

Abstract
Inflammation is a cancer hallmark that underlies cancer incidence and promotion, and eventually progression to metastasis. Therefore, adding an anti-inflammatory drug to standard cancer regiments may improve patient outcome. One such drug, aspirin (acetylsalicylic acid, ASA), has been explored for cancer chemoprevention and anti-tumor activity. Besides inhibiting the cyclooxygenase 2-prostaglandin axis, ASA's anti-cancer activities have also been attributed to nuclear factor ĸB (NFĸB) inhibition. Because prolonged ASA use may cause gastrointestinal toxicity, a prodrug strategy has been implemented successfully. In this prodrug design the carboxylic acid of ASA is masked and additional pharmacophores are incorporated.
This protocol describes how we synthesized an aspirin-fumarate prodrug, GTCpFE, and characterized its inhibition of the NFĸB pathway in breast cancer cells and attenuation of the cancer stem-like properties, an important NFĸB-dependent phenotype. GTCpFE effectively inhibits the NFĸB pathway in breast cancer cell lines whereas ASA lacks any inhibitory activity, indicating that adding fumarate to ASA structure significantly contributes to its activity. In addition, GTCpFE shows significant anti-cancer stem cell activity by blocking mammosphere formation and attenuating the cancer stem cell associated CD44+CD24- immunophenotype. These results establish a viable strategy to develop improved anti-inflammatory drugs for chemoprevention and cancer therapy.
Keywords: Cancer Research, Issue 119, inflammation, prodrug, aspirin, fumarate, nuclear factor ĸB, cancer stem cells, breast cancer
Introduction
Inflammation is a hallmark that underlies multiple aspects of tumorigenesis, such as incidence and promotion, and eventually progression to metastasis1. In breast cancer, this is further supported by epidemiological observations showing that regular use of the classical non-steroidal anti-inflammatory drug aspirin (acetyl salicylic acid, ASA) is associated with a reduction in both breast cancer incidence, and risk of metastasis and recurrence2,3. ASA acts primarily by inhibiting cyclooxygenase-2 activity, which is often upregulated in breast cancer4,5. However, the anti-cancer effects of ASA may also be mediated by suppressing aberrant nuclear factor κB (NFκB) signaling6-8. This is important because a deregulated NFκB pathway promotes tumor cell survival, proliferation, migration, invasion, angiogenesis, and resistance to therapy9-11. NFκB pathway activation is also critical for mounting an immune response. Therefore, for anti-cancer therapy where prolonged NFκB inhibition is required, one must consider the detrimental side effects involving long-lasting immune suppression. Hence, ASA may serve as a good starting point for therapeutic optimization.
One limitation for ASA application in cancer therapy is the elevated doses required for cyclooxygenase 2 and NFκB inhibition, which are associated with gastrointestinal toxicity, such as ulcers and stomach bleeding12,13. However, converting ASA into as ester prodrug, may reduce ASA's gastrointestinal toxicity. To further enhance potency and/or add functionality, additional structural elements or ancillary pharmacophores may also be incorporated into ester prodrug design. One such pharmacophore added to enhance ASA potency against the NFκB pathway is fumarate, which we have previously shown to be important for NFκB pathway inhibition14,15.
We synthesized an aspirin-fumarate prodrug15, GTCpFE, and hypothesized that such hybrid molecule would be safe yet potent against the NFκB pathway. We tested its anti-NFκB activity in breast cancer cells and its ability to block breast cancer stem cells (CSCs)15, which rely on NFκB signaling for survival and growth16-21. We find that the potency of GTCpFE against the NFκB pathway is significantly improved over ASA15. In addition, GTCpFE blocks mammosphere formation and attenuates the CSC surface marker CD44+CD24- immunophenotype, indicating that GTCpFE is capable of eradicating CSCs15. These results establish the aspirin-fumarate prodrug as an effective anti-inflammatory agent that can also target breast CSCs. In terms of breast cancer therapy, GTCpFE may have the potential to treat aggressive and deadly disease.
Protocol
1. Synthesis of Aspirin-fumarate Prodrug GTCpFE
Using a plastic plunger syringe, measure 0.81 ml (20 mmol) of methanol and mix it in water (10 ml) in a round bottom flask. Cool the resulting mixture to 0 °C by placing the flask in an ice-water bath. Add 4-hydroxybenzyl alcohol (2.48 mg, 20 mmol) and stir the reaction mixture until the solution is clear.
Prepare a solution of O-acetylsalicyloyl chloride (3.77 mg, 19 mmol) in anhydrous toluene (10 ml) by weighing the desired amount of O-acetylsalicyloyl chloride and dissolving it in the solvent in a separate flask. Using a plastic plunger syringe, add this solution to the mixture prepared in step 1.1, and leave the reaction stirring at 0 °C.
- Monitor the reaction using thin layer chromatography (TLC). The TLC plates should have silica as a stationary phase.
- Prepare a mobile phase composed of 20:80 ethyl acetate (AcOEt)/hexane. In a TLC chamber (or small container) add 5 ml of mobile phase to cover the bottom of the chamber. NOTE: The amount of mobile phase depends on the chamber selected. Always add enough to cover the bottom, but once the TLC is placed inside, the solvent line should not surpass the height where the compounds are spotted.
- Take a sample of the reaction with a syringe (0.2 ml), place it in a small vial, and dilute it with ethyl acetate (2-3 drops). With a TLC spotter, carefully take a sample of the diluted reaction mixture and spot it in the TLC. NOTE: The spots should be 1-2 mm and this should be done at the lower 1/4 inch of the TLC plate.
- On the same TLC, place a spot of a solution of O-acetylsalicyloyl chloride (0.2 mg in 0.5 ml of ethyl acetate) as a comparison. Once the spots are dry, place it in the chamber and let it run until the solvent front has almost reached the top of the TLC.
- Take the TLC out, let it dry and visualize it under a UV lamp. The reaction is complete when the O-acetylsalicyloyl chloride spot disappears from the reaction mixture.
- When the reaction is completed, remove the ice-water bath, and allow the reaction to stir at room temperature for 20 hr.
Filter the precipitate using a Buchner funnel with a fritted disk of medium porosity. Place the solid in a scintillation vial, and leave the compound in vacuo overnight in a desiccator at room temperature. Use phosphorus pentoxide (P2O5) as the desiccant.
Dry a round bottom flask by placing it in an oven at 80 °C for at least two days before setting up this reaction. Alternatively, seal the flask with a septum and pierce it with a needle that is connected to a vacuum pump system. Turn the vacuum pump on and dry the flask using a heat gun. Allow to cool, and repeat this step 2 more times.
- Inflate a balloon using argon gas, and connect it to a plastic syringe (whose plunger has been removed) with a needle. Turn off the vacuum pump and remove the needle attached to it that was inserted in the now dried round bottom flask. Insert the needle connected to the argon balloon through the septum sealing the round bottom flask.
- Add 4-hydroxymethylphenol ester of 2-acetyloxybenzoic acid (100 mg, 0.349 mmol), 4-dimethylaminopyridine (4 mg, 0.033 mmol) and triethylamine (53 mg, 0.523 mmol) to a separate flask, then dissolved them in anhydrous tetrahydrofuran (5 ml). With a plastic syringe attached to a needle, add this solution to the sealed round bottom flask. Cool the mixture down to 0 °C by placing the flask in an ice water bath.
To the previous mixture, add a solution of ethyl fumaroyl chloride (68 mg, 0.419 mmol) in anhydrous tetrahydrofuran (5 ml), drop-wise over a period of 10 min. Stir the resulting solution at 0-5 °C for 2-4 hr.
- Extract the reaction mixture with ethyl acetate (100 ml) and brine (50 ml). NOTE: Brine is a saturated solution of sodium chloride (NaCl). To prepare it, place 200 ml of water in a clean glass container then start adding NaCl (while stirring) until it doesn't dissolve anymore.
- After extraction, remove the aqueous phase and repeat the latter step two more times.
- Dry the mixture by adding sodium sulfate to the organic phase, until the solid does not clump up when the glassware is stirred. Using another Buchner funnel with a fritted disk, filter the mixture to a round bottom flask to remove the sodium sulfate, then evaporate to dryness using a rotary evaporator with the temperature set at 40 °C.
Using a TLC, determine a proper mobile phase to use in column chromatography. NOTE: For this experiment a gradient of 20:80 AcOEt/hexane was used.
- Prepare a column with silica gel and the appropriate solvent phase. Once the compound has been added, add a layer of sodium sulfate (or sand) to protect the column as the solvent is added.
- As the column runs, monitor the eluate by TLC. Spot every other test tube as they are collected from the column. When the product of interest elutes, mix all samples containing pure product into a large round bottom flask, and dry them using a rotary evaporator with the bath temperature set at 40 °C. NOTE: Product should appear as a white solid.
To confirm the structure of GTCpFE, collect proton and carbon (1H and 13C, respectively) nuclear magnetic resonance (NMR) spectra as per manufacturer's instructions. NOTE: In this study a 400 MHz FT NMR spectrometer was used to collect the 1H and 13C spectra. Run at least 25 scans at room temperature to obtain enough data resolution. The NMR peaks observed were the following, 1H NMR (CDCl3): δ 1.29 (t, 3 H, 3J = 7.0 Hz, CH3); 2.31 (s, 3 H, CH3); 4.26 (q, 2 H, 3J = 7.0 Hz, COOCH2); 5.25 (s, 2 H, OCH2); 6.89 (s, 2 H, HC=CH); 7.19 (m, 3 H, Ar); 7.42 (m, 3 H, Ar); 7.65 (m, 1 H, Ar); 8.22 (dd, 1 H, 3J = 7.8, 4J = 1.2 Hz, Ar). 13C NMR (CDCl3) δ: 169.70, 164.86, 164.75, 162.89, 151.28, 150.69, 134.76, 134.32, 133.26, 133.16, 132.25, 129.81, 126.26, 124.11, 122.47, 122.04, 66.40, 61.43, 21.05, 14.15.
In addition, collect a high resolution mass spectrum using liquid chromatography mass spectrometry in combination with ion trap and time-of flight technology (LCMS-IT-TOF) for accurate mass measurements as per manufacturer's instructions. NOTE: In this study (M+NH4+) calculated: 430.1496; observed: 430.1477.
2. GTCPFE Inhibits the NFĸB Activity in Breast Cancer Cells
- Cell Culture Conditions
- Maintain human breast cancer cell lines, MCF-7 and MDA-MB-231 as per standard cell culture techniques and propagate in a humidified incubator at 37 °C with 5% CO2.
- Prepare MCF-7 cell medium from Roswell Park Memorial Institute (RPMI) 1640 medium with phenol red supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids, 2 mM L-glutamine, 1% antibiotics penicillin-streptomycin, and 6 ng/ml insulin. Prepare MDA-MB-231 cell medium from Improved Minimum Essential Medium (IMEM) supplemented with 5% FBS, 1% non-essential amino acids, 2 mM L-glutamine, and 1% antibiotics penicillin-streptomycin.
- NFκB-RE Luciferase Reporter Assay
- Trypsinize breast cancer MCF-7 cells using 0.25% trypsin for 5 min at 37 °C, manually count using a hemocytometer and seed in 24-well plates at 90,000 cells per well density.
- The following day, co-transfect cells with plasmid DNA of an NFκB response element (NFκB-RE) luciferase construct, 1 µg/well, along with the promoterless Renilla luciferase construct, 0.2 µg/well. Perform transfections for each treatment group in triplicate.
- Wash cells twice with PBS, and incubate with mixtures of plasmid DNA and 1 µl per well of transfection reagent (e.g. lipofectamine) in serum free media. After 6 hr, change the medium to 1 ml of regular medium (without the antibiotics penicillin and streptomycin).
- After 16 hr, add 1 µl of vehicle or different stock solutions of the GTCpFE or ASA drugs to each well. Dissolve drug stock solutions (100, 50, 20, 10 and 1 mM) in dimethyl sulfoxide at 1,000x concentration. After adding the drugs, incubate cells for 2 hr at 37 °C.
- To activate the NFκB pathway, add the pro-inflammatory cytokine TNFα (10 µg/ml stock solution) into each well for a final concentration of 10 ng/ml and incubate for 4 hr. Include a TNFα alone control. Aspirate the medium and store the cells at -80 °C.
- Measure luciferase using a luciferase reporter assay system according to manufacturer's instructions. NOTE: When using the luciferase assay system (e.g. Dual Luciferase assay system) for the first time, prepare a solution of luciferase assay reagent by re-suspending it in 10 ml of buffer and storing it at -80 °C in 1 ml aliquots.
- Prepare 1x lysis buffer by diluting 5x stock buffer with water. Lyse cells in each well using 100 µl 1x lysis buffer. Incubate the plates on an orbital shaker for 15 min, at medium speed.
- Label one 1.5 ml tube per sample. Thaw luciferase assay reagent to room temperature (50 µl per sample will be needed) and keep in foil. Prepare 1x quenching reagent in a glass vial, 1:49 dilution in buffer and vortex it (50 µl per sample will be needed).
- Add 10 µl of cell lysate to a labeled microcentrifuge tube. Then add 50 µl of luciferase assay reagent and gently vortex. Immediately after, place the tube in a holder and measure the luminescence via the dual luciferase program in the luminometer. Select and press the following: Run Promega Protocols → Dual Glo → OK.
- Add 50 µl of the quenching reagent to the test tube and gently vortex. Measure the luminescence. Repeat these steps for each sample.
- Analyze data using spreadsheet by normalizing NFκB-RE to the Renilla internal control (NFκB-RE/Renilla). Compare inhibitor data to TNFα only control (TNFα only is set at 100%).
- NFκB-Target Gene Transcription
- Seed MCF-7 cells in 6-well plates at 250,000 cells per well in 2 ml medium volume prepared as described in section 2.1.
- The following day, add 2 µl of different GTCpFE 1,000x stocks (100, 50, 20, 10 and 1 mM) for 2 hr prior to adding TNFα 10ng/ml for another 2 hr. Run every treatment in triplicate. Include vehicle and TNFα alone controls.
- Isolate RNA using the guanidinium thiocyanate-phenol-chloroform extraction method according to the instructions of the manufacturer22. Determine RNA concentration (in diethylpyrocarbonate (DEPC)-treated water) and RNA purity using a spectrophotometer. Keep RNA samples on ice or store at -80 °C. Use only RNase-free barrier tips when handling RNA.
- Reverse transcribe 0.5 µg total RNA using a commercially available kit of Moloney murine leukemia virus (M-MLV) reverse transcriptase. NOTE: The kit includes 5x buffer and dithiothreitol (DTT), together with dNTP mixture, and random hexamers mixture.
- Add 0.5 µg of RNA to 200 units of M-MLV, 0.5 mM dNTPs, 100 ng of random hexamers, 10 mM DTT, 1x buffer and DEPC water to a final 10 µl reaction volume. Carry out the reverse transcriptase reaction using a PCR cycler for 50 min at 37 °C, and then 15 min at 70 °C to heat-inactivate the enzyme.
- Dilute the resulting cDNA product to 100 µl with double-distilled water and use 2 µl for each subsequent quantitative polymerase chain reaction (PCR) reaction.
- Mix forward and reverse primers for the gene of interest at 1.25 µM concentration each. Prepare a master mix with 2 µl double distilled water, 1 µl of 1.25 µM primer mix and 5 µl of 2x of the dye to enable detection of the double-stranded DNA. Load the 2 µl of cDNA and 8 µl of master mix into 96-well PCR plates.
- Carry out quantitative PCR using a real time PCR system (40 cycles, 95-60 °C) according to manufacturer's instructions and manually analyze the data. NOTE: All PCR primers used have been validated and reported previously23.
- Calculate gene expression fold change using spreadsheet via the ΔΔCt method, with ribosomal protein 36B4 mRNA serving as the internal control23.
3. GTCpFE Inhibits Breast Cancer Stem Cells In Vitro
- Mammosphere Formation Assay
- Prepare mammosphere (MS) medium by supplementing Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12) phenol red-free medium with 1% methyl cellulose. Allow to dissolve by gentle shaking overnight at 4 °C. Filter sterilize the medium and supplement with B27 1x, 1% penicillin and streptomycin, 5 µg/ml insulin, 1 µg/ml hydrocortisone, and 20 ng/ml recombinant human epidermal growth factor.
- Prepare single cells of MDA-MB-231 cell line by trypsin digestion (trypsin 0.25% for 5 min at 37 °C) of monolayer cultures and filter through mesh sieves. Manually count single dissociated cells and plate in 96-well ultra-low attachment plates at a density of 400 cells/well. The next day, add different concentrations of GTCpFE (e.g., final GTCpFE concentrations: 1, 10, 20, 50 µM) to a final volume is 100 µl. Conduct every treatment in triplicate.
- After 7 days of culture in the incubator, acquire images via an imaging software using an inverted microscope. Manually count the number MS >75 µm in diameter. Compare the drug treatment to vehicle control. NOTE: The diameter of MS >75 µm is determined using the imaging software.
- CD44+CD24- CSC Immunophenotype
- Trypsinize MDA-MB-231 cells with 0.25% trypsin for 5 min at 37 °C. Count using a hemocytometer and seed in 10 cm dishes at 3 million cells per dish in 10 ml medium as described in section 2.1. Add vehicle (10 µl dimethyl sulfoxide) or GTCpFE (10 µl of 50 mM stock) to cells for 72 hr.
- For vehicle or GTCpFE treatment groups, trypsinize cells (as in step 3.2.1) and distribute 1 million cells in 'Test' or 'Control' 5 ml polystyrene tubes containing 2 ml of 1x Hank's Balanced Salt Solution (HBSS) buffer supplemented with 2% FBS.
- To stain for the surface markers CD44 and CD24, spin cells down and add 20 µl of each conjugated antibody and HBSS + 2% FBS to Test tubes for a final 100 µl overall volume (1:5 dilution). Add 100 µl of HBSS + 2% FBS to Control tubes. Include CD44-APC conjugated antibody and CD24-PE antibody single stain controls or the IgG immunoisotype controls.
- Incubate cells in the dark at 4 °C for 30 min. Spin down cells for 5 min at 400 x g and reconstitute in 200 µl HBSS buffer with 2% FBS. Keep cells on ice in the dark.
- Perform Fluorescence-activated cell sorting (FACS) of live cells using a FACS analyzer instrument according to manufacturer's instructions. Collect at least 50,000 events for each tube. Run treatments in triplicate. NOTE: Gating is based on controls from no staining (Control tube), IgG immunoisotypes, and the CD44-APC and CD24-PE single stains.
- Analyze data using an available flow cytometry software. NOTE: The percent of GTCpFE-treated cell with the CD44+CD24- immunophenotype is estimated and compared to vehicle control.
Representative Results
In Figure 1, the chemical structure of aspirin-fumarate prodrug, GTCpFE, and its inhibitory activity on cytokine induced NFĸB pathway in breast cancer cells are indicated. GTCpFE inhibits both NFĸB endpoints, NFĸB-RE luciferase activity (Figure 1B) and expression of NFĸB target genes, such as Intercellular Adhesion Molecule 1 (ICAM1), Chemokine C-C Motif Ligand 2 (CCL2), and Tumor Necrosis Factor (TNF) (Figure 1C) in MCF-7 breast cancer cells. Calculated inhibitory concentration at 50% (IC50) value on both endpoints is ~20 µM. IC50 value is calculated using a graphing software. By comparison ASA itself even at 200 µM (10x IC50 of GTCpFE) shows no inhibitory activity (Figure 1B, blue line) in breast cancer cells. This indicates that the prodrug strategy of adding fumarate pharmacophore to ASA significantly improves its anti-NFĸB activity.
To measure the anti-CSC activity, we used two assays: the mammosphere (MS) formation assay and the population of cells expressing the CD44+CD24- immunophenotype, a bona fide CSC surface marker in breast cancer24. GTCpFE inhibits MS formation of MDA-MB-231 breast cancer cells in a dose dependent manner shown in Figure 2A. Similar to NFĸB pathway inhibition in adherent cultures, the IC50 value for mammosphere formation is ~20 µM. This consistency is expected, given that CSCs rely on NFĸB signaling for survival and propagation16-21. In addition, GTCpFE pre-treatment resulted in a significant depletion of the CD44+CD24- population (Figure 2B) in MDA-MB-231 cells. Together, these results establish GTCpFE's ability to effectively target breast CSCs.
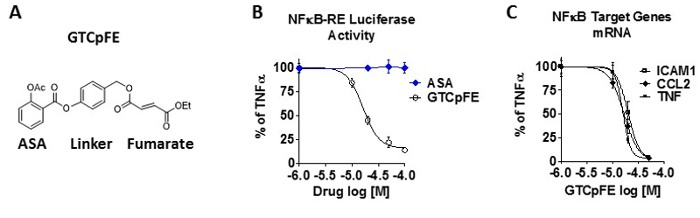 Figure 1: GTCpFE inhibits the NFĸB pathway in breast cancer cells. (A) The chemical structure of the aspirin-fumarate prodrug, GTCpFE. (B-C) MCF-7 cells were pre-treated for 2 hr with various concentrations of GTCpFE, ASA, or vehicle followed by treatment with TNFα (10 ng/ml) for 2-4 hr. (B) NFκB-RE activity was measured by dual luciferase reporter assay. (C) Expression of NFκB target genes, ICAM1, CCL2 and TNF was measured by RT-QPCR. Drug inhibitory activity is plotted as % of TNFα alone. Data are presented as mean ± SEM. This figure has been modified from reference15. Please click here to view a larger version of this figure.
Figure 1: GTCpFE inhibits the NFĸB pathway in breast cancer cells. (A) The chemical structure of the aspirin-fumarate prodrug, GTCpFE. (B-C) MCF-7 cells were pre-treated for 2 hr with various concentrations of GTCpFE, ASA, or vehicle followed by treatment with TNFα (10 ng/ml) for 2-4 hr. (B) NFκB-RE activity was measured by dual luciferase reporter assay. (C) Expression of NFκB target genes, ICAM1, CCL2 and TNF was measured by RT-QPCR. Drug inhibitory activity is plotted as % of TNFα alone. Data are presented as mean ± SEM. This figure has been modified from reference15. Please click here to view a larger version of this figure.
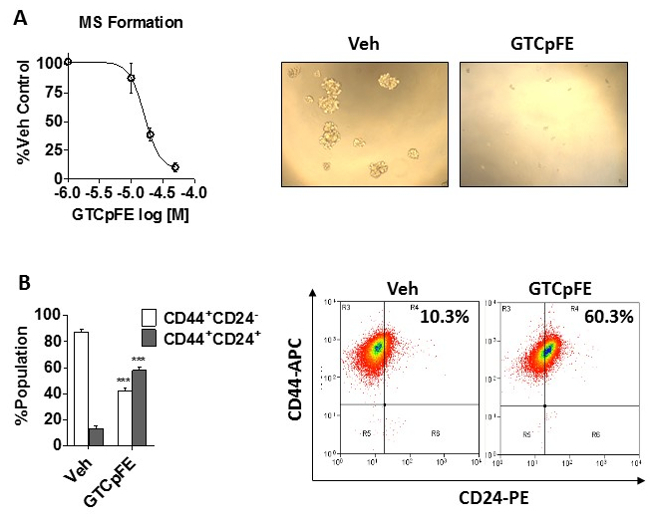 Figure 2:GTCpFE inhibits the mammosphere formation and the CD44+CD24- immunophenotype in breast cancer cells. (A) Mammosphere (MS) formation of MDA-MB-231 cells was measured after treatment with varying concentrations of GTCpFE. Quantitation of MS growth (left) and representative pictures of MS at 20X (right) are shown. The effect of GTCpFE is plotted as % vehicle control. (B) The CD44+CD24- population was determined by FACS analysis of MDA-MB-231 cells treated with 50 µM GTCpFE for 72 hr. Quantitation of each population percentage (left) and representative scatter plots from FACS (right) are shown. Data are presented as mean ± SEM and statistical analysis of 2-way ANOVA followed by Tukey posttest. *** P<0.001. This figure has been modified from reference15.
Figure 2:GTCpFE inhibits the mammosphere formation and the CD44+CD24- immunophenotype in breast cancer cells. (A) Mammosphere (MS) formation of MDA-MB-231 cells was measured after treatment with varying concentrations of GTCpFE. Quantitation of MS growth (left) and representative pictures of MS at 20X (right) are shown. The effect of GTCpFE is plotted as % vehicle control. (B) The CD44+CD24- population was determined by FACS analysis of MDA-MB-231 cells treated with 50 µM GTCpFE for 72 hr. Quantitation of each population percentage (left) and representative scatter plots from FACS (right) are shown. Data are presented as mean ± SEM and statistical analysis of 2-way ANOVA followed by Tukey posttest. *** P<0.001. This figure has been modified from reference15.
Discussion
In this protocol, we demonstrated the synthesis of an ASA prodrug, GTCpFE, where the fumarate pharmacophore was incorporated to improve the anti-NFĸB activity in breast cancer cells. GTCpFE is an effective NFĸB inhibitor, whereas ASA itself is not, even at much higher concentrations. The fumarate moiety has anti-inflammatory properties as shown by its ability to inhibit NFĸB signaling in a variety of cell lines and tissues14,25-29. The prodrug strategy described herein, is amendable to other malignancies where multiple inflammatory pathways are active and contribute to the pathology. Therefore, GTCpFE may be the prototype for developing new fumarate-based anti-inflammatory and anti-CSC class of drugs. Besides ASA, other non-steroidal anti-inflammatory drugs may also be used. In this case, the synthetic route should be re-designed as well. Ensuring that the new prodrug maintains its anti-NFĸB and anti-cyclooxygenase activity via the assays describe herein is critical.
GTCpFE abrogated MS formation and inhibited the CD44+CD24- immunophenotype. Together these findings suggest that GTCpFE may also be a promising, clinically relevant anti-inflammatory molecule for eradicating breast CSCs by exploiting CSC's reliance on multiple inflammatory pathways, including the NFĸB pathway and the cyclooxygenase 2-prostaglandin E2 axis16-21,30. Targeting breast CSCs is important because they are thought to contribute to therapy resistance, recurrence and metastasis31-34.
The anti-CSCs properties described here, the mammosphere formation and the CD44+CD24- immunophenotype, are based on typical in vitro assays to assess effect on stemness. Mammosphere formation is a functional assay because it exploits the unique property of stem-like/progenitor cells to survive and grow in serum-free suspension, while more differentiated cells undergo anoikis and die in these conditions35,36. However, because a reduction in mammosphere formation may represent (i) increased apoptosis, (ii) decreased proliferation of early progenitor cells or stem cells, (iii) a reduction in stem cell self-renewal, or (iv) interference with anchorage-independent growth, it is best used in conjunction with other anti-CSC assays. The choice of the well-established breast CSC CD44+CD24- immunophenotype is based on the pioneering study by Clarke and colleagues who used breast cancer xenografts to isolate a population of cells capable of initiating tumors in immunodeficient mice24. Similar to the CD44+CD24- immunophenotype, expression of other cell surface markers can be used to isolate stem cells via flow cytometry, but the choice of marker can greatly vary depending on tissues or species (human vs murine model). Alternative assays to measure effects on breast CSCs in vitro are (i) the side population technique, which is based on the ability of stem cells to exclude vital dyes via transmembrane transporters37, and (ii) the ALDEFLUOR assay, which is based on the enzymatic activity of aldehyde dehydrogenase 1 (ALDH1)38. Lastly, a suppression of pluripotency genes, transcription factors, or key stemness pathways may also be indicative of anti-CSCs effects. Promising in vitro anti-CSC properties should be followed up by in vivo characterization. The "gold standard" for assaying anti-CSC properties is in vivo tumorigenicity, wherein the ability of drug-treated cells to initiate or seed a xenograft tumor is examined39,40.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by grants provided by the National Institutes of Health (NIH), R01 CA200669 to JF and R01 CA121107 to GRJT, and by a postdoctoral fellowship grant from Susan G. Komen for the Cure to IK (PDF12229484).
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Cuzick J, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10:501–507. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- Terry MB, et al. Association of frequency and duration of aspirin use and hormone receptor status with breast cancer risk. JAMA. 2004;291:2433–2440. doi: 10.1001/jama.291.20.2433. [DOI] [PubMed] [Google Scholar]
- Wang D, Dubois RN. Cyclooxygenase-2: a potential target in breast cancer. Semin Oncol. 2004;31:64–73. doi: 10.1053/j.seminoncol.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Howe LR. Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res. 9. 2007;9:210. doi: 10.1186/bcr1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77–80. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]
- Pierce JW, Read MA, Ding H, Luscinskas FW, Collins T. Salicylates inhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J Immunol. 1996;156:3961–3969. [PubMed] [Google Scholar]
- Frasor J, El-Shennawy L, Stender JD, Kastrati I. NFkappaB affects estrogen receptor expression and activity in breast cancer through multiple mechanisms. Mol Cell Endocrinol. 2014;418:235–239. doi: 10.1016/j.mce.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012;12:121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- Scarpignato C, Hunt RH. Nonsteroidal antiinflammatory drug-related injury to the gastrointestinal tract: clinical picture, pathogenesis, and prevention. Gastroenterol Clin North Am. 2010;39:433–464. doi: 10.1016/j.gtc.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Sostres C, Gargallo CJ. Gastrointestinal lesions and complications of low-dose aspirin in the gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2012;26:141–151. doi: 10.1016/j.bpg.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Kastrati I, et al. Dimethyl Fumarate Inhibits the Nuclear Factor kappaB Pathway in Breast Cancer Cells by Covalent Modification of p65 Protein. J Biol Chem. 2016;291:3639–3647. doi: 10.1074/jbc.M115.679704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastrati I, et al. A novel aspirin prodrug inhibits NFkappaB activity and breast cancer stem cell properties. BMC Cancer. 2015;15:845. doi: 10.1186/s12885-015-1868-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Luo JL, Karin M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl Acad Sci U S A. 2007;104:15852–15857. doi: 10.1073/pnas.0706728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, et al. The canonical NF-kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res. 2010;70:10464–10473. doi: 10.1158/0008-5472.CAN-10-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkaya H, Liu S, Wicha MS. Regulation of cancer stem cells by cytokine networks: attacking cancer's inflammatory roots. Clin Cancer Res. 2011;17:6125–6129. doi: 10.1158/1078-0432.CCR-10-2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinohara K, et al. ErbB receptor tyrosine kinase/NF-kappaB signaling controls mammosphere formation in human breast cancer. Proc Natl Acad Sci U S A. 2012;109:6584–6589. doi: 10.1073/pnas.1113271109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendellen MF, Bradford JW, Lawrence CL, Clark KS, Baldwin AS. Canonical and non-canonical NF-kappaB signaling promotes breast cancer tumor-initiating cells. Oncogene. 2014;33:1297–1305. doi: 10.1038/onc.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, et al. NF-kappaB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat Commun. 2013;4:2299. doi: 10.1038/ncomms3299. [DOI] [PubMed] [Google Scholar]
- Rio DC, Ares M, Hannon GJ, Nilsen TW. Purification of RNA using TRIzol (TRI reagent) Cold Spring Harb Protoc. 2010. [DOI] [PubMed]
- Frasor J, et al. Positive cross-talk between estrogen receptor and NF-kappaB in breast cancer. Cancer Res. 2009;69:8918–8925. doi: 10.1158/0008-5472.CAN-09-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandermeeren M, et al. Dimethylfumarate is an inhibitor of cytokine-induced nuclear translocation of NF-kappa B1, but not RelA in normal human dermal fibroblast cells. J Invest Dermatol. 2001;116:124–130. doi: 10.1046/j.1523-1747.2001.00211.x. [DOI] [PubMed] [Google Scholar]
- Loewe R, et al. Dimethylfumarate inhibits TNF-induced nuclear entry of NF-kappa B/p65 in human endothelial cells. J Immunol. 2002;168:4781–4787. doi: 10.4049/jimmunol.168.9.4781. [DOI] [PubMed] [Google Scholar]
- Seidel P, et al. Dimethylfumarate inhibits NF-{kappa}B function at multiple levels to limit airway smooth muscle cell cytokine secretion. Am J Physiol Lung Cell Mol Physiol. 2009;297:L326–L339. doi: 10.1152/ajplung.90624.2008. [DOI] [PubMed] [Google Scholar]
- Wilms H, et al. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation. 2010;7:30. doi: 10.1186/1742-2094-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, et al. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor kappaB (NF-kappaB) and extracellular signal-regulated kinase 1 and 2 (ERK1/2) and mitogen stress-activated kinase 1 (MSK1) signaling. J Biol Chem. 2012;287:28017–28026. doi: 10.1074/jbc.M112.383380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HJ, Reinhardt F, Herschman HR, Weinberg RA. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012;2:840–855. doi: 10.1158/2159-8290.CD-12-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- Diehn M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollier BG, Evans K, Mani SA. The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J Mammary Gland Biol Neoplasia. 2009;14:29–43. doi: 10.1007/s10911-009-9110-3. [DOI] [PubMed] [Google Scholar]
- Velasco-Velazquez MA, Popov VM, Lisanti MP, Pestell RG. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J Pathol. 2011;179:2–11. doi: 10.1016/j.ajpath.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dontu G, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charafe-Jauffret E, et al. Cancer stem cells in breast: current opinion and future challenges. Pathobiology. 2008;75:75–84. doi: 10.1159/000123845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton H, Titley I, Vivanco M. Growth and differentiation of progenitor/stem cells derived from the human mammary gland. Exp Cell Res. 2004;297:444–460. doi: 10.1016/j.yexcr.2004.03.029. [DOI] [PubMed] [Google Scholar]
- Ginestier C, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324:1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]