A. M. Lambowitz. 1999. RNA and protein catalysis in group II intron splicing and mobility reactions using purified components. Biochemistry 38:9069-9083. [DOI] [PubMed] [Google Scholar]
- 24.Silver, J., T. Naudru, K. Fujita, and R. Repaske. 1993. An RT-PCR assay for the enzyme activity of reverse transcriptase capable of detecting single virions. Nucleic Acids Res. 21:3593-3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steitz, T. A. 1999. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 274:17395-17398. [DOI] [PubMed] [Google Scholar]
- 26.Stenesh, J., and B. A. Roe. 1972. DNA polymerase from mesophilic and thermophilic bacteria I—purification and properties of the enzyme. Biochem. Biophys. Acta 272:156-166. [Google Scholar]
- 27.Studier, F. W., A. H. Rosenberg, J. J. Dunn, and J. W. Dubendorff. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185:60-89. [DOI] [PubMed] [Google Scholar]
- 28.Takami, H., C.-G. Han, Y. Takaki, and E. Ohtsubo. 2001. Identification and distribution of new insertion sequences in the genome of alkaliphilic Bacillus halodurans C-125. J. Bacteriol. 183:4345-4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taube, R., S. Loya, O. Avidan, M. Perach, and A. Hizi. 1998. Reverse transcriptase of mouse mammary tumor virus: expression in bacteria, purification, and biochemical characterization. Biochem. J. 329:579-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties, and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toor, N., G. Hausner, and S. Zimmerly. 2001. Coevolution of group II intron RNA structures with their intron-encoded reverse transcriptases. RNA 7:1142-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vielle, C., and G. J. Zeikus. 2001. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65:1-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voisset, C., R. R. Tonjes, P. Breyton, B. Mandrand, and G. Paranhos-Baccala. 2001. Specific detection of RT activity in culture supernatants of retrovirus-producing cells, using synthetic DNA as competitor in polymerase enhanced reverse transcriptase assay. J. Virol. Methods 94:187-193. [DOI] [PubMed] [Google Scholar]
- 34.Wilhelm, M., M. Boutabout, and F.-X. Wilhelm. 2000. Expression of an active form of recombinant Ty1 reverse transcriptase in Escherichia coli: a fusion protein containing the C-terminal region of the Ty1 integrase linked to the reverse transcriptase-RNase H domain exhibits polymerase and RNase H activities. Biochem. J. 38:337-342. [PMC free article] [PubMed] [Google Scholar]
- 35.Xiong, Y., and T. H. Eickbush. 1990. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 9:3353-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequence of M13amp18 and pUC19 vectors. Gene 33:103-119. [DOI] [PubMed] [Google Scholar]
- 37.Yeo, C. C., J. M. Tham, M. W.-C. Yap, and C. L. Poh. 1997. Group II intron from Pseudomonas alcaligenes NCIB 9867(P25X): entrapment in plasmid RP4 and sequence analysis. Microbiology 143:2833-2840. [DOI] [PubMed] [Google Scholar]
- 38.Zimmerly, S., G. Hausner, and X.-C. Wu. 2001. Phylogenetic relationships among group II intron ORFs. Nucleic Acids Res. 29:1238-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zimmerly, S., J. V. Moran, P. S. Perlman, and A. M. Lambowitz. 1999. Group II intron reverse transcriptase in yeast mitochondria: stabilization and regulation of reverse transcriptase activity by intron RNA. J. Mol. Biol. 289:473-490. [DOI] [PubMed] [Google Scholar]