

ABSTRACT
Autophagy and apoptosis are critical for controlling Toxoplasma gondii (T. gondii) infection. T. gondii infection during pregnancy can damage the fetus and cause birth defects; however, the molecular mechanisms of this process are poorly understood. This study aims to determine the activities of autophagy and apoptosis as well as their regulatory mechanisms during T. gondii infection by using human umbilical cord mesenchymal stem cells (hUC-MSCs) as a model of congenital diseases. LC3B, a hallmark protein of autophagy was incrementally upregulated with the infection duration, whereas p62 was downregulated in T. gondii-infected hUC-MSCs. Concurrent to this result, the invasion of T. gondii into hUC-MSCs increased in a time-dependent manner. The expression levels of Bcl−2 family proteins including Bcl−2, Bcl−xL, Bim, Bax, Bid and Bak were not altered; however, Mcl−1 levels in hUC-MSCs were dramatically decreased upon T. gondii infection. In addition, at 24 h post-infection, cleaved PARP and cleaved caspase-3 protein levels were elevated in hUC-MSCs. Importantly, Mcl−1 overexpression reduced the levels of autophagy- and apoptosis-related proteins in T. gondii-infected hUC-MSCs. Mcl−1 proteins were primarily expressed in the fraction containing mitochondria and strongly interacted with Beclin-1 under normal conditions; however, these interactions were remarkably attenuated by T. gondii infection. These results suggest that mitochondrial Mcl−1 is an essential signaling mediator regulating the activation of autophagy and apoptosis during T. gondii infection.
KEYWORDS: autophagy, apoptosis, cell death, Beclin-1, Mcl−1, Toxoplasma gondii, umbilical cord mesenchymal stem cell
Introduction
Toxoplasma gondii (T. gondii) is a ubiquitous and obligate intracellular protozoan parasite that can infect all nucleated cells from a wide range of host species. Although T. gondii infection of immunocompetent humans is usually asymptomatic, this pathogen can cause life-threatening diseases in fetuses, the newborns of infected mothers, and immunocompromised patients with dormant parasites that become activated.1,2 T. gondii has a variety of transmission methods, including foodborne transmission, animal-to-human (zoonotic) transmission, mother-to-child (congenital) transmission, blood transfusion and organ transplantation. Infection in a mother during the early stages of pregnancy may result in the death of the fetus and abortion, whereas an infection later in the pregnancy can cause fetal damage, stillbirth or live birth of an infant with damage to diverse tissues and organs.3 However, there are a limited number of studies that have focused on the pathogenic mechanisms of this detrimental damage caused by T. gondii infection during pregnancy.4
Human mesenchymal stem cells (hMSCs) are self-renewing multipotent cells that are a promising source of cells for cell-based therapeutics and regenerative medicine.5,6 The proliferative potential of human umbilical cord tissue-derived mesenchymal stem cells (hUC-MSCs) is greater than that of bone marrow-derived mesenchymal stem cells.7 Previous studies have demonstrated that multipotent stem cells from umbilical cord tissue can be induced to differentiate into osteogenic, adipogenic and chondrogenic tissues.5,8 However, it has been reported that haematopoietic stem cell transplantation (HSCT) can lead to severe toxoplasmosis with a high mortality rate even if diagnosed shortly after HSCT.9 In addition, a recent study suggested the clinical significance associated with controlling toxoplasmosis in a patient who underwent allogeneic stem cell transplantation.10 These findings strongly suggest that stem cell transplantation using umbilical cord tissue-derived cells, bone marrow-derived cells or haematopoietic cells may increase the risk of T. gondii infection; thus, it is prudent to understand the effect of T. gondii on stem cells.
Autophagy and apoptosis both are important processes in the host defense system against invading pathogens. Autophagy plays an antimicrobial and antiparasitic role in the activation of host cells to defend against intracellular pathogens; however, these pathogens could take advantage of autophagy in the host cell to promote their proliferation.11 Apoptosis is essential for cell development and tissue homeostasis in eukaryotes and modulates pathogenesis in a variety of diseases.12 It has been reported that T. gondii induces host cell autophagy in both HeLa cells and primary fibroblasts and that host cell autophagy contributes to parasite growth.13 Furthermore, a recent study suggested that T. gondii induces apoptosis of neural stem cells via the endoplasmic reticulum (ER) stress pathway.14 However, the effects and molecular mechanisms of T. gondii in inducing autophagy or apoptosis in hUC-MSCs are unknown.
Stem cells play important roles during embryonic and fetal development.15 As fetal-derived stem cells, hUC-MSCs are considered to be alternative seed cells in several congenital disease models.8,16,17 T. gondii infection in pregnant women can lead to various fetal damages,18 however the detailed pathogenic mechanism during fetal development has not been fully elucidated. Because autophagy and apoptosis are critical for controlling toxoplasmosis, we checked the protein expression levels of autophagy- and apoptosis-related genes to determine whether T. gondii infection leads to induction of either autophagy or apoptosis in hUC-MSCs; furthermore, we also attempted to identify the intracellular signaling pathways in T. gondii-mediated activation of autophagy and/or apoptosis.
Results
Cell characterization and viability
To isolate mesenchymal stem cells, we used flow cytometry to identify various cell surface marker configurations that corresponded to stem cells and confirmed these configurations before the application of the cells used throughout the experiments. hUC-MSCs were negative for CD11b, CD19, CD34 and CD45 (< 5%), positive for CD73, CD90 and CD105 (> 95%) (Fig. 1A) and presented a morphological appearance characteristic of multipotent cells (data not shown). These results suggested that hUC-MSCs used in this study share common characteristics with mesenchymal stem cells. We next infected hUC-MSCs with T. gondii GFP-RH at an MOI of 5 for 4, 8, 12 and 24 h and analyzed cell viability by using the MTS assay. Compared to uninfected control cells, the cell viability was significantly reduced by 21% and 30% in cells infected with T. gondii for 24 and 48 h, respectively (Fig. 1B). We further monitored the microscopic morphology and observed that T. gondii-infected hUC-MSCs showed an elongated morphology compared with that of untreated control cells (Fig. 1C). These results suggest that T. gondii infection of hUC-MSCs resulted in significant cell death in a time-dependent manner.
Figure 1.

Characterization of hUC-MSCs and determination of cell viability after infection with T. gondii. (A) The expression levels of MSC-specific human CD markers were analyzed by flow cytometry. (B) Cell viability was measured by the MTS assays (means ± SD). * P < 0.05, ** P < 0.01 as compared with uninfected control cells. (C) The morphology of hUC-MSCs infected with T. gondii at the indicated time points. Scale bar = 100 μm.
T. gondii induced mTOR-independent autophagy in hUC-MSCs
To investigate the intracellular signaling mechanism that mediates T. gondii-induced cell death of hUC-MSCs, we examined the intracellular survival and proliferative rate of T. gondii at various time points. Tachyzoites of the GFP-RH strain (MOI = 5) were infected in hUC-MSCs for the indicated time periods, and the hUC-MSCs were stained with Texas Red-X phalloidin (red) and DAPI (blue) to identify F-actin and nuclei, respectively. Immunofluorescence microscopy revealed that the number of T. gondii-infected cells was significantly increased in a time-dependent manner (51.4% at 12 h and 62.6% at 24 h; Fig. 2A and B).
Figure 2.

T. gondii infection rates and the expression levels of autophagy-related proteins in hUC-MSCs. (A) hUC-MSCs were infected with GFP-expressing T. gondii at an MOI of 5 for the indicated time durations. Cells were fixed and stained with Texas Red-X phalloidin to label F-actin (red), and nuclei were stained with DAPI (blue). Scale bar = 100 μm. (B) The number of T. gondii-infected cells and the total number of cells were counted to calculate the infection rate. *P < 0.05, **P < 0.01 as compared with cells infected with T. gondii for 4 h. (C) Cell lysates were subjected to western blotting using autophagy-related antibodies (LC3B and p62) and a T. gondii-specific antibody (TP3). (D) The expression levels of p-mTOR, mTOR and its downstream effectors S6K1 (phosphorylation at Thr389) and 4E-BP1 (phosphorylation at Thr37/46) were detected by western blotting. All data shown are representative of 3 independent experiments with similar results.
A recent study indicated that T. gondii-induced activation of host cell autophagy is involved in the enhancement of the intracellular proliferation of T. gondii.11 To examine the roles of autophagy during T. gondii infection, hUC-MSCs were infected with T. gondii (MOI = 5) for various durations, and the protein expression of autophagy-related genes was evaluated. As shown in Figure 2C, infecting hUC-MSCs with the GFP-RH strain led to a progressive increase similar to that of the intracellular parasite content as measured by T. gondii TP3 levels. Consistent with the immunofluorescence results, TP3 protein levels were significantly increased at 24 h post-infection. Similarly, LC3B levels were also gradually increased from 4 to 24 h post-infection; however, the protein expression of p62, which is a hallmark of autophagy activation, was markedly decreased at 12 and 24 h post-infection.
Next, we asked whether T. gondii-mediated induction of autophagy correlates with downregulation of mammalian target of rapamycin (mTOR) activity. The downstream sequelae of mTOR activation include phosphorylation of S6 kinase-1 (S6K) at Thr389 and Ser371 and of 4E-BP1 at Thr37/46.13 As shown in Figure 2D, T. gondii infection of hUC-MSCs did not lead to any alterations in the phosphorylation state of host mTOR, S6K or 4E-BP1, which indicate that T. gondii-induced autophagy is mTOR-independent.
Mcl−1 mediated the T. gondii-induced autophagy in hUC-MSCs
Anti-apoptotic (Bcl−2, Bcl−xL and Mcl−1) and proapoptotic (Bim, Bax, Bid, Bak and Puma) Bcl−2 homologs have been suggested to regulate autophagy.19,20 Therefore, we examined whether any of the Bcl−2 homologs were necessary for T. gondii-induced autophagy. Western blotting analysis revealed that the levels of Bcl−2, Bcl−xL, Bim, Bax, Bid, and Bak were unaffected by T. gondii infection. Interestingly, Mcl−1 protein expression was significantly decreased in a time-dependent manner following T. gondii infection (Fig. 3A). These results indicate that downregulation of Mcl−1 may play an important role in T. gondii-induced autophagy.
Figure 3.
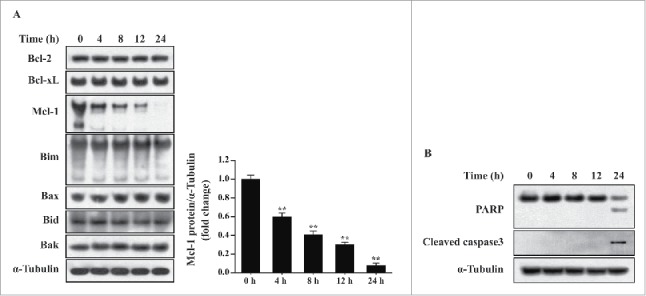
T. gondii-induced Mcl−1 downregulation and apoptosis in hUC-MSCs. hUC-MSCs were infected with T. gondii at an MOI of 5 for the indicated time durations. (A) Left, the protein levels of selected Bcl−2 family members were quantified by western blotting with α-Tubulin as a loading control. Representative blots of 3 independent experiments with similar results are shown. Right, fold changes of Mcl−1 levels in comparison with the results for the cells without T. gondii infection after normalization with the α-Tubulin level are shown (**P < 0.01). (B) The expression levels of PARP and cleaved caspase-3 in hUC-MSCs were determined by using western blotting. Three independent experiments were conducted showing similar results, and one representative blot is shown.
To directly address the consequences of Mcl−1 downregulation on the T. gondii-induced apoptosis of hUC-MSCs, we analyzed protein levels of nuclear poly (ADP-ribose) polymerase (PARP) and cleaved caspase-3 (p19) and found that the cleaved PARP and caspase-3 protein levels were elevated in hUC-MSCs 24 h post-infection (Fig. 3B). These results suggest that T. gondii-induced downregulation of Mcl−1 may be sufficient to induce apoptosis.
Overexpression of Mcl−1 inhibited T. gondii-induced autophagy and apoptosis in hUC-MSCs
To further investigate the role of Mcl−1, we constructed an adenovirus expressing recombinant Mcl−1. After adenoviral infection with hUC-MSCs, Mcl−1 expression was confirmed by qRT-PCR and western blot, which showed that the respective Mcl−1 mRNA and protein levels were significantly increased in hUC-MSCs transduced with adenovirus containing the Mcl−1 gene (Fig. 4A and B). Interestingly, overexpression of Mcl−1 in T. gondii-infected hUC-MSCs dramatically decreased LC3B and cleaved caspase-3 levels but increased the basal levels of p62 (Fig. 4C). In addition, we examined the ultrastructural morphology of Mcl−1-overexpressing hUC-MSCs infected with T. gondii. As shown in Fig. 5, compared with cells treated with control adenovirus (Fig. 5C), the number of autolysosomes (red arrows) was decreased in cells treated with adenovirus expressing Mcl−1 (Fig. 5D). Autophagosomes (red asterisks) were observed in cells treated with adenovirus expressing Mcl−1 (Fig. 5E) and control adenovirus (Fig. 5F). These findings together suggest that overexpression of Mcl−1 can inhibit T. gondii-induced autophagy in hUC-MSCs.
Figure 4.

Overexpression of Mcl−1 inhibited T. gondii-induced autophagy and apoptosis in hUC-MSCs. (A) The Mcl−1 mRNA levels in hUC-MSCs were determined by qRT-PCR after adenoviral infection. * P < 0.05, ** P < 0.01 as compared with Ad/DsRed control groups. (B) The protein levels of Mcl−1 after T. gondii infection were determined by western blotting. (C) hUC-MSCs overexpressing Mcl−1 were infected with T. gondii at an MOI of 5 for 24 h, and the protein levels of Mcl−1 as well as endogenous LC3B, p62 and cleaved caspase-3 protein levels were quantified by western blotting. α-Tubulin was used as a loading control. All data shown are representative of 3 independent experiments with similar results.
Figure 5.

Ultrastructural morphology of normal and T. gondii-infected hUC-MSCs overexpressing Mcl−1. The ultrastructural morphology of hUC-MSCs infected with adenovirus expressing (A) DsRed alone or (B) DsRed-Mcl−1. hUC-MSCs overexpressing (C) DsRed and (D) DsRed-Mcl−1 were infected with T. gondii at an MOI of 5 for 24 h, and autolysosomes (red arrows) were detected by using TEM. hUC-MSCs overexpressing (E) DsRed and (F) DsRed-Mcl−1 were infected with T. gondii at an MOI of 5 for 24 h, and autophagosomes (red asterisks) were detected by TEM.
Mcl−1 regulated T. gondii-induced autophagy of hUC-MSCs by interacting with Beclin-1 in mitochondria
To investigate the cellular localization of Mcl−1 protein, cells were fractionated into the heavy mitochondria (HM) pellet containing mitochondria and the light membrane (LM) pellet containing ER, and protein expression of Mcl−1 was detected by western blotting analysis. As shown in Figure 6A, Bcl−2, Bcl−xL and Beclin-1 are partially localized to both the mitochondria and ER. In contrast, Mcl−1 was primarily localized to the mitochondria, and T. gondii infection remarkably reduced the mitochondrial Mcl−1 protein levels. As Beclin-1, a key upstream regulator of autophagy, can be inhibited upon binding to either Bcl−2 or Bcl−xL,21 we used an immunoprecipitation assay to determine the possibility of a direct interaction between Mcl−1 and Beclin-1. The data showed that Beclin-1 was only co-precipitated with Mcl−1 in the HM fraction containing mitochondria, and T. gondii infection increased the dissociation of Beclin-1 from mitochondrial Mcl−1 (Fig. 6B). These findings suggest that the physical interaction between Mcl−1 and Beclin-1 in the mitochondria may be essential in modulating T. gondii-mediated activation of autophagy.
Figure 6.
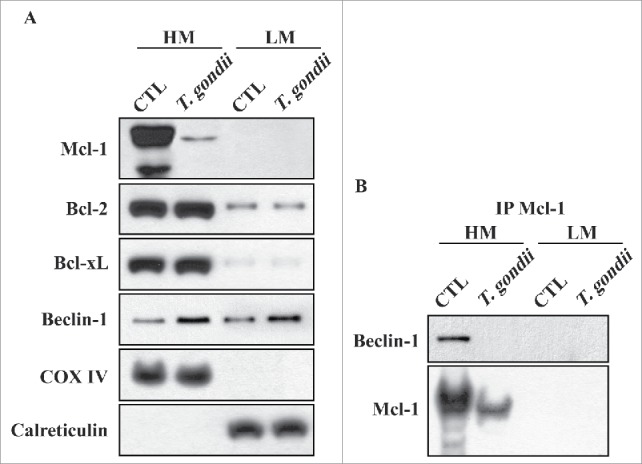
Mcl−1 interacted with Beclin-1 in the mitochondria. (A) hUC-MSCs were infected with T. gondii at an MOI of 5 for 24 h, and the cells were fractionated into the HM (containing mitochondria) and the LM (containing ER). Fractions were analyzed for the expression of Mcl−1, Bcl−2, Bcl−xL, Beclin-1, COX IV (a hallmark of mitochondria) and Calreticulin (a hallmark of ER). (B) Mcl−1 was immunoprecipitated from the HM and LM, and the expression of Mcl−1 and Beclin-1 were analyzed by western blotting. All data shown are representative of 3 independent experiments with similar results.
Discussion
In the present study, we investigated the precise roles of Mcl−1 in controlling T. gondii-induced activation of autophagy and apoptosis in hUC-MSCs. The protein expression of Mcl−1 and the interaction between Mcl−1 and Beclin-1 were dramatically decreased after T. gondii infection, yet overexpression of Mcl−1 decreased autophagy and apoptosis. To the best of our knowledge, this is the first report describing the crosstalk between autophagy and apoptosis in T. gondii-infected hUC-MSCs.
Autophagy is widely regarded to be critical for cell survival under starvation conditions andfor the turnover of dysfunctional organelles.22 We found that autophagy in hUC-MSCs was induced during the early stages (within 8 h) of T. gondii infection, and the level of autophagy was increased with incremental infection periods. Similar results have also shown that T. gondii induces host cell autophagy in both HeLa cells and primary fibroblasts and that host cell autophagy contributes to parasite growth.13 However, it is important to note that the viability of the hUC-MSCs was significantly reduced during the late stages of infection concurrent with the increased autophagy and T. gondii concentration. These data indicate that T. gondii reduced the viability of hUC-MSCs based on increments of autophagy; thus, hUC-MSCs were induced toward autophagic cell death (also known as Type II programmed cell death) after T. gondii infection. Concomitant with this, Ghosh et al.23 reported that autophagy is the primary mechanism of programmed cell death in human foreskin fibroblast cells and Vero cells infected with T. gondii.
T. gondii is able to either promote or inhibit the host cell's apoptotic process depending on the host cell type, infection stage and virulence.24 In previous reports, T. gondii was described as inhibiting host cell apoptosis through PI3 kinase/Akt-dependent Bad phosphorylation,2 whereas T. gondii appeared to activate some apoptotic signals in astrocytes shortly after infection.24 Furthermore, Bcl−2 homologs have recently been suggested to regulate autophagy.20 In this study, the viability of hUC-MSCs was significantly decreased at late infection stage; therefore, we measured the levels of cleaved PARP, cleaved caspase-3 and Bcl−2 family members to evaluate the apoptotic features in hUC-MSCs treated with T. gondii. We found that hUC-MSCs exhibited elevated levels of cleaved PARP and caspase-3 following T. gondii infection. Surprisingly, no obvious expression changes were detected in the Bcl−2 family members including Bcl−2, Bcl−xL, Bim, Bax, Bid and Bak during T. gondii infection, but Mcl−1 was downregulated in a time-dependent manner. Moreover, the T. gondii-induced autophagy and apoptosis in hUC-MSCs was repressed by Mcl−1 overexpression. These results indicate that Mcl−1 plays an important role in the crosstalk between apoptosis and autophagy in T. gondii-infected hUC-MSCs and are consistent with a previous study showing that Mcl−1 is one of the earliest proteins from the Bcl−2 family to respond to stress and is rapidly degraded following stress to allow apoptosis to proceed.25 These data also provide strong evidence supporting the hypothesis that autophagy and apoptosis may be triggered by common upstream signals that either link or polarize the cellular responses, which can occasionally result in combined autophagy and apoptosis.26
Recently, Beclin-1 has been described as a critical factor for the cross-regulation between apoptosis and autophagy.27 It has been shown that Beclin-1 possesses a Bcl−2 homology 3 domain (BH3-only), which is required for binding to the BH3 receptor domain on Bcl−2 and Bcl−xL.20 In this study, we attempted to detect an interaction between Mcl−1 and Beclin-1 by immunoprecipitation and found that Mcl−1 was specifically bound to Beclin-1 in the mitochondrial fraction of uninfected hUC-MSCs but not in T. gondii-infected hUC-MSCs. These results suggest that Mcl−1 strongly interacts with Beclin-1, and the T. gondii-induced autophagy and apoptosis in hUC-MSCs manifests by profoundly reducing Mcl−1 protein expression as well as the interaction between Mcl−1 and Beclin-1. Our data confirmed previous results stating that Mcl−1 downregulation during the autophagy process is an early event for the induction of apoptosis25 and that inhibition of Mcl−1 results in the disruption of the Beclin 1/Mcl−1 complex.28
Among the many signaling pathways in autophagy, its classical regulation is governed by the mTOR pathway, which negatively regulates this process.29 Based on this, we measured the phosphorylation status of mTOR and its downstream effectors S6K1 and 4E-BP1 in hUC-MSCs undergoing T. gondii-induced autophagy. The results showed that there were no alterations in the phosphorylation levels of mTOR, S6K or 4E-BP1, suggesting that T. gondii-induced autophagy in hUC-MSCs is mTOR-independent. There are many mTOR-independent pathways involved in regulating autophagy such as the cAMP/Epac/PKCε, Ca2+/calpain, inositol and JNK1/Beclin-1/PI3KC3 signaling pathways.29 Whether these signaling pathways are involved in the regulation of autophagy in T. gondii-infected hUC-MSCs requires further investigation.
Conclusively, autophagy and apoptosis are highly conserved and genetically regulated intracellular processes that are equally important in development and normal physiology and can be activated by various stimuli, including intracellular parasites.14,30 In the current study, T. gondii induced autophagy in hUC-MSCs during the early stages of infection and induced apoptosis at late stages; thus, both autophagy and apoptosis are involved in the T. gondii-induced death process of hUC-MSCs. Mcl−1 has a key role in determining cell fate by coordinately regulating apoptosis and autophagy based on its interaction with Beclin-1 in the mitochondria (Fig. 7). Although future studies are need to determine the roles of Mcl−1 and Beclin-1 as well as their related signaling networks in T. gondii-mediated cell death, the results presented in the current study are helpful to understand the mechanism of fetal damage by T. gondii in congenital toxoplasmosis.
Figure 7.
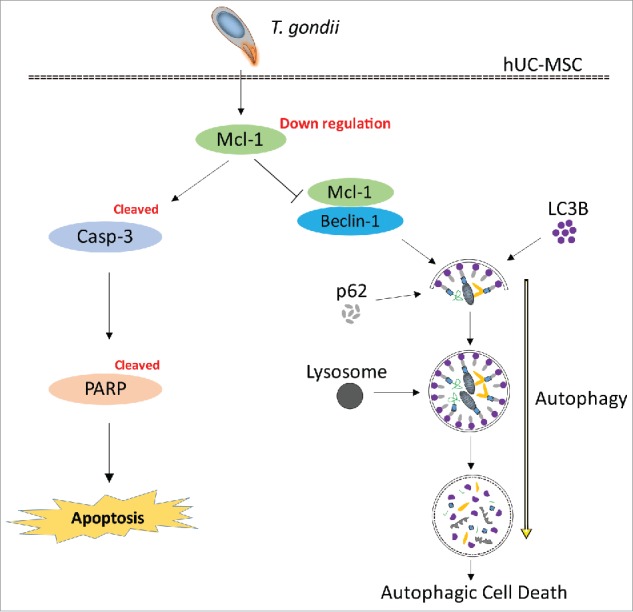
Suggested pathway of Mcl−1-controlled autophagy and apoptosis in hUC-MSCs infected with T. gondii. During the early infection stage (within 8 h), T. gondii reduces the protein levels of Mcl−1 in the mitochondria, which is followed by interruption of the Mcl−1/Beclin-1 complex. Consequently, there is accelerated formation of autolysosomes and autophagosomes. During the late stages of infection (up to 24 h), T. gondii down-regulates the protein levels of Mcl−1 in hUC-MSCs and activates caspase-3, which leads to PARP cleavage and the eventual induction of apoptosis.
Methods
Isolation and culture of hUC-MSCs
hUC-MSCs were isolated and cultured as described previously.31 The umbilical cords used in this study were provided by volunteers who gave informed consent. This protocol was approved by the Ethical Committee of the Affiliated Hospital of Guangdong Medical University in China. hUC-MSCs were cultured in low-glucose Dulbecco's modified Eagle's medium (L-DMEM) supplemented with 10% fetal bovine serum (FBS) (both from Gibco, Grand Island, NY, USA) at 37°C in a humidified atmosphere containing 5% CO2. After 48 h, the nonadherent cells were removed by washing the culture with phosphate-buffered saline (PBS), and the medium was changed 3 times a week until the adherent cells reached approximately 80% confluence.
Phenotypic analysis
hUC-MSCs were harvested at passage 3 (P3) and washed twice with PBS. Then, the cells were characterized by analyzing the expression of the markers CD73, CD90, CD105, CD11b, CD19, CD34, and CD45 by using a FACSCanto II flow cytometer (all detection agents and instruments from BD Biosciences, San Jose, CA, USA). The data were analyzed by using FlowJo software (Tree Star, Ashland, OR, USA).
T. gondii maintenance
Tachyzoites of the GFP-expressing T. gondii GFP-RH strain were maintained using described previously methods with minor modifications.2 Briefly, the human retinal pigment epithelial cell line ARPE-19 (American Type Culture Collection, Rockville, MD, USA) was cultured in a 1:1 mixture of Dulbecco's modified Eagle medium (DMEM) and nutrient mixture F12 (DMEM/F12) supplemented with 10% heat-inactivated FBS and antibiotics/antimycotics (all from Invitrogen, Carlsbad, CA, USA).
The ARPE-19 cells were infected with the GFP-RH strain of T. gondii at a multiplicity of infection (MOI) of 5 and incubated at 37°C and 5% CO2 for 2–3 d. Following spontaneous rupture of the host cell, the parasites and host cellular debris were washed in cold PBS. The final pellet was resuspended in cold DMEM, and the resuspension was passed through a 5.0 μm pore filter (Millipore, Billerica, MA, USA).
Cell viability assay
hUC-MSCs were either mock-infected or infected with T. gondii at an MOI of 5 for 4, 8, 12 or 24 h, after which the cell viability was estimated using the CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI, USA) following the manufacturer's instructions.
Immunofluorescence assay
To evaluate the T. gondii infection rate, GFP-RH parasites at an MOI of 5 were incubated with hUC-MSC monolayers on glass coverslips for 4, 8, 12 and 24 h. Following treatment, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS (PBST) for 10 min. The coverslips were then washed with PBST and stained with Texas Red-X phalloidin (Life Technologies, Carlsbad, CA, USA) to label F-actin. Finally, the coverslips were washed and mounted onto microscope slides using a mounting medium containing DAPI (Vector Laboratories, Burlingame, CA, USA), and the cells were imaged using fluorescence microscopy. The number of parasite-infected cells were counted and reported as the percentage of the total cell count.
Adenovirus
Total RNA was isolated from cultured hUC-MSCs using TRIzol reagent (Invitrogen), of which 2 μg was reverse transcribed by using the PrimeScript 1st Strand cDNA Synthesis Kit (TaKaRa Bio, Shiga, Japan). An aliquot of the RT mixture was subjected to polymerase chain reaction (PCR) with primers that targeted the coding region of human Mcl−1 (5′- GGG GAC AAG TTT GTA CAA AAA AGC AGG CTG CCA CCA TGT TTG GCC TCA AAA GAA ACG CG-3′ and 5′- GAA GAC TTC CCC TGC CCT CTC TTA TTA GAT ATG CCA AAC CAG CTC-3′). The amplified full-length cDNA for Mcl−1 was subcloned into the pAV/DsRed vector containing attL sites that allowed site-specific recombination with a Gateway destination vector (Invitrogen). Replication-incompetent adenoviruses were generated using the ViraPower adenovirus expression system (Invitrogen). Briefly, site-specific recombination between the entry vector and the adenoviral destination vector was achieved by using the LR Clonase II Enzyme mix (Invitrogen). The resulting adenoviral expression vector was then transfected into 293A cells using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). Cells were grown until a cytopathic effect of 80% was observed, at which point they were harvested to prepare the recombinant adenovirus.
Overexpression of Mcl−1 in hUC-MSCs
hUC-MSCs were infected with adenovirus containing either DsRed alone or DsRed-Mcl−1 at an MOI of either 1 or 10. All experiments were performed 48 h after the addition of virus.
Quantitative real-time polymerase chain reaction (qRT-PCR)
After relative treatments, hUC-MSCs were dissolved in TRIzol reagent, and total RNA was extracted according to the manufacturer's instructions. To evaluate Mcl−1 gene expression, 2 μg of total RNA was reverse transcribed and then subjected to PCR with the following primer sets: Mcl−1, 5′-AAT CGG ACT CAA CCT CTA CTG TG-3′ and 5′- CTC CTT CTC CGT AGC CAA AAG T-3′; and HPRT1: 5′-GAC CAG TCA ACA GGG GAC AT-3′ and 5′-CTG CAT TGT TTT GCC AGT GT-3′. qRT-PCR was conducted using SYBR-Premix Ex Taq II (Takara Bio) on an ABI 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with HPRT1 as an internal control. The reactions were conducted in triplicate, and the data were analyzed using the 2−ΔΔCt method.
Western blotting
The protein samples were separated by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA) for the detection of relevant proteins by using the following primary antibodies:mouse anti-Toxoplasma gondii (TP3), mouse anti-α-Tubulin (both from Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-p62 (Sigma-Aldrich, St. Louis, MO, USA), rabbit anti-LC3B, rabbit anti-p-mTOR, rabbit anti-mTOR, rabbit anti-p-4E-BP1, rabbit anti-Bcl2, rabbit anti-Bcl−xL, rabbit anti-Mcl−1, rabbit anti-Bim, rabbit anti-Bax, rabbit anti-Bid, rabbit anti-Bak, rabbit anti-PARP, rabbit anti-cleaved caspase-3, rabbit anti-Beclin-1, rabbit anti-COX IV and rabbit anti-Calreticulin (all from Cell Signaling Technology, Danvers, MA, USA). The blots were developed using an enhanced ECL chemiluminescence detection kit (GE Healthcare, Little Chalfont, UK). Integrated image densities of the western blots were analyzed using the ImageJ 1.48v software (NIH, Bethesda, MD, USA).
Subcellular fractionation
Isolation of the mitochondrial and ER subcellular fractions were performed as described previously with minor modifications.32 Briefly, cells were washed with ice-cold PBS, suspended in a hypotonic extraction buffer comprising 250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA and protease inhibitor cocktail and homogenized by passing the suspension through a 26 gauge syringe needle. The homogenates were first centrifuged at 800 g for 10 min at 4°C, and the subsequent supernatant was centrifuged at 10,000 g for 20 min at 4°C. The pellet corresponded to the mitochondrial fraction (heavy membrane, HM), and the resultant supernatant was further centrifuged at 100,000 g for 1.5 h at 4°C. The final pellet corresponded to the ER-enriched fraction (light membrane, LM). The quality of the fractionation experiments was confirmed by assessing the presence of COX IV for the mitochondrial fraction and Calreticulin for the ER fraction.
Transmission electron microscopy (TEM)
hUC-MSCs were plated onto Aclar discs rather than glass coverslips, infected with recombinant adenovirus for 48 h, and treated with either mock-infected or infected with GFP-RH for 24 h at 37°C. The samples were then fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) at 4°C overnight followed by another fixation in 1.0% osmium tetroxide in the same buffer for 1 h. The samples were dehydrated in a gradient ethanol series and subjected to different concentrations of EPON812 resin (TAAB, Berkshire, UK) mixed with acetone at ratios of 2:1 and 1:2 of acetone:EPON812, as well as treatment with 100% EPON812. Afterwards, the samples were embedded and cured at 40°C for 12 h and 60°C for 24 h. Ultrathin sections (approximately 70 nm in thickness) were cut with a Leica EM UC7 ultramicrotome (Leica Microsystems, Wetzlar, Germany) and stained with uranyl acetate/lead citrate for visualization under a JEM-1400 transmission electron microscope (JEOL, Tokyo, Japan) at 80 kV.
Immunoprecipitation
The HM and LM subcellular fractions were incubated with anti-Mcl−1 antibody (Cell Signaling Technology) for 3 h at 4°C, after which 30 µl of Protein A/G Plus-Agarose beads (Santa Cruz Biotechnology) was added. After an overnight incubation at 4°C, the beads were precipitated and washed 3 times with RIPA buffer (Santa Cruz Biotechnology). Immunoprecipitated proteins were separated via SDS-PAGE under reducing conditions and then electrophoretically transferred to a PVDF membrane. Proteins of interest were detected with either an anti-Beclin-1 or anti-Mcl−1 antibody (both from Cell Signaling Technology) followed by development using an enhanced ECL chemiluminescence detection kit (GE Healthcare).
Statistical analyses
The results are expressed as the means ± standard deviation (SD) of at least 3 independent experiments unless otherwise indicated. The statistical significance of the data was determined by either an unpaired Student's t-test or one-way ANOVA. Values of P < 0.05 were considered as statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Natural Science Foundation of China (81301449 and 81300368), the Special Funds of Public Interest Research and Capacity Building of Guangdong Province in China (2015A030302078), the Foundation for Returned Overseas Scholars of Guangdong Medical College in China (B2012077), the Zhanjiang Municipal Governmental Specific Financial Fund Allocated for Competitive Scientific & Technological Projects (2014A06005) and the Doctor Scientific Research Fund of the Affiliated Hospital of Guangdong Medical University (BJ201508).
References
- [1].Tanaka N, Ashour D, Dratz E, Halonen S. Use of human induced pluripotent stem cell-derived neurons as a model for cerebral Toxoplasmosis. Microbes Infect 2016; 18:496-504; PMID:27083472; http://dx.doi.org/ 10.1016/j.micinf.2016.03.012 [DOI] [PubMed] [Google Scholar]
- [2].Quan JH, Cha GH, Zhou W, Chu JQ, Nishikawa Y, Lee YH. Involvement of PI 3 kinase/Akt-dependent Bad phosphorylation in Toxoplasma gondii-mediated inhibition of host cell apoptosis. Exp Parasitol 2013; 133:462-71; PMID:23333591; http://dx.doi.org/ 10.1016/j.exppara.2013.01.005 [DOI] [PubMed] [Google Scholar]
- [3].Hampton MM. Congenital toxoplasmosis: a review. Neonatal Netw 2015; 34:274-8; PMID:26802827; http://dx.doi.org/ 10.1891/0730-0832.34.5.274 [DOI] [PubMed] [Google Scholar]
- [4].Chen JL, Ge YY, Zhang J, Qiu XY, Qiu JF, Wu JP, Wang Y. The dysfunction of CD4(+)CD25(+) regulatory T cells contributes to the abortion of mice caused by Toxoplasma gondii excreted-secreted antigens in early pregnancy. PLoS One 2013; 8:e69012; PMID:23874852; http://dx.doi.org/ 10.1371/journal.pone.0069012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barry FP, Murphy JM. Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol 2004; 36:568-84; PMID:15010324; http://dx.doi.org/ 10.1016/j.biocel.2003.11.001 [DOI] [PubMed] [Google Scholar]
- [6].Jorgensen C, Gordeladze J, Noel D. Tissue engineering through autologous mesenchymal stem cells. Curr Opin Biotechnol 2004; 15:406-10; PMID:15464369; http://dx.doi.org/ 10.1016/j.copbio.2004.08.003 [DOI] [PubMed] [Google Scholar]
- [7].Li X, Xu Z, Bai J, Yang S, Zhao S, Zhang Y, Chen X, Wang Y. Umbilical cord tissue-derived mesenchymal stem cells induce T lymphocyte apoptosis and cell cycle arrest by expression of indoleamine 2, 3-dioxygenase. Stem Cells Int 2016; 2016:7495135; Article ID 7495135; PMID:27418932; http://dx.doi.org/ 10.1155/2016/7495135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liang L, Han ZC. Umbilical cord mesenchymal stem cells: biology and clinical application In: Stoltz JF, ed. Regenerative Medicine and Cell Therapy. Amsterdam (Netherlands): IOS Press; 2012. p. 62-70 [Google Scholar]
- [9].Martino R, Maertens J, Bretagne S, Rovira M, Deconinck E, Ullmann AJ, Held T, Cordonnier C. Toxoplasmosis after hematopoietic stem cell transplantation. Clin Infect Dis 2000; 31:1188-95; PMID:11073751; http://dx.doi.org/ 10.1086/317471 [DOI] [PubMed] [Google Scholar]
- [10].Kerl K, Ehlert K, Brentrup A, Schiborr M, Keyvani K, Becker K, Rossig C, Groll AH. Cerebral toxoplasmosis in an adolescent post allogeneic hematopoietic stem cell transplantation: successful outcome by antiprotozoal chemotherapy and CD4+ T-lymphocyte recovery. Transpl Infect Dis 2015; 17:119-24; PMID:25581774; http://dx.doi.org/ 10.1111/tid.12344 [DOI] [PubMed] [Google Scholar]
- [11].Gao D, Zhang J, Zhao J, Wen H, Pan J, Zhang S, Fang Y, Li X, Cai Y, Wang X, et al.. Autophagy activated by Toxoplasma gondii infection in turn facilitates Toxoplasma gondii proliferation. Parasitol Res 2014; 113:2053-8; PMID:24696274; http://dx.doi.org/ 10.1016/j.micinf.2016.03.012 [DOI] [PubMed] [Google Scholar]
- [12].Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol 2013; 87:1157-80; PMID:23543009; http://dx.doi.org/ 10.1007/s00204-013-1034-4 [DOI] [PubMed] [Google Scholar]
- [13].Wang Y, Weiss LM, Orlofsky A. Host cell autophagy is induced by Toxoplasma gondii and contributes to parasite growth. J Biol Chem 2009; 284:1694-701; PMID:19028680; http://dx.doi.org/ 10.1074/jbc.M807890200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhou J, Gan X, Wang Y, Zhang X, Ding X, Chen L, Du J, Luo Q, Wang T, Shen J, et al.. Toxoplasma gondii prevalent in China induce weaker apoptosis of neural stem cells C17.2 via endoplasmic reticulum stress (ERS) signaling pathways. Parasit Vectors 2015; 8:73; PMID:25649541; http://dx.doi.org/ 10.1186/s13071-015-0670-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Weissman IL. Stem cells: units of development, units of regeneration, and units in evolution. Cell 2000; 100:157-68; PMID:10647940; http://dx.doi.org/ 10.1016/S0092-8674(00)81692-X [DOI] [PubMed] [Google Scholar]
- [16].Zhang Y, Hao H, Liu J, Fu X, Han W. Repair and regeneration of skin injury by transplanting microparticles mixed with Wharton's jelly and MSCs from the human umbilical cord. Int J Low Extrem Wounds 2012; 11:264-70; PMID:23089966; http://dx.doi.org/ 10.1177/1534734612463577 [DOI] [PubMed] [Google Scholar]
- [17].Shi W, Nie D, Jin G, Chen W, Xia L, Wu X, Su X, Xu X, Ni L, Zhang X, et al.. BDNF blended chitosan scaffolds for human umbilical cord MSC transplants in traumatic brain injury therapy. Biomaterials 2012; 33:3119-26; PMID:22264526; http://dx.doi.org/ 10.1016/j.biomaterials.2012.01.009 [DOI] [PubMed] [Google Scholar]
- [18].Melamed J, Dornelles F, Eckert GU. [Cerebral CT scan alterations in children with ocular lesions caused by congenital toxoplasmosis]. J Pediatr (Rio J) 2001; 77:475-80; PMID:14647827 [DOI] [PubMed] [Google Scholar]
- [19].Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/ 10.1016/j.cell.2005.07.002 [DOI] [PubMed] [Google Scholar]
- [20].Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al.. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007; 26:2527-39; PMID:17446862; http://dx.doi.org/ 10.1038/sj.emboj.7601689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J 2011; 278:403-13; PMID:21182587; http://dx.doi.org/ 10.1111/j.1742-4658.2010.07965.x [DOI] [PubMed] [Google Scholar]
- [22].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ghosh D, Walton JL, Roepe PD, Sinai AP. Autophagy is a cell death mechanism in Toxoplasma gondii. Cell Microbiol 2012; 14:589-607; PMID:22212386; http://dx.doi.org/ 10.1111/j.1462-5822.2011.01745.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Contreras-Ochoa CO, Lagunas-Martínez A, Belkind-Gerson J, Díaz-Chávez J, Correa D. Toxoplasma gondii invasion and replication within neonate mouse astrocytes and changes in apoptosis related molecules. Exp Parasitol 2013; 134:256-65; PMID:23538030; http://dx.doi.org/ 10.1016/j.exppara.2013.03.010 [DOI] [PubMed] [Google Scholar]
- [25].Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, Opferman JT, Slack RS. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J 2011; 30:395-407; PMID:21139567; http://dx.doi.org/ 10.1038/emboj.2010.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741-52; PMID:17717517; http://dx.doi.org/ 10.1038/nrm2239 [DOI] [PubMed] [Google Scholar]
- [27].Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 2010; 29:1717-9; PMID:20101204; http://dx.doi.org/ 10.1038/onc.2009.519 [DOI] [PubMed] [Google Scholar]
- [28].Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS, Cheng AL, Chen PJ, Chen KF. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis 2013; 4:e485; PMID:23392173; http://dx.doi.org/ 10.1038/cddis.2013.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans 2013; 41:1103-30; PMID:24059496; http://dx.doi.org/ 10.1042/BST20130134 [DOI] [PubMed] [Google Scholar]
- [30].Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis 2008; 13:1-9; PMID:17990121; http://dx.doi.org/ 10.1007/s10495-007-0154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Qiao C, Xu W, Zhu W, Hu J, Qian H, Yin Q, Jiang R, Yan Y, Mao F, Yang H, et al.. Human mesenchymal stem cells isolated from the umbilical cord. Cell Biol Int 2008; 32:8-15; PMID:17904875; http://dx.doi.org/ 10.1016/j.cellbi.2007.08.002 [DOI] [PubMed] [Google Scholar]
- [32].Zhang L, Li L, Leavesley HW, Zhang X, Borowitz JL, Isom GE. Cyanide-induced apoptosis of dopaminergic cells is promoted by BNIP3 and Bax modulation of endoplasmic reticulum-mitochondrial Ca2+ levels. J Pharmacol Exp Ther 2010; 332:97-105; PMID:19841471; http://dx.doi.org/ 10.1124/jpet.109.159103 [DOI] [PMC free article] [PubMed] [Google Scholar]