

Abstract
Trophic factor treatment has been shown to improve the recovery of brain and spinal cord injury (SCI). In this study, we examined the effects of TSC1 (a combination of insulin-like growth factor 1 and transferrin) 4 and 8 h after SCI at the thoracic segment level (T12) in nestin-GFP transgenic mice. TSC1 treatment for 4 and 8 h increased the number of nestin-expressing cells around the lesion site and prevented Wallerian degeneration. Treatment with TSC1 for 4 h significantly increased heat shock protein (HSP)-32 and HSP-70 expression 1 and 2 mm from lesion site (both, caudal and rostral). Conversely, the number of HSP-32 positive cells decreased after an 8-h TSC1 treatment, although it was still higher than in both, non-treated SCI and intact spinal cord animals. Furthermore, TSC1 increased NG2 expressing cell numbers and preserved most axons intact, facilitating remyelination and repair. These results support our hypothesis that TSC1 is an effective treatment for cell and tissue neuroprotection after SCI. An early intervention is crucial to prevent secondary damage of the injured SC and, in particular, to prevent Wallerian degeneration.
Keywords: Wallerian degeneration, Heat shock proteins, IGF-1, Transferrin, Oligodendrocytes, Myelin
Introduction
Spinal cord injury (SCI) causes permanent neurological deficits. Its complex pathophysiology involves hypoxia, ischemia, vascular damage, free radicals production, inflammatory response, necrosis and apoptosis, which contribute to a poor, almost null, recovery [1–4]. SCI causes structural abnormalities, such as reduced axonal density, abnormal myelination, Wallerian degeneration (WD) and glial scar, yet it still exhibits endogenous repair potential [5–8]. SCI leads to molecular and cellular changes which involve both, cytoprotection and cytotoxicity [9, 10]. Heat shock proteins (HSPs) are induced by stressful stimuli. They assist the mechanisms for cellular protection and survival [11]. The expression of HSPs is a first indication of potential cell survival [12]. The expression of HSP-32 and HSP-70 has been used as an indicator of oxidative stress in neurodegenerative disease and both of them are expressed after SCI [13–15]. Neuroprotection of all cells as well as stimulation of neuronal regeneration to restore functional recovery after SCI, are imperative concepts for treatment. Scientific evidence in the past decade proved that neurotrophins and other trophic molecules like transferrin confer neuroprotective effects after brain and SCI [3, 9, 16–19]. Glial-derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (BDNF), for example, have been proven to attenuate blood-spinal cord barrier (BSCB) breakdown, edema formation and cell injury in a rat SCI model [3,10]. Other trophic factors have also been subject to study: insulin-like growth factor 1 (IGF-1) was recently reported to attenuate cell death and enhance axonal outgrowth [10, 20]. Transferrin (Tf), an autocrine/paracrine factor for neural cells, is produced and secreted by oligodendrocytes (OLs) at early stages of postnatal development [21, 22]. This factor is essential for cell survival and differentiation of all mammalian cells [23–25], and it is also important for myelination [22, 26–28]. Our laboratory’s previous studies support the hypothesis that endogenous regeneration of neurons and glial cells after SCI is possible, in a proper environment [15, 29, 30]. Moreover, we have also found that after SC transection in the presence of a hydrogel, the endogenous nestin expressing progenitors remain actively involved in the reconstruction of the spinal cord even 21 months after SCI and gel implantation [31]. Thus, endogenous repair can be enhanced with adequate approaches. For example, a cell transplantation approach is basically a combination therapy where the cells are the first component and the factors they secrete condition the milieu that may support functional recovery. Therefore, if the potential intervention aims to regenerating the spinal cord with endogenous cell populations, the use of specific trophic molecules would enhance such repair.
TSC1, a combination of the trophic factors, IGF-1 and Tf, was previously shown to induce proliferation, migration and specification to the OL lineage of quiescence progenitors in the subventricular zone (SVZ) of the myelin deficient rat mutant [27] and in a mouse model of premature birth [32, 33]. The goal of the present study was to examine the early effects of an acute treatment with TSC1 after severe spinal cord crush. We hereby report that a single TSC1 intervention increased the expression of both, HSP-32 and HSP-70, stimulated a wave of MBP expression with a concomitant and considerable reduced WD. TSC1 also induced fast demyelination of axons while protecting them from being transected as if preparing them to be remyelinated by MBP positive OLs in their vicinity, a phenomenon that would have required longer time points than the currently studied.
Methods
Animals
Six-month-old nestin-GFP transgenic mice were used with an average weight of 28.97 ± 4.85 g. We chose this age because nestin-GFP expression is very low and basically confined to the ependymal canal and therefore, it would not be difficult to detect an increase in nestin-GFP expressing cells. The nestin-GFP transgenic mouse, generated by Yamaguchi and coworkers [34], are bred at UCLA in a restricted access, temperature-controlled vivarium on a 12-h light/dark cycle, with food and water ad libitum. The animals were randomly assigned to five groups; untreated mice (n = 6), SCI mice 4 h (n = 6), SCI mice 8 h (n = 6), SCI + TSC1 mice treated for 4 h (n = 6) and SCI + TSC1mice treated for 8 h (n = 6). All experimental procedures were carried out in accordance with the National Institutes of Health Guide for Care and Use of Animals and were approved by the Institutional Animal Care and Use Committee at UCLA.
Spinal Cord Injury
The surgical procedures were performed under general anesthesia with isoflurane in oxygen-enriched air. The crush model was performed as previously described with minor modifications [35]. Briefly, a severe crush injury was made at the level of T12 using a dissection microscope and a rodent stereotaxic apparatus. Laminectomy was performed in aseptic conditions at the thoracic level T12. The crush injury was made using a number 5 Dumont forceps ground down until the tips contact. Forceps were used to compress the cord laterally from both sides for 5 s. After surgery, mice were maintained in a temperature-controlled environment during recovery and for testing. A single TSC1 administration (2 μl) was performed by delivering it in drops on the dorsal portion of the spinal cord without touching the spinal tissue (Fig. 1a). Samples were collected 4, 8 h or 7 and 14 days after surgery/treatment. A timeline of the injury and therapeutic intervention is shown in Fig. 1b.
Fig. 1.
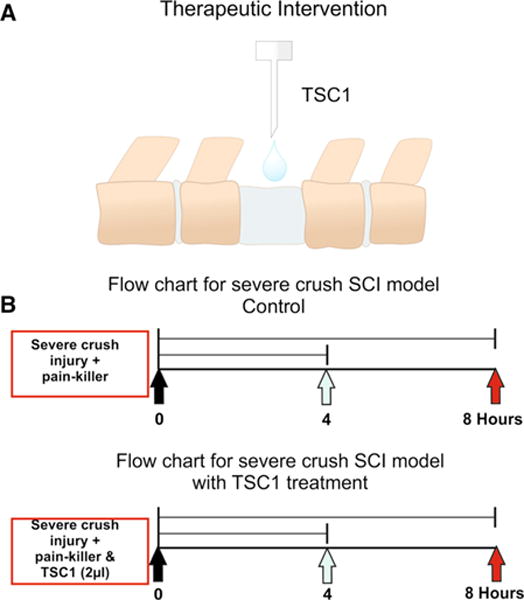
a Schematic representation on TSC1 mode of administration as drops over the lesion at the thoracic level (T12). Nestin-GFP transgenic mice generated by Yamaguchi and co-workers [34] (2000) 6 months of age were used in this study. The surgery included laminectomy and after laminectomy, mice received a severe crush at the lumbar level (L1–L2) followed by the administration of a 2 μl drop of TSC1 as shown in Fig. 1B. This approach allowed immediate contact with the spinal cord tissue and rapid absorption of TSC1. Because the volume is small the full amount of TSC1 reaches the dorsal portion of the spinal cord tissue. b Time line showing when the severe crush was performed as well as, the TSC1 administration
Tissue Preparation and Immunohistochemistry
Deeply anesthetized animals were perfused with 4 % paraformaldehyde. The spinal cords were rapidly removed, post fixed overnight in 4 % paraformaldehyde, and cryopreserved in 30 % sucrose. Thoracic segment of the spinal cord was separated from the other two spinal cord segments (lumbar and cervical). Twenty micrometers cross-sectional and longitudinal sections were obtained using a Micron cryostat. The specific antibodies used in this study are listed in Table 1. Immunohistochemistry was performed according to a previous publication [27]. Serial images stained sections were examined and photographed using the LSM-510 META confocal microscope (Zeiss).
Table 1.
The list of primary and secondary antibodies
| Primary antibodies |
| Anti-heat shock protein-32 (HSP-32, StressGen, Ann Arbor, MC) |
| Anti-heat shock protein-70 (HSP-70, Sigma, Santa Cruz, CA) |
| Anti-chondroitin sulfate proteoglycan (NG2, Chemicon International inc, Temecula, CA) |
| Anti-myelin basic protein (MBP, Biogenesis, UK) |
| Anti-neurofilament-200 (NF-200, Boehringer Mannheim, Germany) |
| Anti-microtubule associated protein-2 (MAP-2, Sigma, Santa Cruz, CA) |
| Anti-glial fibrillary acidic protein (GFAP, Chemicon Interanational inc, Temecula, CA) |
| Secondary antibodies |
| Rabbit anti-mouse IgG Texas Red (Jackson Immunological Research Laboratories, Inc, West Grove, PA) |
| Goat-anti mouse IgG Alexa Fluor 635 (Invitrogen, Carlsbad, CA) |
Cell Counting and Statistical Analysis
Cross-sections at the thoracic (T12) level of the spinal cord were divided in 6 areas for cell counting purposes, as shown in Fig. 1b. The number of nestin-GFP expressing cells, HSP-32 and double-labeled nestin-GFP/HSP-32 were counted in each of the six areas at different levels (Fig. 1c–e). At least 4 consecutive sections/animal and 3 animals/group were used for each marker. Cell count results were expressed as the mean ± SD numbers of positive cells. Cells from the lesion epicenter, as well as 1 and 2 mm above and below the lesion, were counted.
Statistical analysis was performed by One-way analysis of variance (ANOVA), followed by Tukey’s test using the StatPage Software (Statpage.net). The level of significance was chosen as p < 0.05.
Results
To analyze the effects of TSC1 on uncommitted neural progenitors after SCI, we took advantage of the nestin-GFP expressing mice, generated by Yamaguchi and coworkers [34]. These mice express GFP under the control of the nestin-promoter. Nestin is a commonly used marker for neural stem cells (NSCs) also known as NPCs or neural progenitors. This mouse model allowed us to clearly identify changes in the NPCs population in untreated mice, SCI mice (both, 4 and 8 h) and SCI with TSC1 mice (both, 4 and 8 h). We examined the number of cells expressing nestin-GFP at the lesion site, above and below lesion (1 and 2 mm) at thoracic segment T12 of the spinal cord.
Nestin-GFP Expressing Cells were Increased After Treatment with TSC1
We found that nestin-GFP expressing cells were present in all groups with or without TSC1, and that the total number of nestin-GFP expressing cells was already decreased 4 and 8 h after SCI (Fig. 2b). Additionally, the lesion area showed the lowest expression of nestin-GFP expressing cells when compared to 1 mm and 2 mm above and below the lesion. In contrast, we found that animals treated with TSC1 showed a higher total number of nestin-GFP expressing cells than mice with SCI but without TSC1. Four hours after treatment with TSC1, animals showed a significant decrease of nestin-GFP expressing cells, one and two mm below the lesion, when compared to mice 4 h after injury. We also found that there was no significant difference at the lesion level between SCI treated with TSC1 and non-treated mice. Interestingly, 8 h after treatment with TSC1 resulted in a higher number of nestin-GFP expressing cells than in SCI treated animals (Fig. 2b).
Fig. 2.
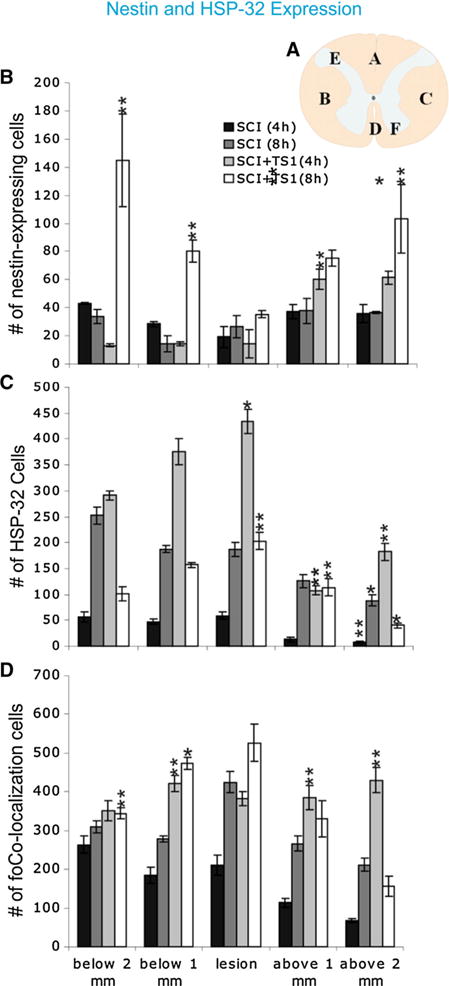
TSC1 increased the number of nestin-expressing cells 4 and 8 h after treatment. a Schematic representation of a transverse section of the spinal cord showing the six different areas used to count cells. The bar graph represents the total number of cells from three separate groups that were counted: nestin-GFP expressing cells (b), HSP-32 positive cells (c), and, co-expression of both markers by the same cell (d). Quantification was performed from coronal spinal cord sections of control (SCI) mice and SCI + TSC1 mice. HSP-32 positive cell numbers reached their highest at the level of the lesion and below (caudal to) the lesion after TSC1 treatment. Nestin-GFP expressing cells in SCI mice were increased after been treated with TSC1 for 4 and 8 h. The number of cells co-expressing nestin-GFP and HSP-32 was increased after treatment with TSC1. Note that the total number of nestin or HSP-32 expressing cells would be the sum of the corresponding bars for each marker and the number represented in the colocalization bar. Bars represent the mean ± SD; *p < 0.05, **p < 0.01 versus respective control
TSC1 Increases HSP-32
In order to determine the effect of TSC1 on cell injury response after SCI, we examined the expression of the stress protein, HSP-32. The total number of HSP-32 positive/nestin negative cells increased both 4 and 8 h after SCI. Interestingly, animals treated with TSC1 after SC1 showed the highest number of HSP-32 positive/nestin negative cells after 4 h and this number considerably decreased 8 h after treatment (Fig. 2c). However, mice receiving the TSC1 treatment showed significantly higher numbers of HSP-32 positive/nestin negative cells than their counterparts both 4 and 8 h after SCI (Fig. 2b). The number of nestin positive/HSP-32 negative cells at the epicenter and the periphery remained virtually constant for mice not treated with TSC1both 4 and 8 h after injury, as well as in TSC1 treated mice 4 h after the intervention. Nonetheless, TSC1 seemed to have rescued nestin-expressing cells as they reached their highest numbers 8 h after treatment (Fig. 2c). We next examined the colocalization of HSP-32 and nestin and it appeared as if most nestin expressing cells co-expressed HSP-32 in all regions examined. At the epicenter, as well as above and below the lesion, this population of cells displayed its lowest number at 4 h with a small but significant increase 8 h after SCI, as well as cells from the SCI + TSC1 group where at the epicenter and 1 mm periphery HSP-32/nestin expressing cells doubled the numbers when compared with 4 h (Fig. 2d). Thus, the number of neural progenitors co-expressing nestin-GFP/HSP-32 cells was significantly increased 4 and 8 h after SCI, but to a lesser extent than in SCI + TSC1 mice both 4 and 8 h still at the epicenter, as well as 1 mm above the lesion. Interestingly, above the lesion, the number of cells showing colocalization of both markers reached its peak 4 h after TSC1 administration, most likely due to migration of these progenitors or perhaps loss of nestin expression of pre-existing progenitors.
HSP-70 Expression in SCI and SCI 1 TSC1
We next examined the expression of HSP-70 by immunofluorescence and found that it was increased in both, 4 and 8 h after SCI. At the lesion site 4 h after injury there were numerous HSP-70 positive cells organized as rows along the gray matter tissue and also some nestin expressing small cells intermingled within the few left white matter fibers (Fig. 3a). Only some nestin positive cells reaching the lesion border where the tissue had lost its cytoarchitecture, co-expressed HSP-70. In this region, the tissue looked like lace or spongy making it difficult to visualize any cells, additionally, there was incipient cavitation. In contrast, mice treated with TSC1 4 h after treatment had preserved cytoarchitecture, white matter tracks were visible where HSP-70 was found in thin cell processes in contrast to SCI mice where the label was found mainly in the cell bodies. The interface between the gray and the white matters was well defined; and in the gray matter spider like structures expressing both HSP-70 and nestin were found, as well as some HSP-70 positive/nestin negative cells. (Figure 3b). Eight hours after SCI the lesion epicenter had lost the vast majority of cells; HSP-70 labeled structures of fibrous nature predominated; only a few nestin expressing cells of enlarged aspect and devoid of cell processes were present (Fig. 3c) and view of the same field at higher magnification in Fig. 3e). At the same time term (8 h), the tissue of mice treated with TSC1 had a beautifully preserved organization where a mesh-like structure appeared to cover the gray matter, while along the white matter tracks interfascicular HSP-70-expressing cells were visible (Fig. 3d). Moreover, nestin-expressing cells or their elongated fibers were arranged along the white matter. In some regions these fibers were perpendicular to axons and appeared to extend over and above white matter thick fibers intensely labeled by HSP-70 (Fig. 3f). Along these nestin labeled fibers, some bipolar nestin positive progenitors seemed to migrate towards or from the middle, where the ependymal canal was located.
Fig. 3.
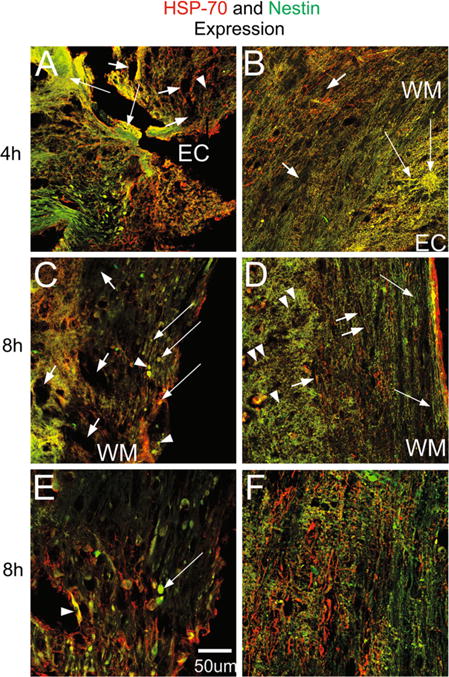
Sagittal views of the spinal cord at the lesion level of SCI and SCI + TSC1 mice at 4 and 8 h after injury. a HSP-70 positive cells and nestin-GFP expressing cells were present at the epicenter (EC) where the tissue that showed sponginess (EC), massive cell loss and cavitation were already visible (short arrows). In most regions nestin expressing cells weren’t distinguishable. HSP-70 labeled the soma of some cells as well as, some thin and hair-like fibers. These cells seemed to migrate in rows towards the lesion in the caudal semi-stump and they would stop at the lesion edge as if there was an imaginary border. A real colocalization of nestin and HSP-70 was not observed. b Mice treated with TSC1 after SCI displayed HSP-70 in the WM both in their thin fibers and in their cell body (long arrows). Numerous HSP +/nestin negative cells were present in the gray matter. Certain large multipolar cells in the WM co-expressed HSP-70 and nestin. c Eight hours after injury caudal and adjacent to the epicenter of the lesion cavitation was considerably reduced and very few cells expressed nestin intensely (arrowheads) while others faintly co-expressed nestin and HSP70 (long arrows). There were also small HSP-70 + positive nestin-negative cells in the white and gray matter. d Eight hours after lesion and TSC1 administration, the cytoarchitecture of both WM and GM were very well preserved. Nestin labeled hairy structures were organized as fishing net over the extent of the gray matter (arrowheads) while, in the WM nestin labeled fibers were parallel to axons and perpendicular to the nestin positive structures in the gray matter (arrows). Hairy fibers were labeled for HSP-70 mainly in the WM and they were devoid of nestin expression. e Eight hours after injury and 1 mm from the lesion epicenter, enlarged green cells were present, and almost no HSP-70 was found. Beyond 1 mm and caudal to de lesion, colocalization of both markers and more HSP-70 were observed. f Eight hours after mice were treated with TSC1 both, the WM and GM were well preserved with a robust expression of HSP-70 in numerous thin and thick fibers (short arrows) in the GM. There were also green small cells around axonal fibers in the WM with numerous thin nestin positive fibers extending perpendicularly to the ependymal canal over the GM towards the WM (arrowheads)
TSC1 Promotes the Emergence of NG2-Expressing OLPs
To determine the impact of TSC1 on OLPs in this model of SCI, we examined the expression of the OLP specific marker neuron-glial antigen-2 (NG2). At the 4 h term after SCI, the tissue looked spongy due to damage and/or loss of cells and structures, and single cells were not distinguishable in the epicenter region (Fig. 4a). In contrast, in mice treated with TSC1 although the tissue exhibited damage after the crush, large NG2 expressing cells appeared to extend long cell processes into the white matter as if they connected with both, WM and GM. NG2 positive cells appeared also along white matter tracks that had partially kept their orientation. In some of these cells, segments of their cell processes were NG2 labeled. Other NG2 positive cells had a spider-like appearance with numerous hairy processes extending over the white matter fibers. Moreover, some cell processes were clearly NG2 positive and they appeared to be underneath the spider-like cells. Small bipolar cells were visible in both, the gray and white matters (Fig. 4b). A closer view of the white matter of SCI + TSC1 mice 4 h after the intervention, unipolar and bipolar nestin expressing progenitors were clearly distinguished from NG2 positive cells (Fig. 4c). A view of a naive mouse SC is shown in panel (D) where NG2 appeared to be absent in both gray and white matter and where the nestin label was visible along blood vessels indicating that the nestin signal belongs to pericytes in the intact spinal cord. Seven days after injury, cells along the ependymal canal expressed intensely the nestin gene and small cells in the gray matter appeared to express both, nestin and NG2 (Fig. 4e). At this time point mice receiving TSC1 also had nestin-expressing cells in the ependymal canal and small cells co-expressing the label were seen in the gray matter (Fig. 4f). The white matter of TSC1 treated mice had been populated by bipolar nestin-expressing cells with long processes extending parallel to axons, some of these nestin positive progenitors appeared to migrate from the gray matter into the white matter, where some tissue loss could be observed (Fig. 4g). The gray matter of the naïve spinal cord did not show NG2 positive cells and nestin was solely expressed in pericytes (Fig. 4h). The gray matter of mice 14d after SC crush continued to deteriorate even within 2 mm away from the epicenter; cavitation, debris and enlarged yellow cells were abundant. The tissue consisted of NG2 labeled fibrous cells and only few faintly labeled nestin expressing cells were intermingled with the NG2 labeled fibers; pericytes were not visible (Fig. 4i). After 14 days of acute treatment with TSC1, the gray matter showed NG2 positive cells labeled in their cell bodies. An interphase-like region of NG2 labeled fibers had formed between the gray and the white matter. In the white matter and beyond that interface numerous bipolar nestin-expressing cells appeared to travel along the NG2 matrix that had formed where usually axons reside. A few of these cells appeared to co-express NG2 and nestin (Fig. 4k). The gray matter of these mice displayed numerous NG2 positive cells with shorter processes than those found in the white matter. These cells were arranged in a gradient-like pattern. At this same time point (14 days) more small cells co-expressed nestin and NG2 (Fig. 4l).
Fig. 4.
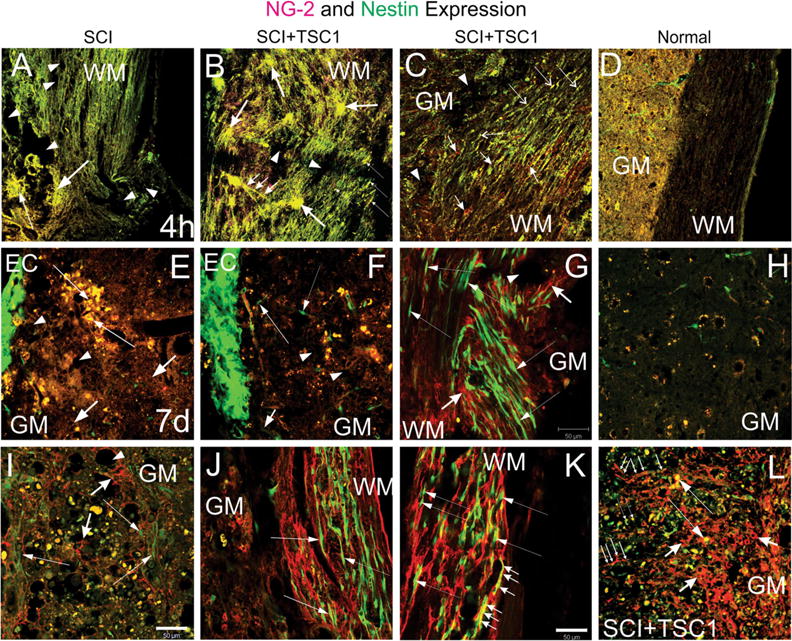
Two types of progenitors were present upon TSC1 intervention. a and b show low magnification views of the lesion epicenter. a Four hours after injury at the level of the lesion, the tissue appeared disrupted and without defined cellular structures that would have expressed nestin (neural progenitors) or OLPs identified by NG2. Moreover, the tissue showed a spongy appearance (arrows), and presented extensive cavitation (arrowheads). b In contrast the tissue from mice receiving TSC1 showed two types of structures: 1 Clusters of cells co-expressing nestin and NG2 (yellow). These cells were characterized by extending several long cell processes that appeared to go above nestin negative NG2 positive structures as a bridge for small cells to migrate towards the WM (small arrows). 2 Cells devoid of their processes (thin, long arrows). There were very small signs of cavitation (dotted areas). c Below the lesion (1.5 mm) along white matter tracks numerous small bipolar nestinansd NG2 expressing cells had appeared (Long and thin arrows). There were also interfascicular cells expressing solely NG2 mainly in their cell body (short arrows). d Low magnification view of a spinal cord of a naïve mouse at the T12 level. Seven days after injury panels e–g. e At higher magnification, the ependymal canal showed intense nestin expression (EC). The tissue adjacent to the ependymal canal had a spongy appearance (short arrows). A few patches of cells co-expressing nestin and NG2 were visible (yellow cells; long, thin arrows). At a longer distance from the EC the yellow cells were disappearing leaving behind only debris. f Seven days after injury and TSC1 treatment, the ependymal canal displayed an intense nestin expression. Beyond the EC few scattered, bipolar nestin-bearing cells appeared to migrate perpendicular to the EC and towards the white matter (long thin arrow). There was some cavitation in the GM (arrowheads). Some NG2 and nestin co-expressing cells presumably had migrated away from the canal (yellow cells). g The WM displayed a plethora of nestin-expressing bipolar cells mostly arranged in rows following the direction of the axons. Thus, nestin-expressing cells that had migrated following those axons directly exposed to the severe crush accumulated where the axons were severed. Long, thin arrows show examples of bipolar cells appearing to migrate along the axons. In areas where the white matter suffered to a lesser extent, nestin-expressing cells with elongated thin processes were also seen (thin, long arrows). Cavitation was less frequent in TSC1 treated mice (arrowhead). NG2 expressing cells had formed a matrix with their cell processes that intermingled with the axons and nestin-expressing cells (short, thick arrows). h Low magnification view of a naïve spinal cord’s gray matter. i Fourteen days after the intervention, the GM of mice that did not receive TSC1 had a lace-like aspect and NG2 labeled fibrous cell processes surrounding the cavities that formed upon tissue loss. Numerous cells that co-expressed both NG2 and nestin were enlarged and seemed to be dying. Very few green cells bearing faint nestin expression were visible at this time point (long, thin arrows). In contrast, mice treated with TSC1 (panel L) was populated of numerous NG2-expressing cells with large cell body and fibrous processes (short, thick arrows) Long. They had formed a dense structure across the GM that appeared to serve as a migratory substrate of both nestin expressing cells (long thin arrows) and NG2/nestin co-expressing cells (thin, long arrows). NG2 expressing cells were much larger than the NG2/nestin co-expressing cells. j Surprisingly, the white matter of mice 14 days after SCI had preserved its structure to some extent and nestin positive cells had reached them and appeared to migrate along axons (thin, long arrows). k In mice treated with TSC1 the white matter was populated by bipolar nestin positive cells intermingled with NG2 intensely labeled cells, the WM structure was nicely preserved allowing for the migration of nestin-expressing neural progenitors (thin, long arrows). Interestingly, bipolar cells co-expressing nestin and NG2 were abundant (small arrows), while in mice without TSC1 bipolar cells in the white matter mostly expressed nestin alone
In order to identify the effects of acute intervention with TSC1 after SC crush and to assess the integrity of the tissue we examined the lesion site and surrounding areas (1 and 2 mm rostral and caudal to the lesion). We used the neuronal/axon marker NF-200 (red) and anti-MBP (blue) to visualize myelin. We found that 4 h after SCI there was severe damage and cavitation surrounding the lesion site (Fig. 5a). The formation of a dense scar was already present at this time point. Caudal to the lesion there was a gradient of naked axons still labeled by the anti-NF antibody in the gray matter, the NF gradient led to MBP-expressing OL labeled in the cell body rather than the fibers. The white matter was positive to MBP and intensely labeled. At a higher magnification of the portion rostral to the lesion, fibers and scar-like tissue were seen across the spinal tissue. MBP-positive cells were clearly seen and bundles of fibers along the white matter extended rostrally (Fig. 5b). As for MBP-labeled fibers they were visible in the white matter intermingled with necrotic bundles. Moreover, farther caudal from the lesion necrotic tissue and cavitation were already noticeable and both, the gray matter and the white matter showed lack of NF and MBP. Nestin-expressing cells were seldom visible in the vicinity of the lesion (Fig. 5c).
Fig. 5.
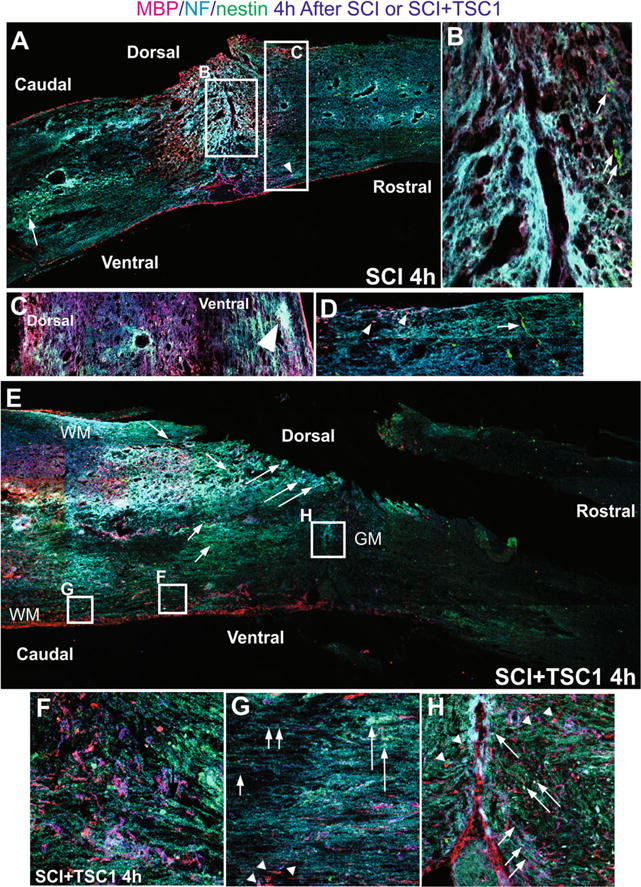
Comparison of mouse spinal cord 4 h after SCI or SCI + TSC1. a The low magnification view of the spine showed fibrosis at the epicenter and surrounding areas that appeared to be severed axons as NF labeled fibers accumulated. Caudal to the lesion cavitation was visible in the gray matter where nestin expressing cells had accumulated and were intermingled with NF labeled fibers and both dorsally and ventrally there was reactive tissue. b At higher magnification the view of the epicenter showed spongy tissue and some MBP positive cells were visible while the neighboring tissue had already become lace-like. Rostrally and adjacent to the epi-center few small nestin positive cells could be seen (arrows). c Higher magnification view of the tissue rostral to the lesion where cavitation had not started and nestin expressing cells appeared to sit on NF labeled axons. d Caudal to the lesion bipolar nestin positive cells appeared to migrate from the ependymal canal [EC] and perpendicular to it across axonal fibers. MBP positive cells were present in this area. e Low magnification view of a mouse spinal cord 4 h after SCI + TSC1, the overall structure of the spinal cord has been preserved. Caudal to the lesion NF labeled cell bodies were present (long arrows) and an extensive scar had not formed. Motoneurons were also present (long arrows). Numerous nestin positive cells single or in rows were frequent (short arrows). f High magnification view of the ventral white matter caudal to the lesion MBP expressing OL and myelin segments are visible intermingled and parallel to the ependymal canal. Numerous nestin expressing cells were also seen. g The white matter showed segments of myelinated axons that were abruptly ended by tissue loss (cavitation; arrowheads). Other axons appeared normally myelinated. Numerous axonal fibers faintly labeled for NF appeared to keep their position and structure of the WM (short arrowheads). h Higher power view of the epicenter at the level of the gray matter (GM). The tissue lost its form but not completely, MBP labeled axons appear to have severed or lost their label. Nonetheless, numerous MBP expressing cells were also in the vicinity of the lesion both caudally and rostrally (arrowheads). Numerous nestin positive small cells surrounding the lesion from the rostral side appeared to migrate towards it (long arrows). In this area the axons had adopted the form of the tissue after the severe crush they were bearing the NF marker (small arrows)
Four hours after SCI + TSC1 the tissue looked more even in terms of MBP and NF staining. There was a thin faint scar but much smaller than in the SCI 4 h term. Cavitation was visible rostral to the lesion in the white matter, but not in the white matter caudal to the lesion (Fig. 5d). In the caudal part to the lesion gray matter there was some necrosis and visible cavitation close to the epicenter. Higher magnification views of the caudal white matter showed numerous MBP positive cells and/or segments intermingled with NF labeled axons and some small, round or bipolar nestin-expressing progenitors all appeared to converge in the vicinity of the lesion (Fig. 5e). Even farther caudal to the lesion numerous naked axons were present bearing the NF label. Although unmyelinated, the vast majority appeared well organized and only a few myelin segments were found (Fig. 5f).
We also examined the spinal cord of mice that underwent SCI without TSC1; 8 h after, the intervention showed massive axonal disruption at the epicenter as well as cavitation in the gray matter caudal to the lesion. NF and MBP immunoreactivity was observed caudal and rostral to the lesion. The white matter showed massive foci of necrotic cells at the epicenter as well as caudal and rostral to the lesion (Fig. 6). Higher power view of the epicenter showed some OL recognized by MBP positive in the gray matter (Fig. 6b). At the level of the epicenter some enlarged OLs were found in the lace-like gray matter. At this time point, cavitation had developed in the white matter, near the epicenter, but there were also nestin-positive cells arranged along NF faintly labeled axons (Fig. 6c). At even higher magnification the tissue loss was clearly visible and the enlarged OLs appeared not to have any cell process, and most of the NF label was practically gone in the epicenter regions (Fig. 6d). Mice that received TSC1 after SC crush displayed considerable cavitation in the white matter, extending into the gray matter at the epicenter and its vicinity. However, the most striking finding was the presence of numerous MBP-positive OL in the gray matter. These cells were bearing the label in their cell bodies (Fig. 6e). Another important aspect was the presence of many nestin-expressing cells either surrounding some of the tissue cavities in the white matter (Fig. 6f), or as a thin extensive network of nestin-labeled cell fibers that appeared to extend over the damaged white and gray matters (Fig. 6h). These thin fibers were perpendicular to axons and appeared to originate from the ependymal canal (EP) across the gray matter. Some necrotic tissue was also visible in the gray matter (Fig. 6h).
Fig. 6.
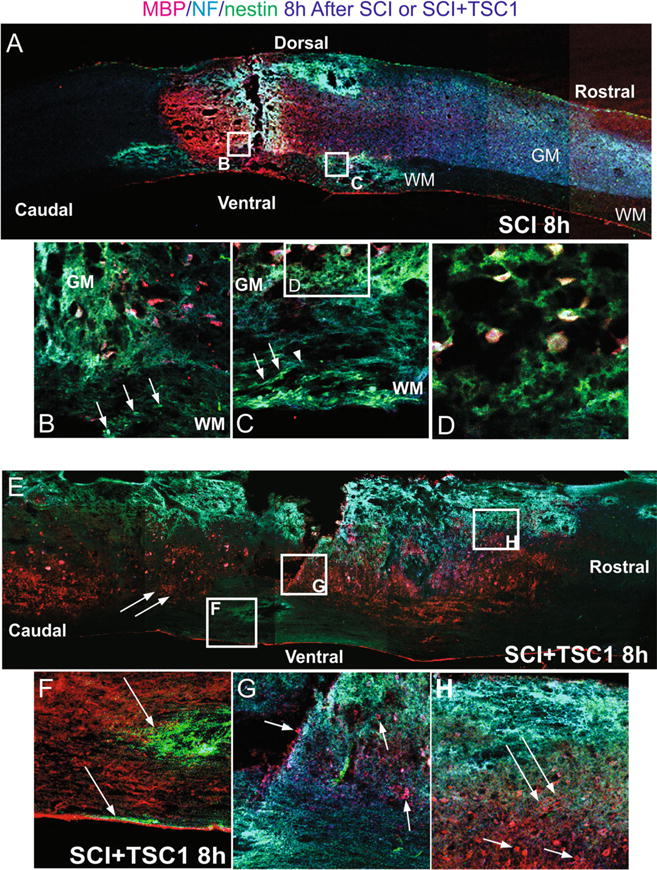
Comparison of mouse spinal cord 4 h after SCI or SCI + TSC1. a The low magnification view of the spine shows fibrosis at the epicenter and surrounding areas that appeared to be severed axons as NF labeled fibers accumulated at both sides, caudal to the lesion there was cavitation mostly in the gray matter where the NF label was arrested. Rostral to the lesion the white matter appeared to have suffered the most showing cavitation dorsally and rostrally. b A high magnification view of the interphase between the white and gray matter adjacent to the epicenter where nestin positive cells were present (arrows). In the gray matter there were some MBP positive cells without cell processes in areas of cavitation. c Rostrally and ventral to the lesion in the white matter nestin expressing cells were seen as if they were using axonal fibers to migrate in the direction of the gray matter (arrows). These cells seem to find the end of the road where the fiber stops at the cavitation edge (arrowhead) and an accumulation of green progenitors was seen in the outer portions of the white matter. d A closer view (even at higher magnification) of the white matter shows MBP positive cells held by thin tissue fibers. Some of these OLs showed a round appearance and no cell processes. Other MBP positive cells appeared bipolar with MBP expressed in their cell body and one nestin positive cell process. Yet, in the gray matter the cavitation was expanded. e There was a striking difference between the untreated and the TSC1 treated spinal cords. TSC1 appears to have preserved the myelin fibers in the white matter neighboring the gray matter. Moreover, numerous OL bearded MBP in their somas. f At the caudal portion of the lesion and ventral to it there were foci formed by cavitation that had been invaded by numerous nestin expressing progenitors (arrow) or in the external portion of the white matter (arrow). But neither MBP nor NF staining was present in the adjacent white matter. g At the epicenter in the gray matter MBP positive cells were visible together with NF immunoreactivity (arrows). Dorsally and rostral to the lesion the WM displayed considerable cavitation the cavitation that extended into the gray matter. Nonetheless, fibrous cells and scar-like structures appeared to be covered by a light and extensive veil of nestin positive thin and hairy fibers shown in h (long arrows). These fibers were located in a radial fashion in an approximately 45 degrees angle from the pia mater in direction to the epicenter. The gray matter contained numerous MBP positive OLs (arrows)
TSC1 Prevents WD Increasing MBP-Expressing Cells After SCI
We further examined the effects of TSC1 1 and 2 weeks after the intervention. Figure 7 shows the comparison of the spinal cord at 7 and 14 days after severe crush and that of one that received TSC1 adjacent to the epicenter. Seven days after severe crush most of the white matter had lost both myelin and MBP labeling; also the NF staining was seen in somata of cells in the gray matter but not in the white matter. Moreover, in the white matter very few NF-labeled fibers were seen. Numerous small nestin-expressing cells had migrated to the epicenter and its vicinity. Nonetheless, almost no myelinated fibers were seen. Some MBP-positive OL were intermingled in the tissue at the epicenter edge and fewer of them were in the gray matter. Their numbers increased as distance to the epicenter shortened (Fig. 7a). Seven days after SCI + TSC1 abundant MBP and NF immunoreactivity were seen and among these cells and fibers some GFP-nestin bipolar cells or dividing cells were also present (Fig. 7b). Fourteen days after SCI cavitation was extensive and numerous MBP-positive cells were labeled solely in the cell body. Moreover, just a few neuronal cell bodies were visible. Disorganized segments of MBP positive fibers were present in the white matter and they were longer and increased in number at what one could call the interface between the white and the gray matter. Nestin-labeled bipolar cells or cells with numerous hairy processes were also present (Fig. 7c).
Fig. 7.
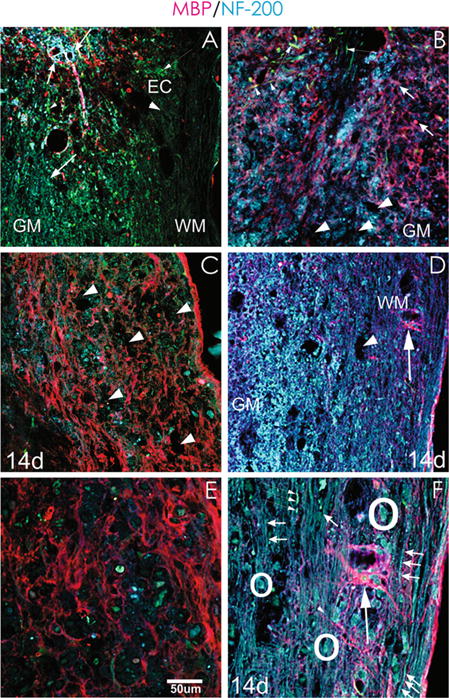
TSC1 prevents Wallerian degeneration after SCI. a Seven days after crush and 1 mm from the epicenter the SC displayed cavitation but, it still had preserved it’s cytoarchitecture, besides at the lesion border where MBP label was seen. Beyond that point between 1 and 2 mm from the lesion almost not MBP could be seen in either the WM or GM. There was not NF label along axons in this region, NF was seen on some cells expressing it in their soma and processes that extended only at the border of the lesion but not beyond it (Thick, short arrows). b At this time point mice treated with TSC1 showed scarce cavitation sites of modest size when compared to panel A. Both MBP and NF labels. Few nestin labeled, bipolar cells and fibers appeared in the vicinity of the lesion (thin arrows). c A week later SCI non-treated mice had more cavitation than seven days after injury (arrows), numerous nestin positive small cells were visible in both the gray and white matter (arrowheads). Some moto-neurons expressing NF in their cell body were also found although devoid of processes. Disorganized or even transected-like myelin structures were abundant in the white matter and less abundant and fainter in the gray matter, some of them appeared to be reactive-like OL. d Fourteen days after injury and TSC1 administration, the GM was populated with numerous NF positive cells and structures and MBP labeled fibers were abundant. MBP staining allowed for the visualization of the nodes of Ranvier along axons (arrows). Some turquoise colored bipolar cells appeared to migrate along a myelinated segment (arrowhead). e At higher magnification cavitation was more evident (14 d after SCI), motoneuron cell bodies in blue were scarce. Green enlarged cells of unhealthy appearance were present and there were also a few smaller, healthy nestin positive cells. In the white matter bundles of axons were disrupted and neuronal cell bodies were clearly compromised. Some very small cells intensely labeled for NF were seen but there was not tissue to support their potential migration. f Higher magnification view of a spinal cord 14 days after but not transected. They extended along the field of view in a parallel arrangement. Other fibers were myelinated and alternating nodes of Ranvier and myelin segments were clearly distinguishable. Neuronal survival or perhaps naisance of new neurons, were facilitated by TSC1 as numerous motoneurons were present. Moreover, only scarce cavitation was found. Interestingly, the colocalization of nestin and NF in some if not most motoneurons at the level of the cell body is evident (arrowheads). OLs with a prominent cell body and at least ten visible large processes extended over long distance reaching out to axons
Mice treated with TSC1 after severe crush and 14 days after the intervention had preserved cytoarchitecture. Numerous cell bodies and long uninterrupted and naked NF-labeled axons were clearly seen. Myelinated fibers were also present and they showed continuity. Among fibers arranged in parallel as expected of the white matter, small and larger neuronal bodies were visible. In the gray matter, NF-labeling predominated but MBP-expressing cells were also seen. Although there was cavitation in both, the white and the gray matters, at large the cytoarchitecture had been preserved. At this time point and at this magnification it was difficult to determine the presence of nestin-expressing cells (Fig. 7D). At a higher magnification, there was extensive cavitation in the SCI mice that did not receive TSC1. These areas devoid of tissue would not offer support for axonal re-growth or for progenitor migration to repopulate and re-construct the spinal cord (Fig. 7E). In contrast, two weeks after SCI + TSC1 the vast majority of axons were present. Some of them were myelinated and many more were unmyelinated, but not transected. Moreover, some bipolar nestin-expressing cells appeared to use naked axons to migrate and a few nestin-positive cells were present between axons. Healthy neuronal cell bodies were also present. The most remarkable finding was that MBP-positive OLs extended numerous long cell processes to reach axons across long distances. This was most likely because axons were still intact and perhaps signaling OLs for re-myelination (Fig. 7F).
Discussion
In the past the concept of “regeneration” after traumatic brain injury or SCI, basically referred to the potential of axonal re-growth from an injured neuron. This phenomenon and sprouting are due to the well-known innate anatomical plasticity of the CNS. After SCI the distal segments undergo WD and the proximal segments do not spontaneously regenerate [36]. Many approaches have been taken to understand why spontaneous regeneration initiates, but is not sustained. Studies describing the mechanisms underlying axonal growth failure including cavitation, inflammation, gliosis, scar formation and lack of trophic factors, have been reviewed and summarized [37]. These and other studies have led to experimental efforts to enhance regeneration of the injured spinal cord. One of these approaches has been the use of growth factors and bridges. Among those factors are NGF, BDNF, NT3, NT4/5, and GDNF [37], and in the brain Platelet derived growth factor-alpha (PDGF-aa), neurotrophin-3 (NT-3), basic fibroblast growth factor (bFGF), and insulin-like growth factor-1 (IGF-1) have been show to enhance re-myelination in a mouse model of cuprizone demyelination [38].
We recently described the use of “TSC1”, a mixture of transferrin and somatomedine C (also known as IGF-1) to promote CNS endogenous repair and myelination of the myelin deficient (md) mutant rat [27], re-myelination of the adult demyelinated mouse brain [39] and rescue of the forming white matter in a mouse model of periventricular leukomalacia resulting from premature birth, where we demonstrated that white matter loss after excitotoxicity can be partially rescued by using TSC1 that conferred neuroprotection to preexisting OLP and regeneration of the WM via OLP proliferation. Moreover, we also showed that an early TSC1 administration maximizes neuroprotection [32].
In all three animal models the mobilization of nestin-expressing progenitors played a major role on the improvement of myelin loss. Therefore, here we sought to determine if TSC1 would be beneficial if administered acutely after SC severe crush.
We believe that SCI should be treated like heart failure or stroke where the patient is treated as soon as possible to optimize the potential outcome and minimize secondary damage. Because the trauma to the tissue was severe we did not want to increase the injury further and therefore, we decided to administer TSC1 without touching the spinal tissue.
It has been reported that HSPs play protective roles and provide tolerance against cellular stress. These proteins are involved in the modulation of proteolytic machinery, preventing protein misfolding and aggregation, thereby accelerating cellular repair processes [40]. After trauma, the secondary injury to the spinal cord is the induction of oxidative stress by initiation of free radical reaction and lipid peroxidation. The oxidation of fatty acids, including arachidonic acid, generates oxygen radicals as a byproduct and may be the primary mechanism of free radical generation in the traumatized spinal cord [13]. HSP-32 or heme oxygenase (HO)-1 is expressed in many kinds of cells (e.g. astrocytes, microglia, macrophages, neutrophils or neurons) which are stimulated by pro-inflammatory cytokines after SCI [41, 42] HSP-32 is important to control acute inflammation [14, 19]. It is well known that SCI induces oxidative stress response that elicits the upregulation of HSP-32 and HSP-70 [11, 19, 42]. This is an important mechanism for the recovery and survival of injured cells; [14, 43, 44]. In the present work, we examined the response of two stress proteins HSP-32, and HSP-70, as biomarkers of the extent of injury, its intensity and the potential for recovery in the presence of TSC1. We found that mice that were treated with TSC1 after severe crush showed higher number of cells expressing HSP-32 than in mice with SCI without TSC1, suggesting that in the presence of TSC1 these cells had activated their machinery to survive the insult in a more efficient manner. Therefore, TSC1 increased the number of cells that expressed HSP-32 allowing them to survive at the lesion epicenter, 1 and 2 mm caudal to (or below) the lesion. These HSP-32 expressing cells included a subpopulation of nestin-expressing neural progenitors. These data confirm our previous work where HSP-32 was scarce and delayed in a rat model of SC hemi-section [15]. Nonetheless, in rats that underwent reconstruction of the lesion motoneurons and cells from the ependymal canal appeared to be the main populations expressing HSP-32.
The affymetrix profile of expression for HSP-32 has been reported to increase 11.4 times 24 h after injury and 70 times after 48 h [45] which suggests that higher levels would be achieved in the presence of TSC1 resulting in reduction of cell death.
Another stress protein, HSP-70 is involved in the repair or degradation of polypeptides that become denatured under stress [46]. It recognizes and binds to nascent polypeptide chains as well as partially folded proteins, preventing their aggregation and misfolding. It also interacts with HSP-40 to promote cellular protein folding and repair misfolded proteins [13, 40, 46, 47]. In contrast to HSP-32, only a few nestin-expressing cells co-expressed HSP70 indicating that cells from defined lineages responded to SCI by expressing HSP-70 in mice with SC crush without TSC1, treatment. In mice treated with TSC1 HSP-70 was conspicuous mainly in long and thick fibers in the white matter (perhaps myelinated axons or in myelin itself), and in the gray matter in neuronal cell bodies at the epicenter and below the lesion confirming that the neuronal population had a propensity to up-regulate HSP-70 in response to TSC.
TSC1 Promoted Survival and Migration of Nestin Expressing Cells
Nestin is an intermediate filament protein that is known as a neural stem/progenitor cell marker. It is expressed in undifferentiated central nervous system (CNS) cells during development, but also in the normal adult uncommitted progenitors CNS [48, 49] and in CNS tumor cells where the degree of nestin expression correlates with the malignancy of the CNS tumor [50]. Thus, nestin is generally recognized as a marker protein of undifferentiated CNS cells at the stage that precedes exit from the cell cycle and commitment of the mature progeny to a specific lineage. Moreover, GFP-expression and nestin protein co-expression have been studied in the dentate gyrus of the adult nestin promoter–GFP mice created by [34, 51]. In fact there are two types of nestin-GFP-bearing ptogenitor cells in these mice even if uninjured, some that co-labeled with an anti-nestin antibody and an anti-GFAP antibody. These cells expressing nestin, GFP and GFAP were described to often come in contact with blood vessels [52, 53]. Other cells co-expressed PSA-NCAM, nestin and GFP but not GFAP as visualized by their respective antibodies [52].
It has also been shown that after focal cerebral ischemia induced in rats nestin is expressed by reactive astrocytes [54] in the peri-infarct zone during the first 3 days while, in the ischemic core nestin expression was observed only in the microvasculature and in larger caliber cells, suggesting that nestin may be involved in the regulation of cell structure remodeling after ischemia [54]. Nonetheless, in a separate manuscript we have shown that with the TSC1 combination nestin–expressing cells become—MBP or Tf positive cells [27]. In addition we have shown that nestin and GFAP do not colocalized upon TSC1 administration into the de-myelinated brain parenchyma [33].
In this study, we showed that the number of nestin-GFP expressing cells was decreased in both SCI and SCI + TSC1 mice both at 4 and 8 h after treatment at the lesion site as well as 1 and 2 mm rostral and caudal to it. Having counted separately nestin-expressing cells from those co-expressing HSP-32 and nestin, we determined that only a small number of nestin-expressing cells was unable to express HSP-32. Nonetheless, the number of nestin-positive/HSP-32 negative was higher in mice treated with TSC1 than in those that underwent SCI without TSC1 treatment. Previous studies have shown that 1 day after a minimal SCI, the expression of nestin is induced achieving the peak of expression by 7 days after injury [55]. Taking both, their data and ours together we could deduct that nestin expressing cells are not numerous at the epicenter and around the lesion at 4 and 8 h, because they had not yet migrated that far. We previously showed that TSC1 elicits the mobilization and migration of nestin-expressing cells from the subventricular regions of the brain to the parenchyma in the neonate dys-myelinated, uninjured rat brain [27]. In another more recent paper using a mouse model of excitotoxicity we have shown that the nestin-expressing cell population regenerates in two manners: it mobilizes preexisting dormant nestin-expressing cells as a consequence of the insult, and those nestin-expressing cells had also regenerated via cell proliferation when mice were treated acutely with TSC1. Moreover, a delayed administration of TSC1 after excitotoxicity reduced ventriculomegaly but not as much as when NMDA and TSC1 were injected simultaneously [33] indicating that there is a window of opportunity that is widely open just after the insult and that it gradually closes allowing a less robust regeneration upon TSC1 administration. In other words, the beneficial effects of TSC1 are considerably reduced as a function of time; the longer it takes prior to its administration, the least optimal its effects swill be. Therefore, TSC1 appears to be the proper combination of factors to activate and produce adequate numbers of nestin- expressing cells in a sustained manner (up to 2 weeks) after SCI.
IGF-1 is a pleiotropic factor with diverse roles in the CNS [56]. It is a potent proliferation agent for neural-stem cell precursors and neuroblasts and it promotes subsequent steps in the differentiation of neurons, astrocytes and OLs. Besides proliferation, IGF-1 has been shown to promote myelination [57, 58]. IGF-1 is widely distributed in the fetal and neonatal CNS, but it is restricted in the adult CNS [59]. Similarly, IGF-1 receptor expression is heterogeneous during early stages of CNS development, but the expression levels decline postnatally [60] reaching low levels in the aging brain. IGF-1 is also known to prevent apoptosis [61].
IGF-1 not only works on neurons, but also controls the balance between OLP proliferation and differentiation in CNS [62–64]. Moreover, Gage and collaborators [64] showed that IGF-I alone instructs the differentiation of multipotent adult rat hippocampus-derived neural progenitor cells into OsL. OL differentiation by IGF-I appears to be mediated through an inhibition of bone morphogenetic protein signaling. Furthermore, overexpression of IGF-I in the hippocampus leads to an increase in OL markers. Thus, demonstrating that IGF-I, dictates the fate choice of multipotent adult neural progenitor cells to an OL lineage.
Transferrin is the main iron carrier glycoprotein in mammals, behaving as a paracrine factor. It plays an important role in metabolic activity in all mammalian cells and prevents cell death after stress or injury [17, 65]. Cell culture studies showed that immature neurons require transferrin and IGF-1 for survival as they become post-mitotic [66]. Tf is also needed by developing OLPs [67]. We have demonstrated that it is synthetized and secreted by OL [68] and that it is an essential factor for the up-regulation of the MBP gene [22]. We have previously reported that low levels of Tf in the CNS contribute to myelin deficiency and that correction of Tf levels in cell culture [68] or in vivo, leads to myelination [22]. We have also shown that Tf synergizes with IGF-1 to enhance myelinogenesis in the md mutant rat [26] and the adult demyelinated brain [26].
Although the molecular mechanisms underlying TSC1 neuroprotection against cell death were not examined in this study, it has been reported both in vitro and in vivo that IGF-1 may directly prevent cell death and apoptosis of cells after injury [69–71]. In addition, Tf has been identified as a stabilizing factor of cytoskeletal proteins in nerve fibers and it also prevents the disruption of the axonal transport and myelin degeneration [71]. Therefore, the neuroprotective effects of TSC1 on axon preservation may be due, at least in part, to uninterrupted circulation of nutrients and the preservation of axonal cytoskeleton.
NG2-expressing cells also known as “polydendrocytes” comprise an abundant glial population widely distributed throughout the developing and the mature CNS. The NG2 proteoglycan is characteristically expressed by OLPs in the normal CNS. OLs are particularly sensitive to stress resulting in cell death in the white matter after hypoxic or ischemic insults of premature infants and in some types of Multiple Sclerosis lesions where OLPs die. Polydendrocytes differentiate into OLs in vitro and have often been equated with OLPs and they appear to be the primary source of re-myelinating cells in demyelinated lesions [72]. The NG2 proteoglycan binds to OMI/HtrA2, a mitochondrial serine protease which is released from damaged mitochondria into the cytosol in response to stress. In the cytosol, OMI/HtrA2 initiates apoptosis by proteolytic degradation of anti-apoptotic factors. OLPs in which NG2 has been downregulated by siRNA, or using OLPs from the NG2-knockout mouse showed an increased sensitivity to oxidative stress evidenced by increased cell death. The proapoptotic protease activity of OMI/HtrA2 in the cytosol can be reduced by its interaction with NG2 [73]. Another study supporting this idea has shown that human glioma expressing high levels of NG2 are less sensitive to oxidative stress than those with lower NG2 expression. Moreover, reducing NG2 expression by siRNA increases cell death in response to oxidative stress. Binding of NG2 to OMI/HtrA2 may thus help protect cells against oxidative stress-induced cell death in our experimental paradigm [73].
NG2 has also been described as an inhibitor of axonal growth based on the use of the NG2 molecule rather than NG2-expressing cells [74]. But, when using NG2-expressing cells rather than the isolated NG2 proteoglycan in coculture experiments, Yang and coworkers showed that NG2 positive cells derived from postnatal rat brain promoted axon outgrowth on hippocampal neurons rather than inhibiting it [74]. In fact, growth cones of hippocampal neurons preferentially formed contacts with NG2 cells, whereas coculture with MAG-expressing cells or mature OLs led to growth cone retraction. In addition, cocultures of neurons and astrocytes promoted less axon outgrowth than NG2 cells what led the authors to conclude that NG2 cells are responsible for the axonal growth-promoting effects [74]. These authors also ascertained that NG2 cells express laminin and fibronectin at the cell surface [74], extracellular molecules known to exert growth-promoting effects and which the authors suggested to be responsible for the extensive contacts between growth cones and NG2-positive cells that were observed in the developing corpus callosum in vivo. Using the myelin deficient (md) mutant rat, we have previously demonstrated that upon exposure to TSC1, nestin expressing cells rapidly mobilize from the SVZ into the brain parenchyma, some proliferate and others do not. Then 3 days later in the brain of the md animal, these cells had migrated widely and dispersed throughout the caudate putamen, and they all expressed the early OL marker transferrin (Tf) [27]. With time these cells progressed in the OL lineage by acquiring more OL markers such as PDGF receptor alpha, NG2 and myelin markers. Two weeks after a single administration of Tf + IGF-1 (TSC1) md rats showed myelinated fibers both, in the lateral and contralateral hemispheres [26]. In the same paper we showed that Tf upregulates MBP gene expression and that in combination with IGF-1 the effect is synergistic enhancing myelination. This information together with the findings [64] discussed above leave no doubt that the NG2-expressing cells in this study were OLs.
The fundamental features of WD include demyelination, myelin clearance and axonal degeneration or transection. These are some of the components of secondary effects after SCI [75]. It has been reported that demyelination occurs over the first week after SCI and the next step is remyelination which is completed between 2 and 4 weeks [76, 77]. It is possible that surviving NG2-positive cells were induced to divide early after injury by non-myelinated axons within the injury area. Moreover, the timing of NG2-positive cells expression suggests a possible role for these cells in remyelinating the injured area [78]. Therefore, NG2 seems to play an important role to achieve remyelination after SCI.
Wallerian degeneration is a component of the secondary phase of SCI where there is cellular damage and cell death, demyelination and axonal degeneration [78].
Since TSC1 contains IGF1, it may be a potential action of TSC1 on promoting mitochondria homeostasis and energy management, which in turn, can play a role on reducing tissue damage. According to the diagram, TSC1 can reduce mitochondrial damage involving cytochrome C release. These in turn can act on reducing apoptosis by inactivation of caspase-9. This mechanisms work in conjunction with the many roles discussed for transferrin synergizing their effect and emphasizing the suitable combination between IGF-1 and transferrin for neural repair purposes (see Fig. 8 in Appendix).
Fig. 8.
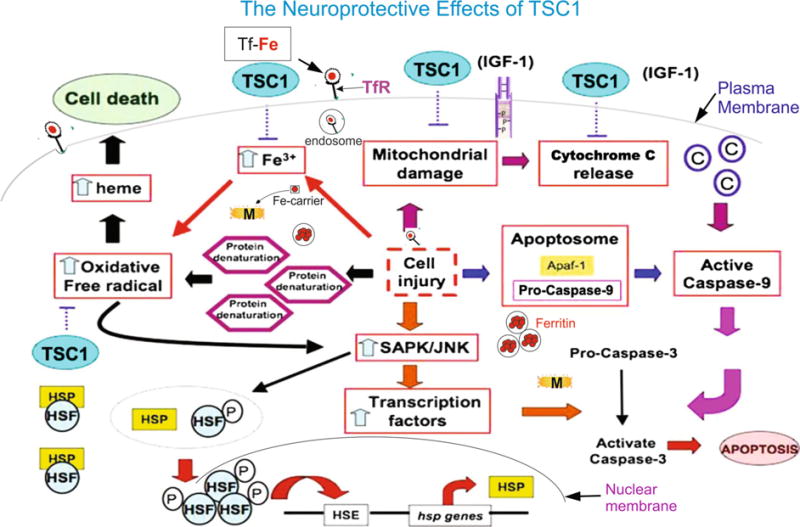
Schematic representation of some mechanisms that may be modulated by TSC1 with respect to apoptosis and iron homeostasis
Conclusion
Conditioning the cellular micro-environment with different factors, such as brain-derived neurotrophic factor (BDNF), or epidermal growth factor receptor (EGFR) [38, 79] to overcome injury or disease, is an ideal approach. What is critical for future therapeutic interventions based on endogenous neural stem cells/progenitors, it is the specific choices on the combination of factors and the time of their administration. We have previously reported that the endogenous regeneration potential the spinal cord possesses can be sustained and brought to functional recovery of hindlimb movement by implanting a hydrogel to provide support to newly formed cells and cells migrating towards the lesion, thus rebuilding the lost tissue after double transaction [80]. The present study demonstrates that the contribution of TSC1 to recovery of the tissue after SCI is on one hand the preservation of cells and the cytoarchitecture of the spinal cord and, on the other, it promotes the regeneration of the cell populations that were lost as a consequence of the crush itself and the secondary damage. Interestingly, TSC1 appears to have accelerated demyelination while preserving the axons from transection, a phenomenon that took approximately 1 week to clear pre-existing damaged myelin. Moreover, the remyelination process appears to have actively started during the second week after the administration of TSC1 when OLs appeared to be contacting numerous axons suggesting that within one more week most axons would have been myelinated by endogenous OLs. While this study did not include examination of the origin of these re-myelinating OLs (i.e. preexisting OL progenitors or, nestin-derived OLPs, or both), we can say that TSC1 acutely applied just once, on the surface of the spinal cord, has long-lasting effects on the spinal cord by attenuating cell and myelin loss in certain regions, supporting demyelinated axons and accelerating remyelination and recruitment of nestin-expressing progenitors at all time points studied. Considering that here we used 6 months old mice that would be equivalent to 20–35 years of age in humans, these results augur well for a successful outcome on the clinical use of TSC1 as soon as possible after the lesion occurs. Moreover, the use of a combinatorial therapy that would include a support such as a hydrogel as we previously reported, where nestin-expressing cells continue to migrate 21 months after spine reconstruction [80] together with TSC1 as described herein, and an assisted exercise therapy program that would enhance even more the numbers of nestin-positive cells and OLPs as we previously reported [81], appears to be the most promising approach currently available to prevent WD and enhance a robust functional recovery from SCI even in mature adults.
Acknowledgments
We are grateful to Dr. Michael Sofroniew and his team for teaching us the spinal cord injury technique. We also thank Dr. Nuanchan Jutapakdeegul for their contribution to improve the quality of the article, Dorwin Birt for help with computer support and figure composites. This work was supported by NIH Grants: HD004612, HD006576 and scholarship from the University Development Program (UDP), Thailand.
Appendix
There are various signaling pathways involved in IGF-1 normal activity in the CNS. Ligand binding to IGF-1 receptors (IGF1Rs) triggers receptor autophosphorylation and association with docking proteins (not shown; for review see [82]. Upon cell injury (center of diagram) apoptosome formation is triggered by the release of cytochrome c from damaged mitochondria in response to either intrinsic or extrinsic cell death stimuli. The cytochrome binds to ATP-dependent proteolysis factor 1(Apf-1) monomeres which then create APF heptameres or heptamere apoptosomes. Recruitment of pro-caspase-9, either in the apoptosome or binding directly to Apf1, activates caspase 9, cleaves pro-caspase-3 and activates it. Caspase-3 breaks down cellular components for their recycling. Based on the reports from Wood and collaborators one can infer that in our experimental paradigm exogenous IGF-1 contained in TSC1 may prevent mitochondrial damage and cytochrome release blocking the caspase chain of events that lead to apoptosis (see text for details). The amount of iron (left part of the diagram) in cells is controlled by the cell surface transferrin receptor (TfR)-mediated uptake of iron as transferrin-iron. [83] TfR synthesis is regulated by interaction of the iron regulatory protein (IRP) with the iron-responsive element (IRE) present on the 3′ untranslated region of TfR mRNA. IRPs serve as sensors of cellular iron. [83–85]. The cellular oxidative damage caused by reactive oxygen species and reactive nitrogen species is critically controlled by cellular iron homeostasis [85]. Exposure to H2O2, enhances the expression of TfR mRNA, where TfR appears to be the link between oxidative stress and TfR-mediated iron uptake. Transferrin (Tf) (left part of the diagram) is the iron carrier glycoprotein for all normal cells of mammalians. In circulation, it is saturated to around 30 % in adults. Thus, allowing for the chelation of iron radicals released after cell injury. In the CNS, Tf is synthesized by OLs and choroid plexus. Iron (Fe) is found in OLs and myelin in high density and both iron and Tf are required for myelin production [22, 85, 86]. As discussed by [87] and other authors, excess and/of free iron can be cytotoxic, by catalyzing the production of hydoxyl radicals from hydrogen peroxide [88]. Increased iron concentrations in the brain have been shown in neurodegenerative diseases, e.g. Alzheimer’s disease and Parkinson’s disease [89, 90] where oxidative stress has been proposed as a pathogenic mechanism of neurodegeneration. Conversely, brain Tf levels decrease with age and the decrease is dramatic when Alzheimer’s and Parkinson’s disease are superimposed on the aging process [91]. This stoichiometry of the iron related proteins Tf, ferritin and transferrin receptor (TFR) is essential to maintain CNS function. Upon OLPs and or myelin breakdown free iron is released. In our experimental model, Tf may act as a transient chelator while “Ferritin”, the iron storage protein, is synthesized (circle with red dots) helping prevent oxidative stress and lipid peroxidation. Heat shock factors (HSF) are transcriptional activators of heat shock HSP (P) genes [92]. These activators bind specifically to Heat Shock sequence Elements (HSE) throughout the genome [93–98]. The Heat shock factor (HSF/HSF1) mediates the stress-induced expression of heat shock or stress proteins (HSPs). HSF/HSF1 is inactive in unstressed cells and it is activated during stress. Activation includes its hyperphosphorylation. Twelve Serine residues are phosphorylated in heat-activated HSF1 from which phosphorylation of HSF1 residue Ser326 plays a critical role in the induction of the factor’s transcriptional competence by heat stress and chemical stress. The functional role of other newly identified phosphor Ser residues remains unknown [99]. The Heat Shock sequence Element is highly conserved from yeast to humans. Heat shock factors (HSF) are transcriptional activators of heat shock genes. These activators bind specifically to Heat Shock sequence Elements (HSE) throughout the genome [93]. Although not shown in the diagram, eat shock proteins bind to the misfolded proteins and dissociate from HSF-1. This allows HSF1 to form trimers and translocate to the cell nucleus and activate transcription [7]. Its function is not only critical to overcome the proteotoxic effects of thermal stress, but also needed for proper animal development and the overall survival of cancer cells [100, 101]. It is worth mentioning that we have previously shown a synergistic effects of TSC1 as a neuroprotective agent. In the present work, we showed that a single dose of TSC1 was necessary and sufficient to increase the number of HSP-32-expressing cells most likely by HSF phosphorylation, hence promoting mitochondria homeostasis and energy management (Fig. 8).
References
- 1.Nelson E, Gertz SD, Rennels ML, Ducker TB, Blaumanis OR. Spinal cord injury. The role of vascular damage in the pathogenesis of central hemorrhagic necrosis. Arch Neurol. 1997;34:332–333. doi: 10.1001/archneur.1977.00500180026005. [DOI] [PubMed] [Google Scholar]
- 2.Lu J, Ashwell KW, Waite P. Advances in secondary spinal cord injury: role of apoptosis. Spine. 2000;25(14):1859–1866. doi: 10.1097/00007632-200007150-00022. [DOI] [PubMed] [Google Scholar]
- 3.Chang H, Wo JP, Tzeng SF. Neuroprotection of glial cell line-derived neurotrophic factor in damaged spinal cords following contusive injury. J Neuro Res. 2002;69:397–405. doi: 10.1002/jnr.10303. [DOI] [PubMed] [Google Scholar]
- 4.McDonald JW, Sadowsky C. Spinal-cord injury. Lancet. 2002;359:417–425. doi: 10.1016/S0140-6736(02)07603-1. [DOI] [PubMed] [Google Scholar]
- 5.Beattie MS, Bresnahan JC, Komon J, et al. Endogenous repair after spinal cord contusion injuries in the rat. Exp Neurol. 1997;148(2):453–463. doi: 10.1006/exnr.1997.6695. [DOI] [PubMed] [Google Scholar]
- 6.Grossman SD, Rosenberg LJ, Wrathall JR. Relationship of altered glutamate receptor subunit mRNA expression to acute cell loss after spinal cord contusion. Exp Neurol. 2001;168(2):283–289. doi: 10.1006/exnr.2001.7629. [DOI] [PubMed] [Google Scholar]
- 7.McGraw J, Hiebert GW, Steeves JD. Modulating astrogliosis after neurotrauma. J Neurosci Res. 2001;63(2):109–158. doi: 10.1002/1097-4547(20010115)63:2<109::AID-JNR1002>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg LJ, Zai LJ, Wrathall JR. Chronic alterations in the cellular composition of spinal cord white matter following contusion injury. Glia. 2005;49(1):107–120. doi: 10.1002/glia.20096. [DOI] [PubMed] [Google Scholar]
- 9.Sharma HS, Gordh T, Wiklund L, Mohanty S, Sjoquist PO. Spinal cord injury induced heat shock protein expression is reduced by an antioxidant compound H-290/51. An experimental study using light and electron microscopy in the rat. J Neural Trans. 2005;113:521–536. doi: 10.1007/s00702-005-0405-2. [DOI] [PubMed] [Google Scholar]
- 10.Sharma HS. A select combination of neurotrophins enhances neuroprotection and functional recovery following spinal cord injury. Ann N Y Acad Sci. 2007;1122:95–111. doi: 10.1196/annals.1403.007. [DOI] [PubMed] [Google Scholar]
- 11.Yenari MA. Heat shock proteins and neuroprotection. Adv Exp Med Biol. 2002;513:281–299. doi: 10.1007/978-1-4615-0123-7_10. [DOI] [PubMed] [Google Scholar]
- 12.Houenou LJ, Li L, Lei M, Kent CR, Tytell M. Exogenous heat shock cognate protein Hsc 70 prevents axotomy-induced death of spinal sensory neurons. Cell Stress Chaperones. 1996;1(3):161–166. doi: 10.1379/1466-1268(1996)001<0161:ehscph>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mautes AE, Bergeron M, Sharp FR, Panter SS, Weinzierl M, Guenther K, Noble LJ. Sustained induction of heme oxygenase-1 in the traumatized spinal cord. Exp Neurol. 2000;166(2):254–265. doi: 10.1006/exnr.2000.7520. [DOI] [PubMed] [Google Scholar]
- 14.Carama NOS, Soares MP. Heme oxygenase-1 (HO-1), a protective gene that prevents chronic graft dysfunction. Free Radic Biol Med. 2005;38(4):426–435. doi: 10.1016/j.freeradbiomed.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 15.Woerly S, Awosika O, Zhao P, Agbo C, Gomez-Pinilla F, de Vellis J, Espinosa-Jeffrey A. Expression of heat shock protein (HSP)-25 and HSP-32 in the rat spinal cord reconstructed with Neurogel. Neurochem Res. 2005;30(6–7):721–735. doi: 10.1007/s11064-005-6866-8. [DOI] [PubMed] [Google Scholar]
- 16.Kitagawa H, Hayashi T, Mitsumoto Y, Koga N, Itoyama Y, Abe K. Reduction of ischemic brain injury by topical application of glial cell line-derived neurotrophic factor after permanent middle cerebral artery occlusion in rats. Stroke. 1998;29(7):1417–1422. doi: 10.1161/01.str.29.7.1417. [DOI] [PubMed] [Google Scholar]
- 17.Lee TT, Green BA, Dietrich WD, Yezierski RP. Neuroprotective effects of basic fibroblast growth factor following spinal cord contusion injury in the rat. J Neurotrauma. 1999;16(5):347–356. doi: 10.1089/neu.1999.16.347. [DOI] [PubMed] [Google Scholar]
- 18.Watabe K, Ohashi T, Sakamoto T, Kawazoe Y, Takeshima T, Oyanagi K, Inoue K, Eto Y, Kim SU. Rescue of lesioned adult rat spinal motoneurons by adenoviral gene transfer of glial cell line-derived neurotrophic factor. J Neurosci Res. 2000;60:511–519. doi: 10.1002/(SICI)1097-4547(20000515)60:4<511::AID-JNR10>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 19.Sharma HS, Nyberg F, Gordh T, Alm P, Westman J. Neurotrophic factors influence upregulation of constitutive isoform of heme oxygenase and cellular stress response in the spinal cord following trauma. Amino Acids. 2000;19:351–361. [PubMed] [Google Scholar]
- 20.Ozdinler PH, Macklis JD. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nature Neurosci. 2006;9:1371–1381. doi: 10.1038/nn1789. [DOI] [PubMed] [Google Scholar]
- 21.Espinosa de los Monteros A. Chiapelli F, Fisher RS, de Vellis J. Transferrin: an early marker of oligodendrocytes in culture. Int J Dev Neurosci. 1988;6:167–175. doi: 10.1016/0736-5748(88)90042-1. [DOI] [PubMed] [Google Scholar]
- 22.Espinosa de los Monteros A. Kumar S, Zhao P, Huang CJ, Nazarian R, Pan T, Scully S, Chang R, de Vellis J. Transferrin is an essential factor for myelination. Neurochem Res. 1999;24(2):235–248. doi: 10.1007/s11064-004-1826-2. [DOI] [PubMed] [Google Scholar]
- 23.Morgan EH. Molecular iron processing. J Gastroenterol Hepatol. 1996;11:1027–1030. doi: 10.1111/j.1440-1746.1996.tb00028.x. [DOI] [PubMed] [Google Scholar]
- 24.Morgan EH, Moos T. Mechanism and developmental changes in iron transport across the blood–brain barrier. Dev Neurosci. 2002;24:106–113. doi: 10.1159/000065699. [DOI] [PubMed] [Google Scholar]
- 25.Moos T. Brain iron homeostasis. Dan Med Bull. 2002;49:279–301. [PubMed] [Google Scholar]
- 26.Espinosa-Jeffrey A, Zhao PM, Awosika O, Huang A, Chang R, de Vellis J. Transferrin regulates transcription of the MBP gene and its action synergizes with IGF-1 to enhance myelino-genesis in the md rat. Dev Neurosci. 2002;24(2–3):227–241. doi: 10.1159/000065698. [DOI] [PubMed] [Google Scholar]
- 27.Espinosa-Jeffrey A, Zhao P, Awosika W, Wu N, Macias F, Cepeda C, Levine M, de Vellis J. Activation, proliferation and commitment of endogenous, stem/progenitor cells to the oligodendrocyte lineage by a combination of neurotrophic factors in a rat model of dysmyelination. Dev Neurosci. 2006;28(6):488–498. doi: 10.1159/000095111. [DOI] [PubMed] [Google Scholar]
- 28.Paez PM, Garcia CI, Soto EF, Pasquini JM. Apotransferrin decreases the response of oligodendrocyte progenitors to PDGF and inhibits the progression of the cell cycle. Neurochem Inter. 2006;49:359–371. doi: 10.1016/j.neuint.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Woerly S, Doan VD, Evans-Martin F, Paramore CG, Peduzzi JD. Spinal cord reconstruction using NeuroGel implants and functional recovery after chronic injury. J Neurosci Res. 2001;66(6):1187–1197. doi: 10.1002/jnr.1255. [DOI] [PubMed] [Google Scholar]
- 30.Woerly S, Doan VD, Sosa N, de Vellis J, Espinosa-Jeffrey A. Prevention of gliotic scar formation by NeuroGel allows partial endogenous repair of transected cat spinal cord. J Neu-rosci Res. 2004;75(2):262–272. doi: 10.1002/jnr.10774. [DOI] [PubMed] [Google Scholar]
- 31.Espinosa-Jeffrey A, Oregel K, Wiggins L, Valera R, Bosnoyan K, Agbo C, Awosika O, Zhao P, de Vellis J, Woerly S. Strategies for endogenous spinal cord repair: HPMA hydrogel to recruit migrating endogenous stem cells. Adv Exp Med Biol. 2012;760:25–52. doi: 10.1007/978-1-4614-4090-1_3. [DOI] [PubMed] [Google Scholar]
- 32.Espinosa-Jeffrey A, Barajas S, Arrazola A, Taniguchi A, Zhao P, Bokhoor P, Holley S, Dejarme D, Chu B, Cepeda C, Levine M, Gressens P, Feria-Velasco A, de Vellis J. White matter loss in a mouse model of periventricular leukomalacia is rescued by trophic factors. Brain Sci. 2013;3(4):1461–1482. doi: 10.3390/brainsci3041461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Espinosa-Jeffrey A, Arrazola A, Chu B, Taniguchi A, Barajas S, Bokhoor P, Garcia J, Feria-Velasco A, de Vellis J. J Neurosci Res. 2015 doi: 10.15761/imm.1000228. (accepted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamaguchi M, Saito H, Suzuki M, Mori K. Visualization of neurogenesis in the central nervous system using nestin promoter-GFP transgenic mice. Neuroreport. 2000;11:1991–1996. doi: 10.1097/00001756-200006260-00037. [DOI] [PubMed] [Google Scholar]
- 35.Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blesch A, Tuszynski MH. Spinal cord injury: plasticity, regeneration and the challenge of translational drug development. Trends Neurosci. 2009;32:41–47. doi: 10.1016/j.tins.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 37.Yoon C, Tuszynski MH. Frontiers of spinal cord and spine repair: experimental approaches for repair of spinal cord injury. Adv Exp Med Biol. 2012;760:1–15. doi: 10.1007/978-1-4614-4090-1_1. [DOI] [PubMed] [Google Scholar]
- 38.Kumar S, Biancotti JC, Matalon R, de Vellis J. Lack of aspartoacylase activity disrupts survival differentiation of neural progenitors and oligodendrocytes in a mouse model of Canavan disease. J Neurosci Res. 2009;87(15):3415–3427. doi: 10.1002/jnr.22233. [DOI] [PubMed] [Google Scholar]
- 39.Espinosa-Jeffrey A, Hitoshi S, Zhao P, Awosika O, Agbo C, Olaniyan E, Garcia J, Valera R, Thomassian A, Chang-Wei R, Yamaguchi M, de Vellis J, Ikenaka K. Functional central nervous system myelin repair in an adult mouse model of demyelination caused by proteolipid protein overexpression. J Neurosci Res. 2010;88(8):1682–1694. doi: 10.1002/jnr.22334. [DOI] [PubMed] [Google Scholar]
- 40.Richter-Landsberg C, Goldbaum O. Stress proteins in neural cells: functional roles in health and disease. Cell Mol Life Sci. 2003;60:337–349. doi: 10.1007/s000180300028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panahian N, Maines MD. Site of injury-directed induction of heme oxygenase-1 and -2 in experimental spinal cord injury: differential functions in neuronal defense mechanisms? J Neurochem. 2001;76:539–554. doi: 10.1046/j.1471-4159.2001.00023.x. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Tachibana T, Dai Y, Kondo E, Fukuoka T, Yamanaka H, Noguchi K. Heme oxygenase-1 expression after spinal cord injury: the induction in activated neutrophils. J Neurotrauma. 2002;19:479–490. doi: 10.1089/08977150252932424. [DOI] [PubMed] [Google Scholar]
- 43.Goldbaum O, Richter-Landsberg C. Stress proteins in oligodendrocytes: differential effects of heat shock and oxidative stress. J Neurochem. 2001;78(6):1233–1242. doi: 10.1046/j.1471-4159.2001.00507.x. [DOI] [PubMed] [Google Scholar]
- 44.Cizkova D, Lukacova N, Marsala M, Kafka J, Lukac I, Jergova S, Cizek M, Marsala J. Experimental cauda equina compression induces HSP70 synthesis in dog. Physiol Res. 2005;54:349–356. [PubMed] [Google Scholar]
- 45.Carmel JB, Galate A, Soteropoulos P, Tolias P, Recce M, Young W, Hart RP. Spinal cord injury: plasticity, regeneration and the challenge of translational drug development. Physiol Genom. 2001;7:201–213. doi: 10.1152/physiolgenomics.00074.2001. [DOI] [PubMed] [Google Scholar]
- 46.Ulrich HF. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–580. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 47.Michels AA, Kanon B, Konings AWT, Ohtsuka K, Bensaude O, Kampinga HH. Hsp70 and Hsp40 chaperone activities in the cytoplasm and the nucleus of mammalian cells. J Giol Chem. 1997;272:33283–33289. doi: 10.1074/jbc.272.52.33283. [DOI] [PubMed] [Google Scholar]
- 48.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255(5052):1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 49.Steinert PM, Chou YH, Prahlad V, Parry DA, Marekov LN, Wu KC, Jang SI, Goldman RD. A high molecular weight intermediate filament-associated protein in BHK-21 cells is nestin, a type VI intermediate filament protein. Limited co-assembly in vitro to form heteropolymers with type III vimentin and type IV alpha-internexin. J Biol Chem. 1999;274(14):9881–9890. doi: 10.1074/jbc.274.14.9881. [DOI] [PubMed] [Google Scholar]
- 50.Tohyama T, Lee VM, Rorke LB, Marvin M, McKay RD, Tro-janowski JQ. Nestin expression in embryonic human neuroepithelium and in human neuroepithelial tumor cells. Lab Invest. 1992;66(3):303–313. [PubMed] [Google Scholar]
- 51.Suzuki S, Namiki J, Shibata S, Mastuzaki Y, Okano H. The neural stem/progenitor cell marker nestin is expressed in proliferative endothelial cells, but not in mature vasculature. J Histochem Cytochem. 2010;58(8):721–730. doi: 10.1369/jhc.2010.955609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukuda S, Kato F, Tozuka Y, Yamaguchi M, Miyamoto Y, Hisatsune T. Two distinct subpopulations of nestin-positive cells in adult mouse dentate gyrus. J Neurosci. 2003;23(28):9357–9366. doi: 10.1523/JNEUROSCI.23-28-09357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 54.Shin YJ, Kim HL, Park JM, Cho JM, Kim SY, Lee MY. Characterization of nestin expression and vessel association in the ischemic core following focal cerebral ischemia in rats. Cell Tissue Res. 2013;351(3):383–395. doi: 10.1007/s00441-012-1538-x. [DOI] [PubMed] [Google Scholar]
- 55.Mothe AJ, Tator CH. Proliferation, migration, and differentiation of endogenous ependymal region stem/progenitor cells following minimal spinal cord injury in the adult rat. Neuroscience. 2005;131:177–187. doi: 10.1016/j.neuroscience.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 56.Fernandez AM, Torres-Aleman I. The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci. 2012;13(4):225–239. doi: 10.1038/nrn3209. [DOI] [PubMed] [Google Scholar]
- 57.McMorris FA, Duboi-Dalcq M. Insulin-like growth factor I promotes cell proliferation and oligodendroglial commitment in rat glial progenitor cells developing in vitro. J Neurosci Res. 1988;21:199–209. doi: 10.1002/jnr.490210212. [DOI] [PubMed] [Google Scholar]
- 58.McMorris FA, McKinnon RD. Regulation of oligodendrocyte development and CNS myelination by growth factors: prospects for therapy of demyelinating disease. Brain Pathol. 1996;6:313–329. doi: 10.1111/j.1750-3639.1996.tb00858.x. [DOI] [PubMed] [Google Scholar]
- 59.Werner H, Woloschak M, Adamo M, She-Orr Z, Roberts CT, LeRoith D. Developmental regulation of the rat insulin-like growth factor I receptor gene. Proc Natl Acad Sci USA. 1989;86:7451–7455. doi: 10.1073/pnas.86.19.7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Madathil SK, Evans HN, Saatman KE. Temporal and regional changes in IGF-1/IGF-1R signaling in the mouse brain after traumatic brain injury. J Neurotra. 2010;27:95–107. doi: 10.1089/neu.2009.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anderson MF, Aberg MA, Nilsson M, Eriksson PS. Insulin-like growth factor-I and neurogenesis in the adult mammalian brain. Brain Res Dev Brain Res. 2000;134:115–122. doi: 10.1016/s0165-3806(02)00277-8. [DOI] [PubMed] [Google Scholar]
- 62.Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dubois-Dalcq Murray K. Why are growth factors important in oligodendrocyte physiology? Pathol Biol (Paris) 2000;48:80–86. [PubMed] [Google Scholar]
- 64.Hsieh J, Aimone JB, Kaspar BK, Kuwabara T, Kakashima K, Gage FH. IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J Cell Biol. 2004;164:111–122. doi: 10.1083/jcb.200308101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Espinosa de los Monteros A. Pena LA, de Vellis J. Does transferrin have a special role in the nervous system? J Neurosci Res. 1989;24:125–136. doi: 10.1002/jnr.490240202. [DOI] [PubMed] [Google Scholar]
- 66.Aizenman Y, Weichsel ME, de Vellis J. Changes in insulin and transferrin requirements of pure brain neuronal cultures during embryonic development. Proc Natl Acad Sci USA. 1986;83:2263–2266. doi: 10.1073/pnas.83.7.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saneto RP, de Vellis J. Characterization of cultured rat oligodendrocytes proliferating in a serum-free, chemically defined médium. Proc Natl Acad Sci USA. 1985;82:3509–3513. doi: 10.1073/pnas.82.10.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Espinosa de los Monteros A. Zhang M, Gordon MN, Kumar S, Scully S, de Vellis J. The myelin-deficient rat mutant: partial recovery of oligodendrocyte maturation in vitro. Dev Neurosci. 1990;12:326–339. doi: 10.1159/000111861. [DOI] [PubMed] [Google Scholar]
- 69.Barres BA, Jacobson MD, Schmid R, Sendtner M, Raff MC. Does oligodendrocyte survival depend on axons? Curr Biol. 1993;3:489–497. doi: 10.1016/0960-9822(93)90039-q. [DOI] [PubMed] [Google Scholar]
- 70.Guan J, Bennet L, Gluckman PD, Gunn AJ. Insulin-like growth factor-1 and post-ischemic brain injury. Prog Neurobio. 2003;70:443–462. doi: 10.1016/j.pneurobio.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 71.Aquino JB, Musolino PL, Coronel MF, Villar MJ, Setton-Avruj CP. Nerve degeneration is prevented by a single intraneural apotransferrrin injection into colchicines-injured sciatic nerves in the rat. Brain Res. 2006;30:80–91. doi: 10.1016/j.brainres.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 72.Nishiyama A, Komitova M, Suzuki R, Zhu X. Polyden-drocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- 73.Maus F, Sakry D, Binamé F, Karram K, Rajalingam K, Watts C, Heywood R, Krüger R, Stegmüller J, Werner HB, Nave KA, Krämer-Albers EM, Trotter J. The NG2 proteoglycan protects oligodendrocyte precursor cells against oxidative stress via interaction with OMI/HtrA2. PLoS ONE. 2015;10:e0137311. doi: 10.1371/journal.pone.0137311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang Z, Suzuki R, Daniels SB, Brunquell CB, Sala CJ, Nish-iyama A. NG2 glial cells provide a favorable substrate for growing axons. J Neurosci. 2006;14:3829–3839. doi: 10.1523/JNEUROSCI.4247-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bareyre FM, Schwab ME. Inflammation, degeneration and regeneration in the injury spinal cord: insights from DNA microarrays. Trend Neurosci. 2003;26:555–563. doi: 10.1016/j.tins.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 76.Totoiu MO, Keirstead HS. Spinal cord injury is accompanied by chronic progressive demyelination. J Comp Neurol. 2005;486:373–383. doi: 10.1002/cne.20517. [DOI] [PubMed] [Google Scholar]
- 77.McTigue D, Wei P, Stokes BT. Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J Neurosci. 2001;21:3392–3400. doi: 10.1523/JNEUROSCI.21-10-03392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sekhon LH, Fehlings MG. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine. 2001;26:S2–S12. doi: 10.1097/00007632-200112151-00002. [DOI] [PubMed] [Google Scholar]
- 79.Aguirre A, Gallo V. Reduced EGFR signaling in progenitor cells of the adult subventricular zone attenuates oligodendrogenesis after demyelination. Neuron Glia Biol. 2007;3:209–220. doi: 10.1017/S1740925X08000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Espinosa de los Monteros A. Zhao PM, de Vellis J. In vitro injury model for oligodendrocytes: development, injury, and recovery. Micro Res Tech. 2001;52:719–730. doi: 10.1002/jemt.1056. [DOI] [PubMed] [Google Scholar]
- 81.Krityakiarana W, Espinosa-Jeffrey A, Ghiani C, Zhao M, Topaldjikian N, Gomez-Pinilla F, Yamaguchi M, Kotchabhakdi N, de Vellis J. Voluntary exercise increases oligodendrogenesis in spinal cord. Int J Neurosci. 2010;120(4):280–290. doi: 10.3109/00207450903222741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Katic M, Kahn CR. The role of insulin and IGF-1 signaling in longevity. Cell Mol Life Sci. 2005;62(3):320–343. doi: 10.1007/s00018-004-4297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Klausner RD, Ashwell G, van Renswoude J, Harford JB, Bridges KR. Binding of apotransferrin to K562 cells: explanation of the transferrin cycle. Proc Natl Acad Sci USA. 1983;80:2263–2266. doi: 10.1073/pnas.80.8.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klausner RD, Van Renswoude J, Ashwell G, Kempf C, Schechter AN, Dean A, Bridges KR. Receptor-mediated endocytosis of transferrin in K562 cells. J Biol Chem. 1983;258:4715–4724. [PubMed] [Google Scholar]
- 85.Pantopoulos K, Hentze MW. Rapid responses to oxidative stress mediated by iron regulatory protein. Embo J. 1995;14:2917–2924. doi: 10.1002/j.1460-2075.1995.tb07291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hill JM, Switzer RC., 3rd The regional distribution and cellular localization of iron in the rat brain. Neuroscience. 1984;11(3):595–603. doi: 10.1016/0306-4522(84)90046-0. [DOI] [PubMed] [Google Scholar]
- 87.Takeda A, Takatsuka K, Connor JR, Oku N. Abnormal iron delivery to the bone marrow in neonatal hypotransferrinemic mice. Biometals. 2002;15:33–36. doi: 10.1023/a:1013133217085. [DOI] [PubMed] [Google Scholar]
- 88.Zaleska MM, Floyd R. Regional lipid peroxidation in rat brain in vitro: possible role of endogenous iron. Neurochem Res. 1985;10:397. doi: 10.1007/BF00964608. [DOI] [PubMed] [Google Scholar]
- 89.Double KL, Gerlach M, Youdim MB, Riederer P. Impaired iron homeostasis in Parkinson’s disease. J Neural Transm Suppl. 2000;60:37–58. doi: 10.1007/978-3-7091-6301-6_3. [DOI] [PubMed] [Google Scholar]
- 90.Pinero DJ, Hu J, Connor JR. Alterations in the interaction between iron regulatory proteins and their iron responsive element in normal and Alzheimer’s diseased brains. Cell Mol Biol (Noisy-le-grand) 2000;46:761–776. [PubMed] [Google Scholar]
- 91.Loeffler DA, Connor JR, Juneau PL, Snyder BS, Kanaley L, DeMaggio AJ, Nguyen H, Brickman CM, LeWitt PM. Transferrin and iron in normal, Alzheimer’s disease, and Parkinson’s disease brain regions. J Neurochem. 1995;65:710–716. doi: 10.1046/j.1471-4159.1995.65020710.x. [DOI] [PubMed] [Google Scholar]
- 92.Clos J, Westwood JT, Becker PB, Wilson S, Lambert K, Wu C. Molecular cloning and expression of a hexameric Drosophila heat shock factor subject to negative regulation. Cell. 1990;63(5):1085–1097. doi: 10.1016/0092-8674(90)90511-c. [DOI] [PubMed] [Google Scholar]
- 93.Guertin MJ, Lis JT. Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet. 2010;6(9):e1001114. doi: 10.1371/journal.pgen.1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guertin MJ, Petesch SJ, Zobeck KL, Min IM, Lis JT. Drosophila heat shock system as ageneral model to investigate transcriptional regulation. Cold Spring Harb Symp Quant Biol. 2010;75:1–9. doi: 10.1101/sqb.2010.75.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sorger PL. Heat shock factor and the heat shock response. Cell. 1991;65(3):363–366. doi: 10.1016/0092-8674(91)90452-5. [DOI] [PubMed] [Google Scholar]
- 96.Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993;259(5100):1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- 97.Rabindran SK, Giorgi G, Clos J, Wu C. Molecular cloning and expression of a human heat shock factor, HSF1. Proc Natl Acad Sci USA. 1991;88(16):6906–6910. doi: 10.1073/pnas.88.16.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Prahlad V, Morimoto RI. Intergrating the stress response: lessons for neurodegenerative diseases from C. elegans. Trends Cell Biol. 2008;19(2):52–61. doi: 10.1016/j.tcb.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guettouche T, Boellmann F, Lane WS, Richard Voellmy. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005;6:4. doi: 10.1186/1471-2091-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Salamanca HH, Fuda N, Shi H, Lis JT. An RNA aptamer perturbs heat shock transcription factor activity in drosophila melanogaster. Nucleic Acids Res. 2011;39(15):6729–6740. doi: 10.1093/nar/gkr206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Salamanca HH, Antonyak MA, Cerione RA, Shi H, Lis JT. Inhibiting heat shock factor 1 in human cancer cells with a potent RNA aptamer. PLoS One. 2014;9(5):e96330. doi: 10.1371/journal.pone.0096330. [DOI] [PMC free article] [PubMed] [Google Scholar]