

Abstract
The soft-agar overlay technique was originally developed over 70 years ago and has been widely used in several areas of microbiological research, including work with bacteriophages and bacteriocins, proteinaceous antibacterial agents. This approach is relatively inexpensive, with minimal resource requirements. This technique consists of spotting supernatant from a donor strain (potentially harboring a toxic compound(s)) onto a solidified soft agar overlay that is seeded with a bacterial test strain (potentially sensitive to the toxic compound(s)). We utilized this technique to screen a library of Pseudomonas syringae strains for intraspecific killing. By combining this approach with a precipitation step and targeted gene deletions, multiple toxic compounds produced by the same strain can be differentiated. The two antagonistic agents commonly recovered using this technique are bacteriophages and bacteriocins. These two agents can be differentiated using two simple additional tests. Performing a serial dilution on a supernatant containing bacteriophage will result in individual plaques becoming less in number with greater dilution, whereas serial dilution of a supernatant containing bacteriocin will result a clearing zone that becomes uniformly more turbid with greater dilution. Additionally, a bacteriophage will produce a clearing zone when spotted onto a fresh soft agar overlay seeded with the same strain, whereas a bacteriocin will not produce a clearing zone when transferred to a fresh soft agar lawn, owing to the dilution of the bacteriocin.
Keywords: Infection, Issue 119, bacteria, bacteriophage, bacteriocin, tailocin, antimicrobial agent, bacterial antagonism, PEG precipitation
Introduction
Recently, there has been significant interest in deepening our understanding of microbial ecology (particularly the microbiomes of various environments), as well as new antibacterial compounds to use in combating antibiotic resistant pathogens1,2. An interconnecting theme between these interests is understanding antagonistic interactions among bacterial strains in their natural environment. There are numerous ways in which bacteria antagonize competitors3. Bacteriocins, a diverse group of proteinaceous, antibacterial compounds, have long been studied for their role in mediating interbacterial antagonism, with two of the most studied species being Pseudomonas aeruginosa4,5 and Escherichia coli6, important human pathogens. In addition to bacteriocins, induced prophages can also act as anticompetitor agents, allowing a strain to invade into a niche that is already colonized7. Pseudomonas syringae is a plant pathogen known to produce an array of antimicrobial agents, including single protein bacteriocins8, bacteriophage tail-derived bacteriocins9 (termed tailocins), as well as non-proteinaceous secondary metabolites10. Recently there has been interest in understanding how these antimicrobials influence the ecology of this organism, as well as how they can be harnessed to control plant disease11.
A widely utilized method for studying both bacteriocins and bacteriophage is the soft-agar overlay technique. This method was first described by Gratia in 1936 to aid in enumerating bacteriophage12,13.
Here we describe the application of the soft-agar overlay method, in combination with targeted genetic manipulation and polyethylene glycol (PEG) precipitation, in distinguishing among three different antimicrobial agents (a bacteriophage, a high molecular weight bacteriocin, and a low molecular weight bacteriocin) produced by a single bacterial strain. The benefit of this approach is that it is relatively simple and cheap, which is why, despite it being decidedly 'low-tech', it is still widely utilized.
Protocol
1. Preparation of Supernatant to Be Tested for Activity
Prepare a 0.5 mg/ml stock solution of mitomycin C by dissolving the appropriate amount into sterile 0.1 M MgSO4 buffer (e.g. 1 mg mitomycin C/2 ml buffer). Store stock at 4 °C in a light protected container (mitomycin C is light sensitive).
Inoculate a single colony of P. syringae into 3 ml of King's medium B (KB) broth14. Incubate over night with shaking (200 rpm) at room temperature (21-25 °C).
The following morning, dilute the broth culture 1/100 into 3 ml of fresh KB broth. Incubate for 3-4 hr with shaking at room temperature. Add mitomycin C (0.5 µg/ml, final concentration). Incubate the culture over night with shaking at room temperature. NOTE: Mitomycin C causes double stranded DNA breaks, thus stimulating the cell's SOS response, leading to the production of both bacteriophage and bacteriocins.
Pellet 1-2 ml mitomycin C induced cultures by centrifugation at 20,000 x g for 5 min in a bench top microcentrifuge.
- Remove and sterilize culture supernatants either by passing the supernatant through a 0.22 µm pore size filter or by treating the supernatant with chloroform (100 µl chloroform per 1 ml supernatant).
- If using chloroform, vortex the mixture for 15 sec and let incubate at room temperature for 1 hr. Following incubation, centrifuge the mixture at 20,000 x g for 5 min. NOTE: Chloroform efficiently kills cells by solubilizing their membranes. The benefit of using chloroform over filter sterilization is that it is cheaper and easier to scale-up if processing many (>20) samples at a time. There may be a possibility that a given killing activity is lost following chloroform treatment as a result of partitioning into the organic phase.
- Remove the upper, aqueous phase using a 1 ml pipet to a fresh, sterile 1.5 or 2.0 ml microfuge tube. Be careful not to transfer any of the lower chloroform layer (it is better to remove less of the aqueous phase to avoid carry-over).
- Incubate the transferred supernatant uncapped in a fume hood to allow the residual chloroform to evaporate (several hours). Store supernatants at 4 °C.
2. Separation and Concentration of High Molecular Weight Bactericidal Compounds by Polyethylene Glycol (PEG) Precipitation
To the sterile supernatant, add NaCl and PEG 8000 to 1 M and 10% final concentrations, respectively. Repeatedly invert the sample until both the NaCl and PEG are completely dissolved. Incubate samples in an ice bath for 1 hr or overnight at 4 °C.
Centrifuge samples at 16,000 x g for 30 min at 4 °C. A pellet should form at the bottom of the centrifuge tube. Decant the supernatant and resuspend the pellet in desired volume (such as 1/10 or 1/100 of the original supernatant volume) of buffer (10 mM Tris, 10 mM MgSO4, pH 7.0) by repeated pipetting.
- Remove residual PEG by two sequential extractions with an equal volume of chloroform.
- Combine chloroform with the resuspended pellet and vortex for 10 to 15 sec, then centrifuge the mixture at 20,000 x g for 5 min. Transfer the upper, aqueous phase to a fresh microfuge tube. Repeat this extraction until no white interface between the aqueous and organic phase is visible (usually 2 extractions total). Allow residual chloroform to evaporate from extracted supernatants in a fume hood.
3. Preparation of Overlay and Testing Supernatants for Activity
Inoculate a single colony of a P. syringae strain to be tested for sensitivity into 3 ml of KB, incubate over night with shaking at room temperature.
The following morning, back dilute the culture 1/100 into fresh KB. Incubate 3-4 hr with shaking at room temperature.
Prepare sterilized water agar by autoclaving a suspension 0.35-0.7% (w/v) agar in ultrapure water. NOTE: A stock of soft water agar can be melted in a microwave and reused repeatedly, fresh overlay does not need to be prepared for each experiment. If water agar is reused, it is critical to ensure it is completely melted following microwaving. If not, the overlay will have a grainy texture upon solidifying that will make interpretation difficult. If this occurs, melt the agar for several minutes longer in the microwave than done previously.
Maintain the molten soft agar in a 60 °C water bath prior to use. Using a sterile serological pipet, transfer 3 ml of soft agar to a sterile culture tube. Return the culture tube to water bath to maintain in a molten state.
To pour the overlay, first allow the molten agar to cool (it should feel warm but not hot to touch), but do not allow to solidify.
In a sterile hood, inoculate 100 µl of the tester strain culture into the soft agar and vortex to mix. Rotate culture by hand for 10-15 sec, then pour onto a bottom agar (KB agar). Tilt the plate in all directions to ensure the soft agar evenly covers the bottom agar. NOTE: The bottom agar can be any solidified (1.5% agar) medium on which the test strain grows vigorously. For a standard 100 mm diameter Petri dish, use ~20 ml melted medium. The bottom agar can be prepared several weeks ahead of time (if maintained at 4 °C without drying) or can be prepared the same day. If prepared the same day as performing the overlay, it is best to do so prior to step 3.4.
Cover the plate and allow it 20-30 min to solidify. Be careful not to disturb the plate while solidifying.
Once solidified, spot 2-5 µl of supernatant (generated in sections 1 and 2, above) onto the overlay. Allow the plates to incubate over night at room temperature. Observe and record results the following morning. NOTE: It may be useful to perform and spot from a serial dilution of the supernatant. This will allow researchers to distinguish between bacteriophage and bacteriocin clearing activity. In this case, it is recommended to perform 1:5 or 1:10 dilutions.
Representative Results
The combination of the soft agar overlay and PEG precipitation can be used to identify and characterize different antimicrobial agents produced by the same strain. Figure 1 shows two strains of P. syringae (A and B) that are inhibited by a third strain of the same species. The two strains, however, are inhibited by different bacteriocins. The clearing zone on strain A exhibits a sharp edge, whereas the clearing zone on strain B is larger, and exhibits a non-sharp border. Genetic manipulations within the producing strain that specifically inactivate either the tailocin (tailocin deficient) or low molecular weight bacteriocin (bacteriocin deficient) confirm that strains A and B are sensitive to distinct bacteriocins. Figure 2 shows that bacteriophage-mediated killing can be distinguished from other non-replicative antimicrobials by comparing a dilution series. Because both bacteriocins and prophage can be induced by mitomycin C treatment, activities of these two agents can closely resemble each other (particularly if the activated phage is abundant). In the case of bacteriophage-mediated clearing, dilution of the supernatant will resolve into individual plaques (clearing zones) while bacteriocin-mediated clearing will not resolve into such plaques.
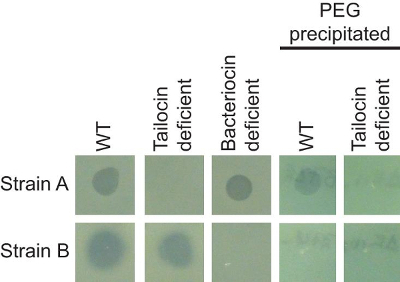 Figure 1: Phenotypic distinction of multiple antimicrobial agents produced by the same strain. Strain A is sensitive to a high molecular weight bacteriocin (tailocin) but not an alternative low molecular weight bacteriocin, as evidenced by lack of clearing in the tailocin deficient strain, but not the bacteriocin deficient strain. Conversely, strain B is insensitive to the tailocin, but is sensitive to the low molecular weight bacteriocin. The tailocin is efficiently recovered following PEG precipitation, while lower molecular weight bacteriocins are not. WT = wild type. Please click here to view a larger version of this figure.
Figure 1: Phenotypic distinction of multiple antimicrobial agents produced by the same strain. Strain A is sensitive to a high molecular weight bacteriocin (tailocin) but not an alternative low molecular weight bacteriocin, as evidenced by lack of clearing in the tailocin deficient strain, but not the bacteriocin deficient strain. Conversely, strain B is insensitive to the tailocin, but is sensitive to the low molecular weight bacteriocin. The tailocin is efficiently recovered following PEG precipitation, while lower molecular weight bacteriocins are not. WT = wild type. Please click here to view a larger version of this figure.
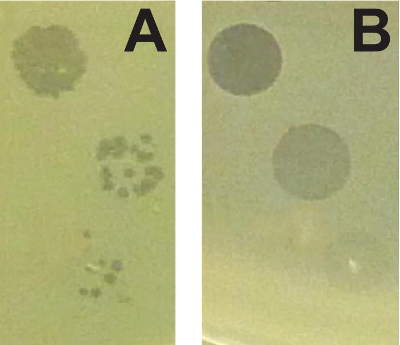 Figure 2: Distinguishing between non-replicative antimicrobials and bacteriophages. (A) Dilution of a high titer of bacteriophages results in individual plaques that become less numerous (high titer top, low titer bottom). (B) Dilution of bacteriocins results in uniformly less clearing that does not resolve into individual plaques. Please click here to view a larger version of this figure.
Figure 2: Distinguishing between non-replicative antimicrobials and bacteriophages. (A) Dilution of a high titer of bacteriophages results in individual plaques that become less numerous (high titer top, low titer bottom). (B) Dilution of bacteriocins results in uniformly less clearing that does not resolve into individual plaques. Please click here to view a larger version of this figure.
Discussion
The soft-agar overlay technique described here has been widely applied for many decades by researchers interested in bacteriophages or bacteriocins. The main benefits of this approach are that it is simple, cheap and relatively easy to interpret. By combining the soft-agar overlay with PEG precipitation and serial dilution, antimicrobial agents can be separated into high vs. low molecular weight agents, and replicative vs. non-replicative agents (i.e. bacteriophage vs. tailocin, respectively). Finally, incorporation of targeted genetic manipulation (deletion and complementation) of putative bacteriocin or prophage loci can firmly establish the identity of a given antagonistic compound. We have recently used this method to describe a new bacteriophage-derived bacteriocin prevalent among Pseudomonas syringae strains9.
The growth media and conditions indicated in this protocol work well for Pseudomonas syringae and would likely suffice for other Gram-negative bacteria that grow vigorously under these conditions. However, researchers using this method will want to optimize the timing, culture medium, induction method, and incubation temperature for their system. The critical parameters include inducing the producing culture while in the logarithmic growth phase, as well as seeding the soft-agar overlay with sufficient logarithmic phase culture to ensure a uniform bacterial distribution, but not excessive culture, which will obscure potential clearing zones. There are many bacteriocins that are not induced as part of the SOS response, but rather are induced through peptide-based quorum sensing systems (particularly Gram-positive bacteriocins15), thus, a researcher may want to screen several different induction methods (such as modifying nutrient content of inducing medium, or allowing for extended incubation of the producing strain).
When using this technique, the soft-agar overlay must be thoroughly melted and maintained at 55-60 °C prior to the addition of the seeding strain. We have had several experiments where the soft-agar was not thoroughly melted (though visually it appeared to be) and resulted in an overlay with a grainy appearance. Results from a grainy overlay can still be generally interpretable, however, this is dependent on the strength of the inhibition (weaker inhibition will be more difficult to observe). A final, confounding variable to consider when using this technique is that the temperature of the molten agar is allowed sufficient time to cool prior to inoculation with the seeding strain, so as to avoid killing the seeding strain. To avoid this issue, we ensure the soft-agar is warm, but not hot, to touch. Along these lines, we have also observed that if we give inadequate time for a seeding culture to acclimate to the molten agar, prior to pouring the overlay, we recover generally poor bacterial growth, which makes interpretation of specific inhibition difficult or impossible to interpret. This is why we allow 10-15 additional seconds of incubation between vortexing and pouring the overlay. The trade-off between maintaining the agar in a completely molten state (hotter is better) but not lethal to the seeding strain (hotter is not better) is one that will likely need to be determined by a researcher through trial and error.
If a PEG precipitation step is used, it would be beneficial to include a negative control where a sterile broth medium is processed identically to the culture supernatant. This will ensure that killing activity is not the result of residual PEG or chloroform within the sample supernatant.
As both bacteriophages and bacteriocins tend to be highly specific, it is not uncommon to encounter no apparent killing activity with a given combination of strains. This can result from a variety of strain-specific resistance mechanisms, one being that the tester strain either lacks or has an unrecognized version of the receptor needed for targeting by a given bacteriocin or bacteriophage.
An alternative method, bacteriocin screening method has been described by Kawai et al., but suffers from drawbacks and has not been widely adopted16. Specifically, this technique requires observing broth samples at time intervals that can be burdensome, and does not allow for the discrimination between bacteriophage-derived and bacteriocin-derived killing activities.
The main limitations of this technique is that it is not well suited for organisms that do not grow robustly (and thus will lack easily discernable clearing zones) or that harbor prophages or bacteriocins that are not robustly produced under laboratory conditions. Adaption of this technique to detect bacteriocins produced in environmental samples would both expand the utility of this technique and facilitate greater insight into the role of bacteriocins within natural settings.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This research was supported by the Agriculture and Food Research Initiative competitive grant 2015-67012-22773 from the USDA National Institute of Food and Agriculture to K.L.H.
References
- Cavera VL, Arthur TD, Kashtanov D, Chikindas ML. Bacteriocins and their position in the next wave of conventional antibiotics. Int J of Antimicrob Agents. 2015;46(5):494–501. doi: 10.1016/j.ijantimicag.2015.07.011. [DOI] [PubMed] [Google Scholar]
- Blaser MJ, Cardon ZG, et al. Toward a Predictive Understanding of Earth's Microbiomes to Address 21st Century Challenges. MBio. 2016;7(3):e00714–e00716. doi: 10.1128/mBio.00714-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8(1):15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel-Briand Y, Baysse C. The pyocins of Pseudomonas aeruginosa. Biochimie. 2002;84(5-6):499–510. doi: 10.1016/s0300-9084(02)01422-0. [DOI] [PubMed] [Google Scholar]
- Ghequire MGK, De Mot R. Ribosomally encoded antibacterial proteins and peptides from Pseudomonas. FEMS Microbiol Rev. 2014;38(4):523–568. doi: 10.1111/1574-6976.12079. [DOI] [PubMed] [Google Scholar]
- Cascales E, Buchanan SK, et al. Colicin Biology. Microbiol and Mol Biol Rev. 2007;71(1):158–229. doi: 10.1128/MMBR.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SP, Le Chat L, De Paepe M, Taddei F. Ecology of Microbial Invasions: Amplification Allows Virus Carriers to Invade More Rapidly When Rare. Curr Biol. 2006;16(20):2048–2052. doi: 10.1016/j.cub.2006.08.089. [DOI] [PubMed] [Google Scholar]
- Grinter R, Roszak AW, Cogdell RJ, Milner JJ, Walker D. The Crystal Structure of the Lipid II-degrading Bacteriocin Syringacin M Suggests Unexpected Evolutionary Relationships between Colicin M-like Bacteriocins. J Biol Chem. 2012;287(46):38876–38888. doi: 10.1074/jbc.M112.400150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockett KL, Renner T, Baltrus DA. Independent Co-Option of a Tailed Bacteriophage into a Killing Complex in Pseudomonas. MBio. 2015;6(4) doi: 10.1128/mBio.00452-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobellis NS, Lavermicocca P, Grgurina I, Simmaco M, Ballio A. Phytotoxic properties of Pseudomonas syringae pv. syringae toxins. Physiol Mol Plant Pathol. 1992;40(2):107–116. [Google Scholar]
- Baltrus DA, Hendry TA, Hockett KL. Ecological Genomics of Pseudomonas syringae. Genomics of Plant-Associated Bacteria. 2014. pp. 59–77.
- Panec M, Katz DS. Plaque Assay Protocols. 2013. Available from: Microbelibrary.org at http://www.microbelibrary.org/component/resource/laboratory-test/3073-plaque-assay-protocols.
- Gratia A. Numerical Relationships between Lysogenic Bacteria and Particles of Bacteriophage. Ann Inst Pasteur. 1936;57:652. [Google Scholar]
- King EO, Ward MK, Raney DE. Two simple media for the demonstration of pyocyanin and fluorescin. J Lab Clin Med. 1954;44:301–307. [PubMed] [Google Scholar]
- Heng NCK, Wescombe PA, Burton JP, Jack RW, Tagg JR. The Diversity of Bacteriocins in Gram-Positive Bacteria. Bacteriocins. 2007. pp. 45–92.
- Kawai Y, Saito T, Uemura J, Itoh T. Rapid Detection Method for Bacteriocin and Distribution of Bacteriocin-producing Strains in Lactobacillus acidophilusGroup Lactic Acid Bacteria Isolated from Human Feces. Biosci Biotechnol Biochem. 2014;61(1):179–182. doi: 10.1271/bbb.61.179. [DOI] [PubMed] [Google Scholar]