

Abstract
Oral ketoconazole therapy is commonly associated with serious hepatotoxicity. Improving ocular drug delivery could be sufficient to treat eye fungal infections. The purpose of this study was to develop optimized ketoconazole poly(lactide-co-glycolide) nanoparticles (NPs) with subsequent loading into in situ gel (ISG) formulation for ophthalmic drug delivery. Three formulation factors were optimized for their effect on particle size (Y1) and entrapment efficiency (Y2) utilizing central composite experimental design. Interaction among components was studied using differential scanning calorimetry (DSC) and Fourier transform infrared (FTIR) spectroscopy. Ketoconazole crystalline state was studied using X-ray powder diffraction. Six different polymeric ISG formulations were prepared and loaded with either optimized NPs or a pure drug. The prepared ISG formulations were characterized for in vitro gelation, drug release and antifungal activity. The permeation through human epithelial cell line was also investigated. The results revealed that all the studied formulation parameters significantly affected Y1 and Y2 of the developed NPs. DSC and FTIR studies illustrated compatibility among NP components, while there was a change from the crystalline state to the amorphous state of the NPs. The in vitro release from the ISG formulations loaded with drug NPs showed sustained and enhanced drug release compared to pure drug formulations. In addition, ISG loaded with NPs showed enhanced anti-fungal activity compared to pure drug formulations. Alginate–chitosan ISG formulation loaded with optimized ketoconazole NPs illustrated higher drug permeation through epithelial cell lines and is considered as an effective ophthalmic drug delivery in the treatment of fungal eye infections.
Keywords: ketoconazole, PLGA, nanoparticles, ISG, ophthalmic delivery
Introduction
Ketoconazole is a synthetic imidazole drug used primarily to treat different types of body fungal infections. The drug works by inhibiting the enzyme cytochrome P450 14α-demethylase, which contributes to the synthesis of the fungal ergosterol.1 Recently, the US Food and Drug Administration (FDA) announced a warning that oral treatment with ketoconazole can cause severe liver damages and also adrenal gland problems.2 Unlike the oral administration, topical drug formulations have not been associated with these adverse effects. When ketoconazole is administered for treatment of fungal eye infections, it is characterized by a very short duration of action since its elimination half-life in the aqueous humor and cornea is ~19 and 43 min, respectively.3,4 Although ketoconazole is characterized by its high lipid solubility (log P=4.74), which may support permeation through biological membranes, especially through the corneal epithelium, the drug high molecular weight (MW; 531.44 Da) hinders its transport. Moreover, it is difficult for the drug to present in a solubilized form in the aqueous corneal surface due to the limited aqueous solubility.5
Ocular drug administration offers lower incidence of systemic drug side effects and drug–drug interactions that are common during systemic treatment. However, due to the unique anatomy and physiology of the eye, ocular drug delivery represents a major challenge during the development of effective ophthalmic preparations. The eye is supported with static and dynamic barriers and efflux pumps that impede drug delivery, particularly to the posterior eye segment.6 Nanoparticulate drug delivery systems have been reported to deliver the administered drug successfully across the different eye segments, including the posterior part.5,7 Polymeric nanoparticles (NPs), especially those that have been developed using biocompatible–biodegradable polymers, such as polycaprolactone, poly(alkyl cyanoacrylate) and poly(lactide-co-glycolide) (PLGA), are a promising ocular delivery system due to enhanced bioavailability and reduced frequency of administration.8,9 PLGA-based NPs are the most suitable polymeric nanoparticulate ocular drug delivery owing to ease of fabrication and approval by the FDA for drug delivery.10,11
Ophthalmic in situ gel (ISG) formulations have been developed to overcome poor drug bioavailability and therapeutic response associated with conventional ophthalmic delivery systems. Ophthalmic solution, suspension and ointment are common examples of conventional ocular dosage forms that are characterized by rapid elimination of the drug from the eyes caused by high tear fluid turnover and dynamics.12 Aqueous solutions of some polymers such as alginic acid, gellan gum, poloxamer and carbopol undergo gelation when dropped into the eye, which leads to prolonged contact time with the eye surface, minimized drug dose frequency and enhanced transcorneal permeation.13–15
The aim of this study was to develop ketoconazole PLGA NPs utilizing novel in vitro solvent exchange technique with subsequent loading into ISG formulation to enhance and sustain the ocular drug delivery. The prepared ISG preparations were characterized for in vitro gelation, drug release, antifungal activity and permeation through human epithelial cell lines.
Materials and methods
Materials
Poly(d,l-lactide-co-glycolide) (ester terminated, inherent viscosity range 0.55–0.75 dL/g) from LACTEL absorbable polymers was purchased from Duracet Corporation (Birmingham, AL, USA). Polyvinyl alcohol (cold water soluble, MW 140,000) was purchased from HiMedia Laboratories Pvt. Ltd. (Mumbai, India). Ketoconazole was a kind gift from Deef Pharmaceutical Industries Co. (Al Qassim, Kingdom of Saudi Arabia [KSA]). Poloxamer 407 was supplied from Xi’an Lyphar Biotech Co., LTD (Xi’an, China). Chitosan of low MW, sodium alginate and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St Louis, MO, USA). Carbopol 940 and hydroxypropyl methyl cellulose (HPMC; MW 86,000, viscosity 4,000 cp [2% solution]) were procured from Acros Organics (Morris Plains, NJ, USA).
Methods
Central composite experimental design
Three formulation factors were optimized for their effects on the particle size (Y1) and drug entrapment efficiency (Y2) of the developed NPs utilizing Statgraphics Centurion XV version 15.2.05 software (StatPoint Technologies, Inc., Warrenton, VA, USA). Table 1 illustrates the independent variables, their levels and studied responses. A preliminary study was conducted to identify the levels of the independent variables.
Table 1.
Independent and dependent variables of ketoconazole PLGA NPs used in the central composite experimental design
| Independent variables | Level
|
||
|---|---|---|---|
| Low | Medium | High | |
| X1 (%) | 3.0 | 4.5 | 6.0 |
| X2 (%) | 4.0 | 5 | 6.0 |
| X3 (%) | 1.0 | 2 | 3.0 |
|
| |||
| Dependent variables | Goal | ||
|
| |||
| Y1 (nm) | Minimize | ||
| Y2 (%) | Maximize | ||
Abbreviations: X1, PLGA in organic phase; X2, organic to aqueous phase; X3, PVA in aqueous phase; Y1, particle size; Y2, entrapment efficiency; PLGA, poly(lactide-co-glycolide); PVA, poly (vinyl alcohol).
Preparation of the formulation
Based on the composition of the 16 formulations, obtained from the experimental design, which is depicted in Table 2, drug NPs were prepared by dissolving the calculated amount of PLGA in the specified volume of DMSO to prepare 3%–6% organic polymer solution. A constant concentration of ketoconazole (0.1% w/v) based on the total formulation was dissolved in this organic solution. Aqueous poly (vinyl alcohol) (PVA) solutions (1%–3%) were prepared by dissolving the specified amount of PVA in distilled water under magnetic stirring. The organic drug polymer solution was gradually added through a micropipette into the aqueous PVA solution, which was placed on a magnetic stirrer. The milky colloidal dispersion obtained was kept stirring for 30 min at 1,200 rpm and then was probe sonicated (Sonics VCX 750 Vibra Cell & Materials INC. [CT, USA]) for 10 min at 60 magnitudes for better colloidal dispersion. Drug-loaded NPs were separated by centrifugation at 15,000 rpm for 1 h at 4°C using Laboratory Centrifuge 3K30 (Sigma, Ostrode, Germany). The precipitated NPs were subjected to two cycles of washing with double distilled water and centrifugation to remove the remaining of PVA and DMSO. Finally, the separated NPs were freeze-dried using Christ Alpha 1-2 LD Plus Lyophilizer (Martin Christ GmbH, Ostrode am Harz, Germany).
Table 2.
Experimental runs along with the observed and fitted values for the studied responses
| Run | X1 (%) | X2 (%) | X3 (%) | Y1 (nm)
|
Y2 (%)
|
||
|---|---|---|---|---|---|---|---|
| Observed (mean ± SD) |
Fitted | Observed (mean ± SD) |
Fitted | ||||
| 1 | 3.0 | 6.0 | 3.0 | 457.0±0.014 | 441.092 | 61.627±8.66 | 61.0401 |
| 2 | 4.5 | 5.0 | 3.682 | 729.0±0.040 | 711.058 | 70.023±6.96 | 67.7879 |
| 3 | 3.0 | 4.0 | 3.0 | 417.0±0.011 | 463.84 | 64.277±0.47 | 66.7802 |
| 4 | 6.0 | 6.0 | 3.0 | 995.0±0.024 | 1,090.13 | 67.354±1.11 | 68.227 |
| 5 | 4.5 | 3.318 | 2.0 | 483.0±0.019 | 526.614 | 83.453±3.08 | 78.0304 |
| 6 | 1.977 | 5.0 | 2.0 | 328.0±0.018 | 302.979 | 55.624±0.44 | 54.9099 |
| 7 | 6.0 | 4.0 | 1.0 | 477.0±0.012 | 520.366 | 69.122±1.19 | 72.6754 |
| 8 | 3.0 | 4.0 | 1.0 | 446.0±0.017 | 378.33 | 64.362±0.54 | 66.4575 |
| 9 | 7.023 | 5.0 | 2.0 | 982.0±0.083 | 968.19 | 69.666±0.78 | 66.182 |
| 10 | 4.5 | 6.682 | 2.0 | 945.0±0.043 | 862.554 | 67.524±3.06 | 68.7485 |
| 11 | 4.5 | 5.0 | 0.318 | 536.0±0.055 | 515.111 | 68.765±4.28 | 66.802 |
| 12 | 6.0 | 6.0 | 1.0 | 962.0±0.066 | 942.618 | 66.912±0.51 | 67.3773 |
| 13 | 3.0 | 6.0 | 1.0 | 356.0±0.017 | 457.081 | 60.911±0.99 | 60.505 |
| 14 | 6.0 | 4.0 | 3.0 | 843.0±0.047 | 769.377 | 69.938±1.19 | 73.3126 |
| 15 | 4.5 | 5.0 | 2.0 | 885.0±0.183 | 893.331 | 67.847±3.10 | 67.5697 |
| 16 | 4.5 | 5.0 | 2.0 | 895.0±0.174 | 893.331 | 66.572±0.96 | 67.5697 |
Abbreviations: X1, PLGA in organic phase; X2, organic to aqueous phase; X3, PVA in aqueous phase; Y1, particle size; Y2, entrapment efficiency; PLGA, poly(lactide-co-glycolide); SD, standard deviation; PVA, poly (vinyl alcohol).
Characterization of the prepared NPs
Measurement of particle size of NPs
Known weight of the freeze-dried NPs was suspended in double distilled water by vortex, and the particle size of the suspended particles was determined by dynamic light scattering using a Zetatrac (Microtrac Inc., York, PA, USA).
Entrapment efficiency
Following centrifugation, the supernatant was filtered through 0.2 µm cellulose acetate membrane filter and the concentration of the drug in the filtrate was determined spectrophotometrically using Jenway 6715 UV–visible spectrophotometer (Jenway, Stone, UK). The absorbance of ketoconazole was recorded against a blank solution of methanol at 231 nm to avoid any interference.16
The entrapment efficiency was calculated using the following equation:
where Dtotal is the total concentration of the drug loaded and Dfree drug represents the concentration of the free drug in the supernatant.
Central composite experimental design, statistical analysis and optimum desirability
The observed values for both Y1 and Y2, tabulated in Table 2, were introduced into the response columns of the Statgraphics optimization software and analyzed to identify the statistical significance of the relationships between the studied variables and obtained responses. A P-value <0.05 was considered significant following statistical analysis of the data. The optimum desirability was calculated, and an optimized formulation was proposed. This formulation, containing the proposed optimum levels for X1, X2 and X3, was prepared and characterized for Y1 and Y2 as previously described, and the differences between predicted and observed values were calculated.
Physicochemical characterization
Differential scanning calorimetry (DSC)
The thermal behavior of pure ketoconazole, PLGA, PVA and the optimized formulation was studied using Shimadzu DSC TA-50 ESI DSC apparatus (Shimadzu, Tokyo, Japan). Aluminum crucibles containing ~2 mg of each sample under a dynamic nitrogen atmosphere (flow rate: 50 mL/min) and at a heating rate of 10°C/min in a temperature range from 25°C to 950°C were used.
Fourier transform infrared (FTIR) spectroscopy
FTIR spectra of the samples used in the DSC technique were recorded using Nicolet iS10 (Thermo Fischer Scientific, Waltham, MA, USA) in the range of 4,000–400 cm−1.
X-ray powder diffraction (XRPD)
The crystalline state of the NPs of the prepared drug compared to that of the pure ketoconazole powder was studied using XRPD (D/max 2500; Rigaku, Tokyo, Japan). The diffraction patterns of the studied samples were recorded at a scan speed of 0.5000 degree/min.
Preparation of ISG formulations
Different ISG base solutions were prepared by simple dispersion of the polymeric material, specifically sodium alginate (1%), chitosan (0.5%), poloxamer 407 (16%) or carbopol 940 (0.5%), in deionized water with continuous mixing on a magnetic stirrer until complete dissolving of the polymer. A polymeric solution of chitosan was prepared by dispersion of the polymer in a slightly acidified solution. HPMC in a concentration of 0.5% (w/v) was added to each solution as a viscosity enhancer. A quantity equivalent of NPs, containing 0.3% ketoconazole, was added to the prepared homogenous polymeric dispersion solutions. For comparative study, the same gel base solutions were prepared and loaded with 0.3% pure ketoconazole. ISG formulations containing mixture of the studied polymers were also prepared. A total of 12 ISG preparations were prepared, and the composition is illustrated in Table 3.
Table 3.
Composition of the prepared ISG formulations, in vitro release parameters, antifungal activities and gelation time
| ISG formula | Composition
|
Release parameters
|
Zone diameter (mm) | In vitro gelation (min) | ||||
|---|---|---|---|---|---|---|---|---|
| Polymer | Concentration (%) | Drug form | JSS (μg/cm2⋅min) | P (cm/h) | D | |||
| 1 | Na alginate | 1 | NPs | 9.88×10−2 | 8.98×10−5 | 7.13×10−6 | 24 | 56 |
| 2 | Na alginate | 1 | Pure drug | 3.73×10−2 | 3.39×10−5 | 9.74×10−7 | 22 | 55 |
| 3 | Carbopol | 0.5 | NPs | 15.04×10−2 | 13.67×10−5 | 1.50×10−5 | 21 | 86 |
| 4 | Carbopol | 0.5 | Pure drug | 12.51×10−2 | 11.37×10−5 | 1.10×10−5 | 20 | 88 |
| 5 | Poloxamer | 16 | NPs | 39.45×10−2 | 35.86×10−5 | 10.9×10−5 | 20 | 65 |
| 6 | Poloxamer | 16 | Pure drug | 45.67×10−2 | 4.15×10−5 | 1.47×10−6 | 18 | 65 |
| 7 | Chitosan | 0.5 | NPs | 75.18×10−2 | 68.35×10−5 | 4×10−4 | 25 | 91 |
| 8 | Chitosan | 0.5 | Pure drug | 75.16×10−2 | 68.33×10−5 | 39.28×10−5 | 22 | 93 |
| 9 | Chitosan–alginate mixture | 0.5–0.5 | NPs | 1.41 | 12.76×10−4 | 13.65×10−4 | 30 | 47 |
| 10 | Chitosan–alginate mixture | 0.5–0.5 | Pure drug | 55.66×10−2 | 50.59×10−5 | 22.11×10−5 | 26 | 48 |
| 11 | Chitosan–poloxamer mixture | 0.5–16 | NPs | 29.89×10−2 | 27.18×10−5 | 6.47×10−5 | 22 | 46 |
| 12 | Chitosan–poloxamer mixture | 0.5–16 | Pure drug | 14.81×10−2 | 13.47×10−5 | 1.54×10−5 | 20 | 47 |
Abbreviations: ISG, in situ gel; JSS, steady-state flux; P, permeability coefficient; D, diffusion coefficient; NP, nanoparticle.
Characterization of the prepared ISG formulations
In vitro gelation
The prepared ISG formulations were characterized for in vitro gelation by mixing 25 µL of each formulation with 7 µL of simulated tear fluid (STF; 0.67 g sodium chloride, 0.2 g sodium bicarbonate and 0.008 g calcium chloride were dehydrated in deionized water quantity sufficient to 100 mL, and the pH was adjusted to 7.4 using hydrochloric acid) at 37°C. A ratio of 25:7 was used to mimic the in vivo ocular conditions since the volume of tear in human beings is estimated to be 7 µL, and the cul-de-sac can transiently contain ~30 µL of fluid.17 The in vitro gelation was evaluated as previously reported,18 by recording the time required for gelation.
In vitro release study
The diffusion of ketoconazole in the form of NPs or pure drug from the prepared ISG formulations was carried out using automated Franz diffusion cell apparatus (Microette Plus™; Hanson Research, Chatsworth, CA, USA). The apparatus is adapted with 1.76 cm2 of diffusion area. A quantity of ISG equivalent to 400 mg, containing 0.3% ketoconazole, was placed in position, donor chamber, and covered with a synthetic nylon membrane of pore size 0.45 µm. STF of pH 7.4 was used as a receiver medium in the receptor chamber, containing 7 mL; the temperature was kept at 32°C±0.5°C and the stirring rate was adjusted at 400 rpm. Samples of 4 mL from the receptor chamber were automatically withdrawn at 0.25, 0.5, 1, 2, 4, 6 and 8 h with replacement and analyzed for drug content spectrophotometrically. The condition of the in vitro release study was adjusted to achieve the sink condition. Blank experiments were also performed at the same wavelength for correction. The release patterns of ketoconazole from the studied ISG formulations were determined by plotting the cumulative amount of the drug permeated (Q) per unit area as a function of time, and the drug steady-state flux (JSS) for each formulation was determined from the slope. The permeability coefficient (Pc) was estimated by dividing the drug flux by the initial drug load (C0). Another plot of the Q versus square root of time was constructed, and the diffusion coefficient (D) was obtained from the equation D = (slope/2C0)2 × π.
Anti-fungal activity
The antifungal activity of the prepared ISG formulations was tested against standard strain of Candida albicans ATCC 76615 that was obtained from the Microbiology Laboratory, King Abdulaziz University Hospital, Jeddah, KSA. Preliminary screening of the antifungal activities was conducted using agar diffusion technique. Briefly, Petri dishes (150 mm) were filled with 50-mL Müller–Hinton agar containing 1 mL fungal culture (1×106 CFU/mL), and the strain was inoculated separately. Holes of 12 mm in diameter were made in the seeded agar plates and filled with 200 µL of each formula. Dishes were then incubated for 4 h at 37°C. Inhibitory activity was defined as the absence of fungal growth in the area surrounding the holes, inhibition zone, that was measured using a caliper.
Transcorneal permeation
The human corneal epithelium represents the main barrier for transcorneal drug permeation. To determine the impact of ocular barriers on the ophthalmic drug delivery, cell culture models can be used.19 In this study, evaluation of ketoconazole transcorneal permeability from ISG formulation loaded with either optimized drug NPs or pure drug (F9 or F10) was performed on human epithelial cell line (Applied Biological Materials Inc., Richmond, BC, Canada). Cells (105) were seeded on T25 flasks for 24 h and then were divided into two groups. Group I was exposed to F9 (0.3 mg/mL ketoconazole in the form of NPs), while group II was exposed to F10 (0.3 mg/mL of pure ketoconazole). Cells were collected after 2, 4, 6, 12 and 24 h (n=3) and washed twice with ice-cold phosphate buffer saline. The collected cell pellets were ruptured by subjecting to two repeated cycles of freezing and thawing, followed by ultrasonication for 10 min. Cell lysates were centrifuged using Laboratory Centrifuge 3K30 at 15,000 rpm for 1 h at 4°C, and the concentration of the drug in the supernatant was determined using high-performance liquid chromatography (HPLC) method previously reported,20 except for slight modifications. An isocratic HPLC method using C18 analytical column (4×250 mm, 5 µm; Thermo Fischer Scientific) was performed. The mobile phase composed of methanol and water, 80:20 (v/v). Its flow rate was adjusted at 1.2 mL/min. The detection wave length was 240 nm. Drug standards in the intracellular concentrate containing known amounts of the drug were prepared and analyzed before determination of the unknown drug concentrations in the samples.
Results and discussion
Preparation of drug NPs based on the in vitro solvent exchange congealing method is a novel approach to develop polymeric drug delivery system by easily achievable and facile technique. The term nanoparticle laden in situ gel has been previously reported to describe the incorporation of PLGA NPs, prepared by the nanoprecipitation process, into ISG formulation suitable for ocular retention.21 In the nanoprecipitation process, the drug and polymer in a specified ratio are dissolved in an organic solvent at room temperature and the resulting polymeric drug solution is slowly injected into aqueous PVA solution while stirring; the organic solvent and some water are evaporated.
In this study, the in vitro solvent exchange congealing technique, which depends on solvent exchange mechanism, was employed to develop PLGA NPs. PLGA is soluble in both water miscible solvents, such as N-methylpyrrolidone and DMSO, and in partially miscible ones, such as triacetin and ethyl acetate.22 When PLGA solution is injected into an aqueous medium, the solvent dissipates into the aqueous environment and the polymer solidifies.23–25 PVA was added to the aqueous phase as a stabilizer to produce small size and stable NPs.26,27 The possible formulation parameters affecting the preparation of the NPs were optimized to achieve the optimum concentration of PLGA in the organic solvent, organic to aqueous phase ratio and PVA concentration in the aqueous phase. Central composite experimental design was used as a tool used to investigate the effects of the independent variables (X1–X3) on the dependent variables (Y1–Y2). The design helps to build second-order quadratic model for the studied variables using the least number of trials without the need to study full factorial experiment. After the design is performed, the data obtained from the experimental work is statistically analyzed to reach the optimum values of the independent variables that achieve the goal of the study.
Effect on particle size
Results of the statistical analysis, using multiple regression analysis and two-way analysis of variance (ANOVA), for the effect of the studied independent variables on particle size indicated that the main effects of X1, X2 and X3 along with their quadratic effects were significantly Y1 as illustrated in Table 4. When the sign of the estimated effect denotes a positive value, this is an indication of a synergistic effect of this variable on the selected response, while antagonistic effect is expected when a negative sign is obtained. F-ratio is used to compare between the actual variation and the expected variation of variable averages; an F-ratio >1 is an indication of a location effect, and hence, the P-value is used to report the significance level. A factor is considered to significantly influence a selected response if the P-value differs from 0 and is <0.05. As the concentration of PVA was increased, the particle size of the prepared particles was increased. This behavior could be attributed to two reasons. First, the increase in particle size is attributed to exceeding the PVA concentration required to provide complete coverage of the surface of the NPs and start formation of PVA micelle molecules. Above this critical micelle concentration, there is a less surfactant surface adsorption since the micelle molecules begin to compete for the adsorption at the solid particles; this effect results in destabilization of the prepared NPs and therefore contributed to increase in the particle size.26–29 Second, increase in the concentration of PVA leads to increase in the viscosity of the aqueous phase, which in turn decreases the rate of PLGA solidification and so formation of large size particles. Results also revealed that, as the concentration of PLGA was increased, the particle size of the prepared NPs was increased. This finding is in good agreement with a previous study that indicated an increase in the average particle size of PLA NPs loaded with 5-fluorouracil when the concentration of the polymer was increased. The authors attributed this finding to high polymer load in the aqueous medium during the solidification process.30 A high percentage of the organic phase also increases the polymer load and so attributed to larger particle size. The standardized Pareto chart that illustrates the effect of X1, X2 and X3; their interaction and the quadratic effect of Y1 confirmed the above finding as depicted in Figure 1. A vertical reference line at a significant P-value of 0.05 is represented in the chart in which an effect that exceeds this line is statistically significant. The polynomial equation that relates the independent variables and the particle size is as follows:
Table 4.
Estimated effects of factors, F-ratio and associated P-values for Y1 and Y2 of ketoconazole PLGA NPs
| Factor | Y1 (nm)
|
Y2 (%)
|
||||
|---|---|---|---|---|---|---|
| Estimated effect | F-ratio | P-value | Estimated effect | F-ratio | P-value | |
| X1 (%) | 395.537 | 72.32 | 0.0001 | 6.70238 | 10.27 | 0.0185 |
| X2 (%) | 199.752 | 18.44 | 0.0051 | −5.51903 | 6.96 | 0.0386 |
| X3 (%) | 116.511 | 6.27 | 0.0462 | 0.58618 | 0.08 | 0.7887 |
| X1X1 (%) | −182.254 | 10.42 | 0.0180 | −4.96651 | 3.82 | 0.0983 |
| X1X2 (%) | 171.75 | 7.99 | 0.0301 | 0.32725 | 0.01 | 0.9086 |
| X1X3 (%) | 81.75 | 1.81 | 0.2272 | 0.15725 | 0.00 | 0.9560 |
| X2X2 (%) | −140.536 | 6.19 | 0.0473 | 4.11522 | 2.63 | 0.1563 |
| X2X3 (%) | −50.75 | 0.70 | 0.4356 | 0.10625 | 0.00 | 0.9702 |
| X3X3 (%) | −198.165 | 12.31 | 0.0127 | −0.194251 | 0.01 | 0.9415 |
Abbreviations: Y1, particle size; Y2, entrapment efficiency; PLGA, poly(lactide-co-glycolide); NP, nanoparticle; X1, PLGA in organic phase; X2, organic to aqueous phase; X3, PVA in aqueous phase; PVA, poly (vinyl alcohol).
Figure 1.

Standardized Pareto charts for the effect of the independent variables on Y1 and Y2.
Abbreviations: Y1, particle size; Y2, entrapment efficiency; X1, PLGA in organic phase; X2, organic to aqueous phase; X3, PVA in aqueous phase; PLGA, poly(lactide-co-glycolide); PVA, poly (vinyl alcohol).
To illustrate the effect of changing two independent variables on Y1, when the third one was kept at the intermediate level, 3D response surface plots were constructed and are represented in Figure 2.
Figure 2.

3D response surface plots for the effect of the independent variables on Y1 (left side) and Y2 (right side).
Note: Units of measure for X1, X2 and X3 are (%), Y1 (nm) and Y2 (%).
Abbreviations: Y1, particle size; Y2, entrapment efficiency; X1, PLGA in organic phase; X2, organic in aqueous phase; X3, PVA in aqueous phase; PLGA, poly(lactide-co-glycolide); PVA, poly (vinyl alcohol).
Effect on entrapment efficiency
Statistical analysis of the obtained data for entrapment efficiency indicated that the main effects of X1 and X2 were significantly affecting Y2 as tabulated in Table 4 and graphically represented in the Pareto chart (Figure 1). As the concentration of the organic phase (DMSO) was increased, the entrapment efficiency was decreased owing to the higher solubility of ketoconazole in DMSO that leads to escape and diffusion of the drug within the organic phase during the solvent exchange process. This behavior was more pronounced when the concentration of the organic phase was increased. The drug entrapment efficiency was increased when the concentration of PLGA was increased, which is in agreement with the previous work of PLA NPs but with water-soluble drug.30 This behavior could be attributed to rapid solidification and formation of the NPs that enclose a higher percentage of the drug when the concentration of PLGA was increased. The polynomial equation that relates the independent variables and the entrapment efficiency is as follows:
To illustrate the effect of changing two independent variables on Y2, when the third one was kept at the intermediate level, 3D response surface plots were constructed and are graphically represented in Figure 2.
To prepare an optimized formulation characterized by smaller particle size and high entrapment efficiency, the optimum desirability that achieves an optimum combination of factor levels was identified. The optimum levels for the independent variables along with predicted, observed and residual values are tabulated in Table 5. The optimized formulation was characterized by a particle size of 331±24 nm and a zeta potential value of 4.11±0.34 mV (n=3). Small zeta potential values were obtained for all the studied formulations (<20), which is an indication of encapsulation of the drug in the prepared PLGA NPs.31 A small value of zeta potential is associated with low particle stability and the tendency of the prepared for aggregation, but separation and lyophilization of the particle with subsequent loading into viscous polymeric solution are expected to retard particle aggregation. Stability of the particles could also be improved by surface modification.27
Table 5.
Multiple response optimization of ketoconazole PLGA NPs
| Factor | Low level | High level | Optimum |
|---|---|---|---|
| X1 (%) | 1.977 | 7.023 | 5.494 |
| X2 (%) | 3.318 | 6.682 | 3.318 |
| X3 (%) | 0.3182 | 3.682 | 1.105 |
|
| |||
| Response | Predicted value | Observed value | Residual |
|
| |||
| Y1 (nm) | 328.0 | 331.0 | +3 |
| Y2 (%) | 78.67 | 77.32 | −1.35 |
Abbreviations: PLGA, poly(lactide-co-glycolide); NP, nanoparticle; X1, PLGA in organic phase; X2, organic to aqueous phase; X3, PVA in aqueous phase; Y1, particle size; Y2, entrapment efficiency; PVA, poly (vinyl alcohol).
Physicochemical characterization
The DSC thermogram of ketoconazole showed a characteristic sharp endothermic peak at 151°C, as illustrated in Figure 3. No characteristic drug peak was observed in the drug-loaded formulation of PLGA NPs, which is an evidence that there was no crystalline drug material in the optimized drug formulation. This is an indication of the change in the drug crystallinity and homogenous dispersion of the drug in the PLGA matrix. The same finding has been previously reported for PLGA NP-loaded capecitabine32 and monensin-loaded PLGA NPs.33 The authors attributed this behavior to the existence of the drug in an amorphous or disordered-crystalline phase, which is distributed as a molecular dispersion or a solid solution state in the polymeric matrix.
Figure 3.

DSC thermograms of pure ketoconazole, PLGA, PVA and optimized NPs loaded with drug.
Abbreviations: DSC, differential scanning calorimetry; PLGA, poly(lactide-co-glycolide); NP, nanoparticle; PVA, poly (vinyl alcohol).
The FTIR spectroscopy of pure ketoconazole displayed a characteristic drug peak of carbonyl group C=O stretching vibration at 1,647.26 cm−1, C–O stretching of the drug aliphatic ether group at 1,031.95 cm−1 and C–O stretching of cyclic ether at 1,244.13 cm−1.34 The spectra of the optimized NPs loaded with the drug exhibited the same characteristic peaks (Figure 4) except for a slight shift or decrease in the intensity of the peaks due to slight overlapping between the drug and the polymer characteristic peaks or due to the drug–polymer ratio.
Figure 4.

FTIR spectroscopy of pure ketoconazole, PLGA, PVA and optimized NPs loaded with drug.
Abbreviations: FTIR, Fourier transform infrared; PLGA, poly(lactide-co-glycolide); NP, nanoparticle; PVA, poly (vinyl alcohol).
XRPD patterns of pure ketoconazole and optimized NP formulation are illustrated in Figure 5. The diffraction spectrum of the pure drug was crystalline in nature as indicated by numerous distinct peaks. The spectrum of drug NPs was different from that of pure ketoconazole, which demonstrated peaks of lower intensity, disappearance and formation of some new peaks. This finding indicates crystalline state transformation of ketoconazole in the NPs into the amorphous form and absence of interaction between the drug and the polymers used.
Figure 5.

X-ray diffraction patterns of pure ketoconazole and optimized NPs loaded with drug.
Abbreviation: NP, nanoparticle.
Characterization of the prepared ISG formulations
Previous studies have indicated that 1% sodium alginate, 16% poloxamer and 0.5% carbopol were suitable as in situ vehicles for ophthalmic delivery.13,14,30 Ionic gelation, thermosensitive transformation and pH-induced transformation are the mechanisms involved during phase transition of alginate, poloxamer and carbopol, respectively. HPMC was added to all the prepared formulation as a viscosity-enhancing agent. Chitosan is a mucoadhesive polymer that possesses viscosity-enhancing properties and is soluble in aqueous acidic solution. The pH of the STF, the presence of alkaline substances in the STF, and addition of HPMC to the chitosan-based ISG formulation could attribute to increase in the formulation viscosity upon contact with the STF. It was noticed that ISG formulations 7 and 8 showed the longest in vitro gelation time among all the studied ISG formulations. ISG formulations containing combination of chitosan and other polymers were also prepared except for carbopol, which was incompatible with chitosan as indicated by formation of white precipitate during preparation. Mixing chitosan with alginate or poloxamer leads to decrease in the time of in vitro gelation. When all the prepared formulation solutions were added to STF, the viscosity was increased owing to gel formation by ionic, thermosensitive or pH interaction. If a higher sheer rate was applied, the high shear rate of eye (4,250–28,500 1/s) during blinking, the viscosity usually decreased due to the pseudoplastic behavior of the formulations as previously described by Nagarwal et al,30 which suggests suitability of these formulations for ocular delivery. Faster gelation of formulations ISG 9 and 10 could be attributed to the presence of double viscosity-enhancing agent, HPMC and chitosan, and to the rapid ionic gelation of alginate with Ca++ in the STF.
The diffusion study revealed superior drug release from NP formulations compared to the same formulations containing pure drug as indicated by the calculated release parameters illustrated in Table 3. Development of drug PLGA NPs results in improvement in the drug permeation, owing to a decrease in the drug particle size. The drug diffusion from ISG formulations 9 and 10 was the highest as indicated in Figure 6, the effect that could be attributed to rapid gelation and easy diffusion of the drug from the chitosan–alginate system.
Figure 6.

Diffusion profile of ketoconazole from the prepared ISG formulations.
Abbreviation: ISG, in situ gel.
Results for the antifungal activity, represented as the inhibition zone diameter, were in direct relation to the release data. A large inhibition zone diameter was noticed in ISG formulation 9, containing drug NPs in the chitosan–alginate mixture, which could be related to high drug permeation from this formulation compared with others with subsequent inhibition effect on the studied fungal strain.
Transcorneal permeation
Fungal eye infections can be very serious if not treated. Infection of the clear, front layer of the eye (the cornea) is known as keratitis, while infection in the interior part of the eye is called endophthalmitis. Eye infections may be attributed to different types of fungi that result in both keratitis and endophthalmitis. Candida albicans, a type of yeast that normally lives on human skin and on the protective mucous membranes lining inside the body, has been reported to be the most common cause of endogenous endophthalmitis. Mycotic keratitis is usually attributed to filamentous fungi such as Lasiodiplodia theobromae, although other species such as Fusarium, Alternaria and Aspergillus have been also mentioned. Keratitis usually occurs in conjunction with trauma to the cornea with vegetable matter. Keratitis that is attributed to yeasts such as Candida is most likely to occur and always happen in abnormal eyes, eg, patients suffering from dry eye, chronic corneal ulceration or corneal scarring.19,35
In this study, the permeation of ketoconazole in the form of drug NPs from the ISG system was studied to evaluate the usefulness of the formulation in treatment of both keratitis and endophthalmitis. Effective permeation of the drug from the human epithelial cells coupled with the antifungal activity will be considered as a successful delivery system in the management of the abovementioned eye inflammation conditions.
Results of the transcorneal permeation indicated higher and sustained drug permeation from formulation F9 compared to F10 as represented in Figure 7. The prepared optimized drug PLGA NPs augmented the drug permeation, owing to the smaller particle size, and sustained the drug release due to entrapping the drug inside the polymeric matrix. It was previously stated that drug release from PLGA polymeric matrix is attributed to diffusion followed by polymer erosion.36 Loading the drug or the prepared optimized NPs into the ISG vehicle represents another factor that could assist in sustaining the drug release, but the effect of the drug release mechanism from PLGA, diffusion and erosion remains superior.
Figure 7.
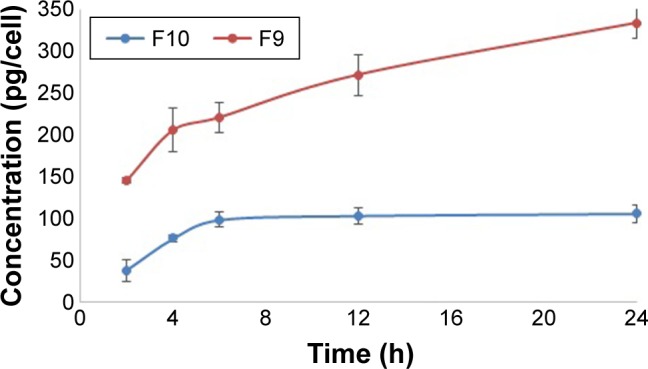
Permeation of ketoconazole through human epithelial cell line.
Conclusion
In this study, development of ketoconazole PLGA NPs based on the solvent exchange technique was successfully achieved after optimization of the formulation factors affecting the development process. Combination of alginate and chitosan was found to be an effective ophthalmic ISG drug delivery system as indicated by in vitro drug permeation and gelation, antifungal activity and transcorneal permeation. Hence, ketoconazole PLGA NPs loaded into the alginate–chitosan ISG formulation could be used as an effective antifungal medication in the treatment of both keratitis and endophthalmitis.
Acknowledgments
This article was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, KSA, under grant no 166-822-D1435. The authors, therefore, acknowledge thanks to DSR for technical and financial support.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Loose DS, Kan PB, Hirst MA, Marcus RA, Feldman D. Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450-dependent enzymes. J Clin Invest. 1983;71(5):1495–1499. doi: 10.1172/JCI110903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.U.S. Food and Drug Administration . FDA Drug Safety Communication: FDA Limits Usage of Nizoral (Ketoconazole) Oral Tablets due to Potentially Fatal Liver Injury and Risk of Drug Interactions and Adrenal Gland Problems. Silver Spring, MD: U.S. Food and Drug Administration; 2013. [Google Scholar]
- 3.De Logu A, Fadda AM, Anchisi C, et al. Effects of in-vitro activity of miconazole and ketoconazole in phospholipid formulations. J Antimicrob Chemother. 1997;40(6):889–893. doi: 10.1093/jac/40.6.889. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Wang L, Gao C, Zhang L, Xia H. Ocular pharmacokinetics of topically-applied ketoconazole solution containing hydroxypropyl beta-cyclodextrin to rabbits. J Ocul Pharmacol Ther. 2008;24(5):501–506. doi: 10.1089/jop.2008.0015. [DOI] [PubMed] [Google Scholar]
- 5.Kakkar S, Karuppayil SM, Raut JS, et al. Lipid-polyethylene glycol based nano-ocular formulation of ketoconazole. Int J Pharm. 2015;495(1):276–289. doi: 10.1016/j.ijpharm.2015.08.088. [DOI] [PubMed] [Google Scholar]
- 6.Gaudana R, Ananthula HK, Parenky A, Mitra AK. Ocular drug delivery. AAPS J. 2010;12(3):348–360. doi: 10.1208/s12248-010-9183-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaur IP, Kakkar S. Nanotherapy for posterior eye diseases. J Control Release. 2014;193:100–112. doi: 10.1016/j.jconrel.2014.05.031. [DOI] [PubMed] [Google Scholar]
- 8.Mandal B, Halder KK, Dey SK, Bhoumik M, Debnath MC, Ghosh LK. Development and physical characterization of chloramphenicol loaded biodegradable nanoparticles for prolonged release. Pharmazie. 2009;64(7):445–449. [PubMed] [Google Scholar]
- 9.Vega E, Gamisans F, García ML, Chauvet A, Lacoulonche F, Egea MA. PLGA nanospheres for the ocular delivery of flurbiprofen: drug release and interactions. J Pharm Sci. 2008;97(12):5306–5317. doi: 10.1002/jps.21383. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed TA, Ibrahim HM, Samy AM, Kaseem A, Nutan MTH, Hussain MD. Biodegradable injectable in situ implants and microparticles for sustained release of montelukast: in vitro release, pharma-cokinetics, and stability. AAPS PharmSciTech. 2014;15(3):772–780. doi: 10.1208/s12249-014-0101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21(23):2475–2490. doi: 10.1016/s0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 12.Madan M, Bajaj A, Lewis S, Udupa N, Baig JA. In situ forming polymeric drug delivery systems. Indian J Pharm Sci. 2009;71(3):242–251. doi: 10.4103/0250-474X.56015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta S, Vyas SP. Carbopol/chitosan based pH triggered in situ gelling system for ocular delivery of timolol maleate. Sci Pharm. 2010;78(4):959–976. doi: 10.3797/scipharm.1001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Başaran B, Bozkir A. Thermosensitive and pH induced in situ ophthalmic gelling system for ciprofloxacin hydrochloride: hydroxypropyl-β-cyclodextrin complex. Acta Pol Pharm. 2012;69(6):1137–1147. [PubMed] [Google Scholar]
- 15.Ma W-D, Xu H, Wang C, Nie S-F, Pan W-S. Pluronic F127-g-poly (acrylic acid) copolymers as in situ gelling vehicle for ophthalmic drug delivery system. Int J Pharm. 2008;350(1–2):247–256. doi: 10.1016/j.ijpharm.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Aggarwal A, Jain S. Physicochemical characterization and dissolution study of solid dispersions of ketoconazole with nicotinamide. Chem Pharm Bull. 2011;59(5):629–638. doi: 10.1248/cpb.59.629. [DOI] [PubMed] [Google Scholar]
- 17.Aldrich DS, Bach CM, Brown W, et al. Ophthalmic preparations. U S Pharmacopeia. 2013;39(5):1–21. [Google Scholar]
- 18.Jaiswal M, Kumar M, Pathak K. Zero order delivery of itraconazole via polymeric micelles incorporated in situ ocular gel for the management of fungal keratitis. Colloids Surf B Biointerfaces. 2015;130:23–30. doi: 10.1016/j.colsurfb.2015.03.059. [DOI] [PubMed] [Google Scholar]
- 19.Barar J, Asadi M, Abdolreza Mortazavi-Tabatabaei S, Omidi Y. Ocular drug delivery; impact of in vitro cell culture models. J Ophthalmic Vis Res. 2009;4(4):238–252. [PMC free article] [PubMed] [Google Scholar]
- 20.Vertzoni MV, Reppas C, Archontaki HA. Optimization and validation of a high-performance liquid chromatographic method with UV detection for the determination of ketoconazole in canine plasma. J Chromatogr B Anal Technol Biomed Life Sci. 2006;839(1–2):62–67. doi: 10.1016/j.jchromb.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Gupta H, Aqil M, Khar RK, Ali A, Bhatnagar A, Mittal G. Nanoparticles laden in situ gel for sustained ocular drug delivery. J Pharm Bioallied Sci. 2013;5(2):162–165. doi: 10.4103/0975-7406.111824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed TA, Ibrahim HM, Ibrahim F, et al. Development of biodegradable in situ implant and microparticle injectable formulations for sustained delivery of haloperidol. J Pharm Sci. 2012;101(10):3753–3762. doi: 10.1002/jps.23250. [DOI] [PubMed] [Google Scholar]
- 23.Ibrahim HM, Ahmed TA, Hussain MD, et al. Development of meloxicam in situ implant formulation by quality by design principle. Drug Dev Ind Pharm. 2014;40(1):66–73. doi: 10.3109/03639045.2012.746360. [DOI] [PubMed] [Google Scholar]
- 24.Al-Tahami K, Singh J. Smart polymer based delivery systems for peptides and proteins. Recent Pat Drug Deliv Formul. 2007;1(1):65–71. doi: 10.2174/187221107779814113. [DOI] [PubMed] [Google Scholar]
- 25.Kurakula M, Ahmed TA. Co-delivery of atorvastatin nanocrystals in PLGA based in situ gel for anti-hyperlipidemic efficacy. Curr Drug Deliv. 2016;13(2):1–11. doi: 10.2174/1567201813666151109102718. [DOI] [PubMed] [Google Scholar]
- 26.Ahmed TA, El-Say KM. Transdermal film-loaded finasteride microplates to enhance drug skin permeation: two-step optimization study. Eur J Pharm Sci. 2016;88:246–256. doi: 10.1016/j.ejps.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 27.Ahmed T. Preparation of finasteride capsules-loaded drug nanoparticles: formulation, optimization, in vitro, and pharmacokinetic evaluation. Int J Nanomedicine. 2016;11:515–527. doi: 10.2147/IJN.S98080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Pan Q, Rempel GL. Micellar nucleation differential micro-emulsion polymerization. Eur Polym J. 2011;47(5):973–980. [Google Scholar]
- 29.Wang Y, Zheng Y, Zhang L, Wang Q, Zhang D. Stability of nanosuspensions in drug delivery. J Control Release. 2013;172(3):1126–1141. doi: 10.1016/j.jconrel.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Nagarwal RC, Kumar R, Dhanawat M, Pandit JK. Modified PLA nano in situ gel: a potential ophthalmic drug delivery system. Colloids Surf B Biointerfaces. 2011;86(1):28–34. doi: 10.1016/j.colsurfb.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 31.Chigumira W, Maposa P, Gadaga LL, Dube A, Tagwireyi D, Maponga CC. Preparation and evaluation of pralidoxime-loaded PLGA nanoparticles as potential carriers of the drug across the blood brain barrier. J Nanomater. 2015;2015:1–5. [Google Scholar]
- 32.Sun S-B, Liu P, Shao F-M, Miao Q-L. Formulation and evaluation of PLGA nanoparticles loaded capecitabine for prostate cancer. Int J Clin Exp Med. 2015;8(10):19670–19681. [PMC free article] [PubMed] [Google Scholar]
- 33.Surolia R, Pachauri M, Ghosh PC. Preparation and characterization of monensin loaded PLGA nanoparticles: in vitro anti-malarial activity against Plasmodium falciparum. J Biomed Nanotechnol. 2011;8(1):1–10. doi: 10.1166/jbn.2012.1366. [DOI] [PubMed] [Google Scholar]
- 34.Kumar P, Mohan C, Kanamsrinivasan Uma Shankar M, Gulati M. Physiochemical characterization and release rate studies of solid dispersions of ketoconazole with pluronic F127 and PVP K-30. Iran J Pharm Res. 2011;10(4):685–694. [PMC free article] [PubMed] [Google Scholar]
- 35.Klotz SA, Penn CC, Negvesky GJ, Butrus SI. Fungal and parasitic infections of the eye. Clin Microbiol Rev. 2000;13(4):662–685. doi: 10.1128/cmr.13.4.662-685.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Husmann M, Schenderlein S, Lück M, Lindner H, Kleinebudde P. Polymer erosion in PLGA microparticles produced by phase separation method. Int J Pharm. 2002;242(1–2):277–280. doi: 10.1016/s0378-5173(02)00187-4. [DOI] [PubMed] [Google Scholar]