

Abstract
Protein-protein interactions are involved in thousands of cellular processes and occur in distinct spatial context. Traditionally, co-immunoprecipitation is a popular technique to detect protein-protein interactions. Subsequent Western blot analysis is the most common method to visualize co-immunoprecipitated proteins. Recently, the proximity ligation assay has become a powerful tool to visualize protein-protein interactions in situ and provides the possibility to quantify protein-protein interactions by this method. Similar to conventional immunocytochemistry, the proximity ligation assay technique is also based on the accessibility of primary antibodies to the antigens, but in contrast, proximity ligation assay detects protein-protein interactions with a unique technique involving rolling-circle PCR, while conventional immunocytochemistry only shows co-localization of proteins.
Nuclear factor 90 (NF90) and RNA-binding motif protein 3 (RBM3) have been previously demonstrated as interacting partners. They are predominantly localized in the nucleus, but also migrate into the cytoplasm and regulate signaling pathways in the cytoplasmic compartment. Here, we compared NF90-RBM3 interaction in both the nucleus and the cytoplasm by co-immunoprecipitation and proximity ligation assay. In addition, we discussed the advantages and limitations of these two techniques in visualizing protein-protein interactions in respect to spatial distribution and the properties of protein-protein interactions.
Keywords: Biochemistry, Issue 119, physical interaction, fluorescence, circular polymerase chain reaction, RNA-binding, trafficking, stress granule
Introduction
Nuclear factor 90 (NF90) is a multi-isoform protein with numerous functions including the response to viral infection, regulation of interleukin-2 post-transcription and regulation of miRNA biogenesis 1-3. RBM3 is an RNA-binding protein, involved in translation and miRNA biogenesis and can be induced by various stressors including hypothermia and hypoxia 4-6. Recently, we found NF90 and RBM3 in a protein complex 7. The interaction of NF90 and RBM3 is essential to modulate protein kinase RNA-like endoplasmic reticulum kinase (PERK) activity in unfolded protein response 7. Both NF90 and RBM3 are located predominantly in the nucleus but a small proportion of NF90 and RBM3 shuttle into the cytoplasm and bind there to each other for specific functions, e.g. to regulate PERK activity. Therefore, it is important to visualize the distribution of NF90-RBM3 interactions in the subcellular compartment, which may indicate their various roles in respective compartment.
Decades ago, yeast two hybrid (Y2H) was developed to detect the interaction between two proteins 8. However, due to artificial construction of fused proteins, false positive results have restricted the application of this method. For a long time, co-immunoprecipitation was the main technique to analyze protein-protein interactions, especially in endogeneous conditions 9. To analyze the co-immunoprecipitated protein complex, Western blot is the most convenient technique, while mass spectrometry is used when super sensitivity and accuracy are desired. In recent years, proximity ligation assay has been developed as a novel method to detect protein-protein interactions in both cells and tissues in situ 10,11.
Here, we compared the most popular co-immunoprecipitation method and relatively novel proximity ligation assay method in capturing NF90-RBM3 interaction in subcellular fractions. We also discussed the advantages and limitations of both techniques.
Protocol
1. Co-immunoprecipitation
Seed HEK293 cells at 2 x 105 cells per well in one 6-well plate in 2 ml Dulbecco's Modified Eagle's Medium (DMEM) supplement with 10% fetal bovine serum (FBS) and 100 U/mL Penicillin-Streptomycin (Pen-Strep).
Grow cells for 48 hr at 37 °C with 5% CO2.
Wash cells with cold phosphate-buffered saline (PBS) three times. Harvest cells by centrifugation at 500 x g for 5 min at 4 °C.
- Prepare nuclear and cytoplasmic fractions using commercial nuclear and cytoplasmic extraction reagents. Follow manufacturer's instruction with some modifications.
- Use 3 x 106 cells for one co-immunoprecipitation experiment (approximately with 90% confluency from 3 wells of one 6-well plate). Add 300 µl cold cytoplasmic extraction solution (CER I) and vortex for 15 sec at the highest speed.
- Incubate for 30 min on ice. During incubation, vortex for 5 s every 10 min.
- Add 16.5 µl cold cytoplasmic extraction solution II (CER II), vortex for 5 sec and incubate for 5 min on ice.
- Vortex for 5 sec and centrifuge at 16,000 x g for 5 min at 4 °C.
- Transfer supernatant (cytoplasmic extract) into a new pre-chilled tube, and keep on ice until used.
- Wash insoluble pellets (containing nuclei) three times with 1 ml cold PBS each time by pipetting up and down five times. Remove PBS after wash.
- Add 150 µl cold nuclear extraction solution (NER), vortex for 15 sec and incubate 1 hr on ice. During incubation, vortex for 15 sec and pipette 10 times with a 200 µl tip every 10 min.
- Vortex for 15 s and centrifuge at 16,000 x g for 10 min at 4 °C.
- Transfer supernatant (nuclear extract) into a new pre-chilled tube, keep on ice until use.
Take out 10% of the volume of nuclear and cytoplasmic extracts each as inputs.
Add cold PBS to the remaining extracts to a final volume of 1 ml. Keep on ice until use. NOTE: Pre-clearing is not essential when using Protein G-conjugated magnetic beads. However, if the background is high, perform pre-clearing by incubating 40 µl Protein G-conjugated magnetic beads (50% slurry) and 1 ml diluted lysate from this step at 4 °C for 30 min on a rotator. Separate supernatant (pre-cleared lysate) from beads with magnetic rack. Discard the beads.
To couple primary antibody with Protein G-conjugated magnetic beads, incubate 40 µl Protein G-conjugated magnetic beads (50% slurry) with 4 µg rabbit polyclonal anti-RBM3 antibody or rabbit IgG (negative control) in 200 µl PBS plus Tween 20 buffer (PBST, 0.02% Tween 20) at room temperature (RT) for 40 min on a rotator.
Separate antibody-coupled beads and supernatant with a magnetic rack, and discard the supernatant. Wash the beads once with 200 µl PBST (0.02%).
Add 1 ml diluted lysates (or if necessary, pre-cleared lysates) to the beads, and incubate on a rotator at 4 °C overnight.
On the next day, separate the beads and supernatant by a magnetic rack, and discard the supernatant. Wash 3 x 10 min with 0.5 ml PBST (0.02%) for each tube at 4 °C on a rotator with a fixed speed of 20 rpm.
Elute proteins from the beads by adding 40 µl sample buffer (1x commercial sample buffer and 50 mM 1,4-dithiothreitol (DTT)). Heat at 70 °C for 10 min. Spin down and transfer liquids to a new 1.5 ml tube.
Dilute input lysates in sample buffer (final concentration: 1x commercial sample buffer and 50 mM DTT) and heat at 70 °C for 10 min. Load inputs and immunoprecipitated proteins from the last step to a precast 4-12% bis-Tris gel. The loading volume of each well should not exceed 20 µl. Perform electrophoresis in 1x commercial running buffer on ice for 35 min. NOTE: Load nuclear and cytoplasmic extracts in a ratio of 1:2 (V/V), which reflects the same initial amount of cells.
Transfer proteins to PVDF membrane in 1x commercial transfer buffer at 30 V for 2 hr at 4 °C.
Block membrane with 5% skimmed milk in PBST (0.1% Tween 20) at RT for 40 min on a shaker.
Incubate membrane with primary antibodies (both diluted in 1:1,000) in PBST (0.1%) at 4 °C overnight.
Wash 3 x 10 min with PBST (0.1%) at RT on a shaker.
Incubate membrane with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit secondary antibodies (both diluted in 1:5,000) in PBST (0.1%) at RT for 1 hr.
Wash 3 x 10 min with PBST (0.1%) at RT on a shaker.
Use 0.2 ml enhanced chemiluminescence (ECL) substrate mixture per cm2 membrane, incubate at RT for 5 min. Avoid all light from this step onwards except red lights.
Discard liquid and expose to an X-ray film in a film cassette. Develop film using an automatic film processing machine.
2. Immunocytochemistry and Proximity Ligation Assay
Seed HEK293 cells at 1.5 x 104 cells per chamber in one 8-chamber poly-D-lysine coated slide in 0.4 ml DMEM supplemented with 10% FBS and 100 U/ml Pen-Strep.
Grow cells for 48 hr at 37 °C with 5% CO2.
Aspirate medium, and fix cells with 4% paraformaldehyde (PFA) for 10 min at RT.
Aspirate PFA, and wash 3 x 10 min with 0.5 ml PBS per chamber.
Permeabilize and block cells with 0.5% Triton X-100 and 5% normal goat serum (NGS) in PBS at RT for 1 hr.
Incubate with primary antibodies in 0.1% Triton X-100 and 5% NGS in PBS on a shaker at 4 °C overnight. Dilute mouse monoclonal anti-NF90 antibody and rabbit polyclonal anti-RBM3 antibody 1:100 in PBS for double-staining. For negative control, omit independently either of the two primary antibodies, and omit both antibodies in a third control. NOTE: Do not use commercial blocking and diluting reagents.
Wash 3 x 10 min with 0.5 ml PBS per chamber.
- Subject to immunocytochemistry or proximity ligation assay protocols
- Immunocytochemistry
- Incubate with 1:500 diluted green fluorescent dye-coupled anti-mouse and red fluorescent dye-coupled anti-rabbit secondary antibodies at RT for 1 hr. Avoid light from this step onwards.
- Counterstain nuclei with 4′, 6-diamidin-2-phenylindol (DAPI) diluted 1:5,000 in PBS at RT for 10 min.
- Wash 3 x 10 min with 0.5 ml PBS per chamber.
- Remove the chambers from the glass slide and dry. Mount with 250 µ, mounting medium per slide.
- Proximity ligation assay
- Prepare proximity ligation assay probes. Mix and dilute two proximity ligation assay probes (anti-mouse and anti-rabbit secondary antibodies attached with different oligonucleotides which can ligate through the addition of two other oligonucleotides in ligation solution) both 1:5 in 0.1% Triton X-100 and 5% NGS in PBS, with a total volume of 320 µl for one 8-chamber slide (approximately 40 µl per cm2). Incubate at RT for 20 min.
- Remove the chambers from the glass slide and add the diluted probes. Incubate in a humidity incubator at 37 °C for 1 hr.
- Prepare ligation solution. Mix 8 µl ligase, 64 µl 5 x ligation stock and 248 µl H2O.
- Tap off the liquid from the slide, and wash 2 x 5 min in 1x Wash Buffer A (provided with the kit).
- Add ligation solution and incubate in a humidity incubator at 37 °C for 30 min.
- Prepare amplification solution. Mix 4 µl polymerase, 64 µl 5x amplification stock and 252 µl H2O. Avoid light from this step onwards.
- Tap off liquid from the slide, and wash 2 x 2 min in 1x Wash Buffer A.
- Add the amplification solution and incubate in a dark humidity incubator at 37 °C for 100 min.
- Tap off the liquid from the slide, and wash 2 x 10 min in 1x Wash Buffer B (provided with the kit). Wash for 1 min in 0.01x Wash Buffer B.
- Dry the slide and mount with 250 µl of a commercial mounting medium (with DAPI) per slide.
Examine fluorescent signals under a microscope using a 10X eyepiece lens and 20X objective lens. Acquire images by a CCD camera.
Representative Results
Figure 1 demonstrates that NF90 and RBM3 are both nuclear proteins and only a small fraction is present in the cytoplasm. Notably, there are three different bands stained positive for RBM3. The smallest just below 20 kDa reflects the correct size of RBM3 (the predicted molecular weight of RBM3 is 17 kDa). The origin of the two other bands remains to be investigated. Co-immunoprecipitation experiments with RBM3 as the bait protein revealed that NF90-RBM3 interactions are predominantly present in the nucleus and a minority in the cytoplasm. Co-immunoprecipitation data supports the localization of each single protein.
As shown in Figure 2A, NF90 and RBM3 are mainly located in the nucleus but also in the cytoplasm in situ. Both proteins show perfect co-localization in both compartments. Proximity ligation assay revealed the pattern of NF90-RBM3 interactions, which is very similar to conventional immunocytochemistry, with most interactions in nucleus in majority of cells. Only a small proportion of cells demonstrated predominantly cytoplasmic distribution of NF90-RBM3 interactions.
Taken together, co-immunoprecipitation and proximity ligation assay techniques reflect basically the same distribution pattern of protein-protein interactions in nuclear and cytoplasmic compartments.
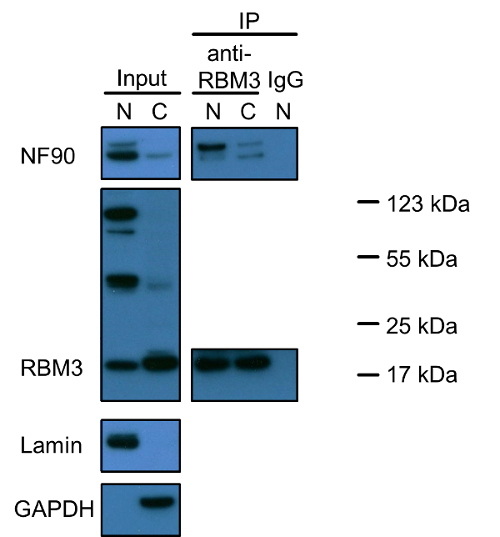 Figure 1: Western Blot Analysis of NF90 and RBM3 and Their Interactions in Nuclear and Cytoplasmic Fractions of HEK293 Cells. Nuclear and cytoplasmic extracts were loaded to SDS-PAGE gel in a ratio of 1:2 (V/V), reflecting the same amount of cells as stated in the extraction protocol. Lamin and GAPDH were used as nuclear and cytoplasmic markers, respectively. Co-immunoprecipitation was performed with anti-RBM3 antibody, or rabbit IgG as negative control. Nuclear and cytoplasmic extracts were also incubated with anti-RBM3 antibody in a ratio of 1:2 (v/v), respectively. Upper band in NF90 blot indicates 110 kDa long isoform NF110. N: nuclear extract; C: cytoplasmic extract, IP: immunoprecipitation. Protein markers were labeled for RBM3 positive bands. Please click here to view a larger version of this figure.
Figure 1: Western Blot Analysis of NF90 and RBM3 and Their Interactions in Nuclear and Cytoplasmic Fractions of HEK293 Cells. Nuclear and cytoplasmic extracts were loaded to SDS-PAGE gel in a ratio of 1:2 (V/V), reflecting the same amount of cells as stated in the extraction protocol. Lamin and GAPDH were used as nuclear and cytoplasmic markers, respectively. Co-immunoprecipitation was performed with anti-RBM3 antibody, or rabbit IgG as negative control. Nuclear and cytoplasmic extracts were also incubated with anti-RBM3 antibody in a ratio of 1:2 (v/v), respectively. Upper band in NF90 blot indicates 110 kDa long isoform NF110. N: nuclear extract; C: cytoplasmic extract, IP: immunoprecipitation. Protein markers were labeled for RBM3 positive bands. Please click here to view a larger version of this figure.
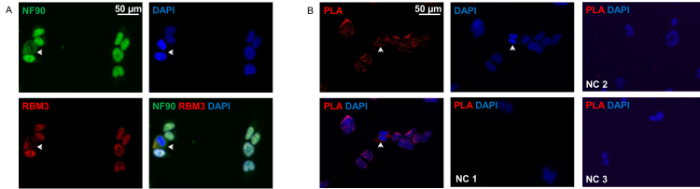 Figure 2: Immunocytochemistry and Proximity Ligation Assay of HEK293 Cells. (A) Double-staining of HEK293 cells with anti-NF90 (green) and anti-RBM3 (red) antibodies. Arrow show cells with clear cytoplasmic co-localization of NF90 and RBM3. Nuclei were counterstained with DAPI (blue). (B) Proximity ligation assay with anti-NF90 and anti-RBM3 antibodies in HEK293 cells. Red fluorescent spots indicate NF90-RBM3 interactions. Arrows show NF90-RBM3 interactions in cytoplasm. Nuclei were counterstained with DAPI (blue). Negative control 1 (NC 1): both primary antibodies were omitted. Negative control 2 (NC 2): RBM3 single primary antibody only. Negative control 3 (NC 3): NF90 single primary antibody only. Please click here to view a larger version of this figure.
Figure 2: Immunocytochemistry and Proximity Ligation Assay of HEK293 Cells. (A) Double-staining of HEK293 cells with anti-NF90 (green) and anti-RBM3 (red) antibodies. Arrow show cells with clear cytoplasmic co-localization of NF90 and RBM3. Nuclei were counterstained with DAPI (blue). (B) Proximity ligation assay with anti-NF90 and anti-RBM3 antibodies in HEK293 cells. Red fluorescent spots indicate NF90-RBM3 interactions. Arrows show NF90-RBM3 interactions in cytoplasm. Nuclei were counterstained with DAPI (blue). Negative control 1 (NC 1): both primary antibodies were omitted. Negative control 2 (NC 2): RBM3 single primary antibody only. Negative control 3 (NC 3): NF90 single primary antibody only. Please click here to view a larger version of this figure.
Discussion
There are several benefits as well as shortcomings for both methods. As a relatively novel technique, an obvious advantage of proximity ligation assay is the feasibility to elucidate protein-protein interactions at single-cell level instead of a batch of heterogeneous cells. Images with high magnitude and resolution (e.g. by confocal microscope) provide the possibility for quantification by counting single fluorescent spots. In contrast, the conventional combination of co-immunoprecipitation technique with Western blot can only semi-quantify protein bands, mainly because a suitable loading control is difficult to determine for IP samples. In addition, when protein-protein interaction is weak or transient, a large amount of biological material, e.g., cells or tissues, is required for co-immunoprecipitation or an artificial overexpressing system with fused tag is applied to enhance the chance to detect the interactions. Alternatively, detection techniques with high sensitivity such as mass spectrometry can improve the experiment quality. However, in respect to the amount of starting material the proximity ligation assay method has clear advantages. Only a few cells are required, provided a high quality of antibodies is given. Furthermore, in tissue samples, in situ visualization of protein-protein interactions in different tissue structures and cell types can be achieved by proximity ligation assay, in a similar pattern as normal immunohistochemistry. In contrast, co-immunoprecipitation is by its nature unsuited to display the spatial distribution of protein-protein interactions.
In cells with large nuclei but small cytoplasmic compartments, the proximity ligation assay method is limited in analyzing nuclear-cytoplasmic distributions and traditional co-immunoprecipitation has its own superiority. For example, in T lymphocyte cell lines, e.g. Jurkat cells, nuclear-cytoplasmic distributions of NF90-RBM3 interaction by proximity ligation assay technique is restricted, because the nucleus occupies almost the whole space inside the cell, and it is difficult to identify the boundary of the cytoplasmic compartment. This can be a common problem for immunocytochemistry in general, when the nucleus-cytoplasm ratio is extremely unbalanced. However, co-immunoprecipitation in this situation is not subject to this limitation.
One special concern regarding proximity ligation assay is whether the signals from the proximity ligation assay represent direct or indirect protein-protein interactions. Both NF90 and RBM3 have RNA-binding properties and their interaction is dependent on RNA, as reported in our previous study 7. Thus, pretreatment of cell lysates with RNase dissolves interaction between NF90 and RBM3 and nothing is detectable by co-immunoprecipitation method 7. However, the proximity ligation assay signal is not affected even if an RNA-dependent protein-protein interaction loses the RNA species, as it is generated when the distance between two proteins is less than 40 nm, which is usually considered as a direct interaction. This property of the proximity ligation assay can overcome technical problems from co-immunoprecipitation, such as RNase release in cell lysate that may abolish the interactions requiring RNA mediation. However, on the other hand, the proximity ligation assay method can also generate false positive signals, if the RNA-dependent protein-protein interaction does not actually exist due to the lack of specific RNA in physiological conditions.
Another issue deserving special attention is the specificity of primary antibodies and the possibility of protein isoforms, precursors and protein aggregates. We observed by Western blot that the long NF90 isoform, NF110, is much less abundant in both nucleus and cytoplasm in HEK293 cells as compared to NF90 (Figure 1). However, co-immunoprecipitation unraveled a higher binding affinity of NF110 to RBM3 particularly in nucleus. Three main bands were discovered in RBM3 blots in nucleus, but only the 20 kDa normal RBM3 band was mainly observed in cytoplasm. Whether the other two 50 kDa and 100 kDa bands reflect RBM3 isoforms, precursors, protein aggregates with bound RBM3 or were due to unspecific background remain to be elucidated. Instead, since the proximity ligation assay is based on immunostaining, the proximity ligation assay cannot distinguish the difference among different protein isoforms, precursors, aggregates or unspecific staining, while co-immunoprecipitation can provide more information on antibody specificity or protein isoforms and precursors than the proximity ligation assay in investigating protein-protein interactions. Regarding RBM3, when considering all three RBM3 bands observed in nucleus, the Western blot result is consistent with a predominant nuclei localization of RBM3 observed by immunostaining.
In conclusion, both co-immunoprecipitation and proximity ligation assay techniques have intrinsic advantages and restrictions. In many cases, they produce consistent results but it is beneficial to use both techniques when systematically investigating certain protein-protein interaction. However, in special cases or with particular purpose, one could show superiority than the other. Recently, proximity ligation assay has been used to screen protein-protein interactions to develop novel prognostic markers 12. Protein-protein interaction is considered as a more reliable biomarker than a single protein. In the future, the proximity ligation assay can potentially become a rapid and reliable way to analyze protein-protein interactions as diagnostic and prognostic biomarkers in clinics, although the identification and characterization of these biomarkers still requires the conventional co-immuoprecipication method.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This study was supported by the Swiss National Science Foundation (SNSF, 31003A_163305).
References
- Patiño C, Haenni AL, Urcuqui-Inchima S. NF90 isoforms, a new family of cellular proteins involved in viral replication? Biochimie. 2015;108:20–24. doi: 10.1016/j.biochi.2014.10.022. [DOI] [PubMed] [Google Scholar]
- Shim J, Lim H, R Yates J, Karin M. Nuclear export of NF90 is required for interleukin-2 mRNA stabilization. Mol Cell. 2002;10(6):1331–1344. doi: 10.1016/s1097-2765(02)00730-x. [DOI] [PubMed] [Google Scholar]
- Sakamoto S, et al. The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol Cell Biol. 2009;29(13):3754–3769. doi: 10.1128/MCB.01836-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresios J, et al. Cold stress-induced protein Rbm3 binds 60S ribosomal subunits, alters microRNA levels, and enhances global protein synthesis. Proc Natl Acad Sci. 2005;102(6):1865–1870. doi: 10.1073/pnas.0409764102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danno S, Itoh K, Matsuda T, Fujita J. Decreased expression of mouse Rbm3, a cold-shock protein, in Sertoli cells of cryptorchid testis. Am J Pathol. 2000;156(5):1685–1692. doi: 10.1016/S0002-9440(10)65039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellmann S, et al. Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J Cell Sci. 2004;117(Pt 9):1785–1794. doi: 10.1242/jcs.01026. [DOI] [PubMed] [Google Scholar]
- Zhu X, Zelmer A, Kapfhammer JP, Wellmann S. Cold-inducible RBM3 inhibits PERK phosphorylation through cooperation with NF90 to protect cells from endoplasmic reticulum stress. FASEB J. 2016;30(2):624–634. doi: 10.1096/fj.15-274639. [DOI] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340(6230):245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Verhelst J, De Vlieger D, Saelens X. Co-immunoprecipitation of the Mouse Mx1 Protein with the Influenza A Virus Nucleoprotein. J Vis Exp. 2015. [DOI] [PMC free article] [PubMed]
- Söderberg O, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2007;3(12):995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Jarvius M, et al. In situ detection of phosphorylated platelet-derived growth factor receptor beta using a generalized proximity ligation method. Mol Cell Proteomics. 2007;6(9):1500–1509. doi: 10.1074/mcp.M700166-MCP200. [DOI] [PubMed] [Google Scholar]
- Liu CH, et al. Analysis of protein-protein interactions in cross-talk pathways reveals CRKL protein as a novel prognostic marker in hepatocellular carcinoma. Mol Cell Proteomics. 2013;12(5):1335–1349. doi: 10.1074/mcp.O112.020404. [DOI] [PMC free article] [PubMed] [Google Scholar]