

Abstract
Mitochondrial dysfunction plays a significant role in the aging process and in neurodegenerative diseases including several hereditary spinocerebellar ataxias and other movement disorders marked by progressive degeneration of the cerebellum. The goal of this protocol is to assess mitochondrial dysfunction in Spinocerebellar ataxia type 1 (SCA1) and assess the efficacy of pharmacological targeting of metabolic respiration via the water-soluble compound succinic acid to slow disease progression. This approach is applicable to other cerebellar diseases and can be adapted to a host of water-soluble therapies.
Ex vivo analysis of mitochondrial respiration is used to detect and quantify disease-related changes in mitochondrial function. With genetic evidence (unpublished data) and proteomic evidence of mitochondrial dysfunction in the SCA1 mouse model, we evaluate the efficacy of treatment with the water-soluble metabolic booster succinic acid by dissolving this compound directly into the home cage drinking water. The ability of the drug to pass the blood brain barrier can be deduced using high performance liquid chromatography (HPLC). The efficacy of these compounds can then be tested using multiple behavioral paradigms including the accelerating rotarod, balance beam test and footprint analysis. Cytoarchitectural integrity of the cerebellum can be assessed using immunofluorescence assays that detect Purkinje cell nuclei and Purkinje cell dendrites and soma. These methods are robust techniques for determining mitochondrial dysfunction and the efficacy of treatment with water-soluble compounds in cerebellar neurodegenerative disease.
Keywords: Medicine, Issue 119, Ataxin-1, Neurodegeneration, Pharmacology, Behavior, Cerebellum, Mouse, Mitochondria, Rotarod, Microscopy, Metabolism, Spinocerebellar ataxia type 1, Purkinje cells
Introduction
Mitochondria are the key producers of adenosine triphosphate (ATP), an essential coenzyme for cellular energy, with the majority of mitochondrial ATP produced through oxidative phosphorylation (OXPHOS) using the electron transport chain. The brain, given its high metabolic demands and its reliance on oxidative phosphorylation for powering neural activity, is highly susceptible to mitochondrial dysfunction. As a result, mitochondrial dysfunction is triggered during the aging process1 and is implicated in the pathogenesis of multiple neurodegenerative diseases 2,3,4. Therefore, it follows that mitochondria are attractive therapeutic targets for neurodegeneration.
In this protocol, we have adopted the use of Spinocerebellar ataxia type 1 (SCA1) as a model neurodegenerative disease for the study of mitochondrial dysfunction and the development of mitochondrial-targeted therapies. SCA1 is caused by a polyglutamine (polyQ) repeat expansion mutation in the ataxin-1 gene product which triggers progressive degeneration of the Purkinje neurons of the cerebellum and neurons of other brain regions. The transgenic mouse line used here (designated as the SCA1 mouse), which expresses a polyQ-mutant ataxin-1 transgene under the control of a Purkinje-cell specific promoter, allows for the targeted analysis of the Purkinje-cell component of SCA1 5. SCA1 mice undergo gradual Purkinje cell degeneration and develop ataxic gait 6.
Mitochondrial complex dysfunction and mitochondrial-targeted treatment efficacy can be evaluated with a battery of molecular and behavioral assays. Mitochondrial complex dysfunction is measured ex vivo by respiration assays that detect altered oxygen consumption within cerebellar tissue in the presence of electron transport chain substrates and inhibitors 7. Respiration assays have previously been used with permeabilized tissue, mitochondrial isolates, and whole tissue 7,8,9. They allow for direct assessment of mitochondrial function unlike morphological data collection methods such as transmission electron microscopy or immunofluorescence staining. The use of whole tissue rather than isolated mitochondria prevents biased selection of healthy mitochondria that may occur during the isolation process 7. When adapted to the protocol as shown, the respiration assay is a valuable method for detecting mitochondrial dysfunction in cerebellar neurodegenerative disease states.
Non-specific activators of metabolism can be used to infer mitochondrial dysfunction in transgenic mice models of neurodegenerative disease and aid in the development of new therapies. Quercetin, coenzyme q10 and creatine have all been shown to ameliorate neurodegenerative disease pathology in patients and in animal models of neurodegenerative disease 10,11,12,13,14,15,16. Here we present a novel metabolic activator, succinic acid, to stimulate metabolism and boost mitochondrial function in neurodegenerative disease. To ensure that the activator is crossing the blood brain barrier, HPLC was employed to detect delivery to neural tissue in treated mice 17.
To evaluate the therapeutic effects of metabolically targeted water soluble compounds such as succinic acid, a battery of behavioral paradigms and immunopathological studies can be used. Due to the motor coordination deficits found in cerebellar neurodegenerative disease, the footprint runway assay, beam assay and accelerating rotating rod assay are used to detect rescue of behavioral pathology 6, 18, 19. These measures are supplemented with immunopathological assessment of cerebellar cytoarchitecture by assessing molecular layer thickness (defined as Purkinje cell dendritic arbor length) and Purkinje cell soma counts within a defined lobule of cerebellar tissue 6, 20, 21. Here we present multiple neuropathological and behavioral methods for detection and treatment of mitochondrial dysfunction with metabolically targeted water soluble compounds.
We use ex vivo analysis of mitochondrial respiration to analyze mitochondrial dysfunction in the SCA1 transgenic mouse. Furthermore, we show that disease symptoms and pathology are improved by the water-soluble mitochondrial booster succinic acid, further implicating mitochondrial dysfunction in SCA1 disease progression.
Protocol
This protocol follows the IACUC guidelines at Skidmore College for working with mice.
1. Treatment with Water-soluble Compounds
Dissolve succinic acid to a concentration of 0.75 mg/mL in cage drinking water. Note that any water-soluble compound of interest at the desired concentration could be substituted at this stage. Stir solution to make sure that the compound is fully dissolved.
After mice reach the desired age of treatment, replace the home cage drinking water of the treatment condition group with the solution in step 1.1.
Measure the weight of both treatment and control group water bottles daily to ensure that both treated and control mice are drinking the same amount of water. Use these measurements to calculate total drug dosage per cage. Continue the treatment throughout the experiment and monitor bottle weight.
2. Footprint Analysis
Place the runway onto the floor or a table in a confined room. Set the runway at a slight upward angle towards the home box lined with bedding and reward food (e.g. sugary cereal). Line the length of the runway with a sheet of white paper. Allow the subject to acclimate in the home box for 5 min.
Remove the subject from the home box and paint left and right hind feet, respectively, with blue and red non-toxic water-based paint. The paint prints will be used to calculate multiple output behavioral measurements.
Place the subject at the beginning of the runway and allow the subject to walk to the home box. Once the subject has reached the home box, close the trap door, and allow the subject to stay within the home box for one minute before returning to the respective home cage. Clean the runway and home box with 30% ethanol and replace the bedding, reward food, and runway paper for the next subject.
Quantify number of strides, width of gate, angle of step, and number of turns taken by subject according to the analytical methods described previously 6. A successful footprint trial is defined as a minimum of 5 sequential footprints in which the subject is walking towards the goal box without turning around or stopping.
3. Beam Analysis
Set up beam apparatus according to the bar cross specifications described previously 6 with the following 6 beams: beam 1 = rectangular, 28 mm width; beam 2 = round, 28 mm diameter; beam 3 = rectangular, 12 mm width; beam 4 = round, 12 mm diameter; beam 5 = rectangular, 6 mm width; beam 6 = round, 6 mm diameter. At one end of the beam, set up a darkened home box with reward food.
To train subjects on the beam, assemble the apparatus with beam 3. Gently place the subject on the end of the beam opposite to the home box, and allow the subject to walk across. Allow each subject up to three 2-min trials to achieve a successful run, defined as crossing the beam and entering the home box within 2 min. In between trials, let the subject rest in the home box for 2 min. In between subjects, clean the beam and home box with 30% ethanol and replace the reward food in preparation for the next subject.
Repeat step 3.2 once per day for the next two days resulting in a total of 3 sequential training days. The testing day, day 4, should sequentially follow the training days.
To test the subjects on day 4, first assemble the apparatus with beam 1 and set up the home box as described in step 3.1. Place a video camera in front of the apparatus and verify that the full beam apparatus can be clearly captured on video. Begin recording and place the first subject on the beam opposite to the home box, allowing up to 3 2-min attempts to successfully complete a trial. In between trials, allow subject to rest for 2 min in the home box.
- Continue to test subject on each of the six beams in numerical order, allowing the subject to rest for 2 minutes in the home box before beginning the next beam. Between subjects, clean each beam and the home box with 30% ethanol and replace the reward food in preparation for each subject.
- Videotape each trial and record the number of successful trials per subject per beam as follows: '1' indicates the subject was successful on the first trial, '2' indicates the subject was successful on the second trial, '3' indicates the subject was successful on the third and last trial, and '4' indicates the subject was not successful on any of the three trials.
Use video recordings of trials measure footslips by scorers who are blinded to the experimental conditions of the subject. Define a footslip as the hindfoot that is in direct view of the camera dipping below a theoretical midline across the length of the beam. Average footslip scores from multiple scorers.
To analyze the number of successful trials and number of footslips data, calculate the mean and standard error within each genotype and experimental condition cohort. Conduct one-way ANOVA and multiple comparisons post-hoc analysis to determine if there is an effect of the treatment and genotype on number of successful trials and number of footslips.
4. Accelerating Rotarod
Acclimate subjects to the assay room 10 min prior to the start of the trial. During this acclimation period, prepare the rotarod apparatus, cleaning each lane of the apparatus with 30% ethanol.
Open the rotarod software and set start settings to 4 rpm, acceleration settings to 5 min and final speed settings to 40 rpm. These settings provide a constant acceleration from 4 - 40 rpm over a period of 5 min. Mice can remain on the rotarod at 40 rpm for an additional 5 min beyond the acceleration period for a maximum run length of 10 min.
Post-acclimation, place subjects onto the rotarod in their respective lanes while the rod is rotating at 4 rpm. Once all subjects are in their respective lanes, begin the first trial. Record the latency to fall using the instrument software; a secondary timer can be employed as a backup.
Following trial completion, allow each subject to rest in their respective lane reservoirs for ten minutes prior to the next trial to prevent fatigue.
Repeat steps 4.3 and 4.4 for a total of four trials.
Repeat steps 4.3 - 4.5 for a total of four consecutive days, continuing treatment throughout the trial period. Upon completion, quantify latency to fall off the rotating rod for each trial per subject 6.
5. Cerebellar Extraction and Fixation
Euthanize mice via carbon dioxide asphyxiation according to institutional IACUC guidelines. Place subject into the carbon dioxide chamber, and release carbon dioxide into the chamber. Watch the animal at all times during the euthanizing step and do not leave the animal unattended.
Once breathing has ceased and subjects show no remaining pain perception by hind foot pressure, remove subject from chamber and make a superior midline incision through the epithelial tissue from the middle of the skull caudally to the base of the skull through epithelial tissue.
Using dissection scissors, cut the skull from the base at the spinal cord through the sagittal suture to the bregma. Cut laterally through each coronal suture before pulling off the frontal, parietal and interparietal bones using blunt forceps.
Use blunt forceps to break off bone lateral to the cerebellum. Use forceps to separate the cerebellum from the colliculi and brainstem and extrude it from the rest of the tissue. Separate the cerebellum into left and right hemispheres with forceps.
Immediately place right cerebelli into liquid nitrogen to save for HPLC analysis and left cerebelli into pre-chilled 4% paraformaldehyde (PFA) for tissue fixation. Harvest any other tissue of interest before severing the aorta and discarding the carcass according to institutional IACUC rules. CAUTION: PFA is a highly toxic, flammable, and corrosive.
Twenty-four hours post-extraction, wash tissue fixed in PFA (4% in PBS) in phosphate buffered saline (PBS) (three times, 5 min/ wash) before placing the tissue into 30% sucrose at 4 °C for up to 2 weeks.
6. Immunopathology Assay
Attach the tubing of a sledge microtome to a sink faucet and a temperature control box. Use the temperature control box to cool the microtome stage to -25 °C. Set the sectioning thickness dial to 50 µm. NOTE: If the dial on the sledge microtome does not reach 50 μm, set the dial to a thinner setting and multiply the thickness by shifting the setting accordingly (for example set the dial to 10 µm and shift five times before sectioning for a total thickness of 50 µm).
- Dab a dime-size portion of Optimal Temperature Cutting (OTC) solution onto the stage. Before the OTC freezes, use forceps to position the hemisphere so that the mid-sagittal cut surface is face down on the OTC layer. Working quickly, gently orient the tissue with forceps so that the lateral tip of the hemisphere is pointed towards the blade, superior to the stage. Allow the OTC to freeze completely.
- Working in layers, add additional OTC around the tissue allowing the OTC to freeze fully before adding the next layer. Add a thin layer of OTC on top of the tissue and freeze.
Section the tissue, distributing equal body weight onto the cutting blade while sectioning. Collect sections using a fine artist paintbrush and place into an appropriately labeled well-plate containing 1x PBS. Between sections, re-set the cutting thickness on the sledge microtome dial, if necessary, to 50 µm.
Replace the PBS in each well of the well-plate with urea solution [0.01 M urea in PBS] and place the plate on ice. When ready, remove the plate from ice and boil sections in urea solution three times for 15 s each time to unmask epitopes for immunolabeling. This step can be easily accomplished by placing the plate on a heating block set to boiling temperature. Between each boiling step, cool the plate containing the tissue sections on ice. NOTE: If performing the urea, blocking, primary antibody, secondary antibody and DAPI steps in a 96-well plate, use 100 μL volumes throughout. If a different sized well is used, adjust the reagent volumes accordingly. In lieu of a heating block, a microwave can be used to boil tissue samples in urea. Microwave samples on a high power setting for three 15 s intervals.
Post boiling, wash sections three times, each time removing present solution, replacing the solution with 1x PBS and agitating for 10 min.
Block sections with donkey serum solution (3% normal donkey serum, 0.3% triton X-100 in PBS) by incubating tissue in serum for one hour with gentle agitation at room temperature. Remove donkey serum solution.
Label sections with 0.2 µg/mL anti-calbindin and 1:2,000 anti-ataxin-1 antibody (11NQ)16 in donkey serum solution and incubate at 4 °C for 72 h with gentle agitation.
Wash sections with PBS as in step 6.5 before incubating sections in appropriate secondary antibody (1:500 Alexa Fluor-488-anti-goat and 1:500 Alexa Fluor-594-anti-rabbit antibodies diluted in donkey serum solution) at 4 °C for 48 h with gentle agitation.
Wash sections three times with PBS as in step 6.5 before labeling cell nuclei with DAPI (4',6-diamidino-2-phenylindol) solution diluted to a final concentration of 0.5 μg/mL in PBS. DAPI incubation should not exceed ten minutes. Remove solution and wash with PBS as in step 6.5.
Mount tissue onto 2% gelatin-coated slides with mounting medium to reduce photobleaching. Coverslip and seal with nail polish.
- With a scanning confocal microscope locate the primary fissure of the cerebellum under brightfield light. With a 10X UIS2 objective, scan and sequentially excite DAPI, AlexaFluor-488 and AlexaFluor-594 channels using a 405 nm diode laser, a 488 nm argon gas laser and a 559 nm diode laser, respectively, in conjunction with a DM488/559 nm dichroic beam splitter. Separate and collect emitted light using SDM490 and SDM560 beam splitters and a BA575-675 bandpass filter, respectively.
- For each channel, construct a z-stack within the primary fissure with z = 20 μm and step = 0.1 μm. Use the post-processing software provided with the confocal microscope to add in a size marker to the final image. NOTE: Alternative lasers, filters and fluorophores can be substituted based on availability. If AlexaFluor-488 or AlexaFluor-594 detection is weak, a gallium phosphide (GaAsP) high-sensitivity detector can be employed to boost the signal if available.
7. Quantifying Molecular Layer Thickness and Purkinje Cell Numbers
Using ImageJ, open a z-stack image of stained primary fissure and split each image into its red, blue and green channels by selecting 'Image' from the Menu Bar, 'Color' from the Image tab and 'Split Channels' from the Color tab.
Create a max Z projection of each channel that was split. When the channel image of interest is open, select 'Image' from the Menu Bar, 'Sections' from the Image tab and 'Z-project' from the 'Sections' tab. Within the 'Z-project' command menu, select 'Max Z projection' and click 'Okay'. Repeat for each channel.
Open the Cell Counter plugin by selecting 'Plugins' from the Menu Bar, 'Analyze' from the Plugins tab and "Cell Counter" from the Analyze tab. NOTE: ImageJ and the ImageJ Cell Counter plugin can be downloaded at the websites provided in the Table of Materials and Equipment.
- Quantify the number of Purkinje cells by counting ataxin-1-labeled nuclei that lie along a pre-determined 200 µm stretch of anterior and posterior lengths of the primary fissure. Use the threshold map by selecting 'Image' from the Menu Bar, 'Adjust' from the Image tab and 'Threshold' from the Adjust tab to make a threshold map of the channel of interest (red in the case of ataxin-1 labeled nuclei).
- Choose a pixel range that can be normalized throughout the quantification (31-225 pixels for example). Discard noise from the images by checking the "Dark Background" box in the Threshold window. Image signal can be inverted for ease of counting.
Quantify molecular layer thickness in the green channel using the 'Split Channels' and 'Threshold Map' tool as in Steps 7.1 and 7.2. Using the size marker on each image as a guide, randomly measure three molecular layer lengths within each of two designated 200 µm stretches of each primary fissure image. Make measurements from the proximal end of the Purkinje dendritic arbor at the base of the Purkinje soma to the distal end of the Purkinje dendritic arbor.
Record Purkinje cell counts and molecular layer thickness as means plus or minus the standard error. Perform one-way ANOVA with multiple comparisons post-hoc analysis to test the effect of treatment conditions or genotype on the number of cells and the length of the dendritic arbor.
8. HPLC Analysis of Cerebellar Succinic Acid Concentrations Post-treatment
- Preparation of Sample for Analysis
- Weigh each harvested right cerebellar hemisphere prior to use in HPLC analysis. Mince hemispheric halves one at a time in a pre-chilled dounce homogenizer and add 0.5 mL of 75:25 (by volume) water:methanol solution to each homogenized half 17.
- Using a narrow pre-chilled homogenizer, grossly break up tissue with 5 dounce strokes. Continue to finely mince each tissue sample with a wider homogenizer, applying 20 dounce strokes.
- Transfer the homogenates to pre-chilled microcentrifuge tube. After each sample, rinse the homogenizer with 0.5 mL of ice-cold water:methanol solution and add the rinse to the microcentrifuge tube.
- Centrifuge homogenate samples at 1,300 x g for 30 min at 25 °C and collect the supernatant in a clean microcentrifuge tube.
- Filter the supernatant through a centrifugal filter unit housed with a regenerated cellulose filter of 10 kilodalton (kDa) nominal molecular weight limit. If not immediately running HPLC analysis, store filtered samples at -80 °C.
- HPLC Instrumentation and Conditions
- Attach an ion exclusion chromatography column with a length of at least 30 cm and a particle size of 7 µm to a high performance liquid chromatograph.
- Prepare and degas a sufficient quantity of 1 mM acetate buffer adjusted to a pH of 5.
- Set the HPLC with a mobile phase velocity of 0.8mL/min. If using a UV-vis detector, select a detector wavelength of 224 nm. Typical elution times for succinic acid under these conditions are between 10 and 12 min.
- Running and Analyzing Standards and Samples
- Prepare a set of standards in acetate buffer mobile phase with succinic acid concentrations between 0 and 250 ppm.
- Run each standard and prepare a calibration curve using the peak area measured from each run.
- Once an accurate standard curve is obtained, run previously prepared samples diluted in acetate buffer. For greater accuracy of measurement, samples can be spiked with known concentrations of succinic acid that have been diluted in the mobile phase. Run all samples in triplicate.
- Analyze HPLC chromatograms to determine the succinic acid concentration of the samples by using the calibration curve and then multiplying by sample volume and dividing by original sample mass.
9. Ex Vivo Analysis of Cerebellar Mitochondrial Functionality Using an Oxygen Electrode Control Unit
- Create a Clark-type electrode using a commercially available system by first applying a drop of potassium chloride (50% KCl w/v) solution on the platinum cathode, cutting a 1.5 cm2 piece of commercial tobacco rolling paper and gently placing the paper over the platinum cathode.
- Cover the cathode and surrounding anode with a 2.5 cm2 piece of polytetrafluoroethylene (P.T.F.E) paper and snuggly secure both membranes with O rings, making sure there are no folds or bubbles within the membranes. Drip 50% KCl solution into the surrounding well.
- Secure the premade electrode into the assay chamber and fill with air-saturated water. Heat the chamber to 30 °C, add a 0.8 cm stir bar to the solution and stir at 70 rpm using the instrument software. Calibrate the chamber by establishing zero oxygen. To do so, serially add a few crystals of the reducing agent sodium dithionite to the solution.
- Once calibration is achieved, wash the chamber once with 70% ethanol, and then wash six times with deionized water before allowing the chamber to acclimate in 1.5 mL of respiration buffer (5 mM MgCl2, 60 mM KCl, 100 mM mannitol, 10 mM KH2PO4, 0.5 mM Na2EDTA, 60 mM Tris-HCl [pH 7.4]) 7. NOTE: all traces of sodium dithionite must be completely removed during the wash steps before attempting to measure oxygen concentrations during the respiration experiments.
Gently homogenize the weighed cerebellar hemispheres in 0.5 mL of homogenate buffer (0.25 M sucrose, 0.5 mM EDTA, 50 mM Tris-HCl [pH 7.4]). Transfer homogenate to a clean 1.5 mL microcentrifuge tube and keep chilled until beginning the assay.
Add the homogenate to the chamber, reaching a final volume of 2 mL. Permeabilize the cerebellar homogenate by adding 40 µL of digitonin or saponin (5 mg/mL); allow homogenized tissue to incubate for 10 min. NOTE: Digitonin and saponin are toxic, and incubation times should be kept to a minimum.
- To begin recording oxygen consumption, determine mitochondrial function through activation and inhibition of the oxidative phosphorylation chain using substrates and inhibitors, respectively (3 min recordings per substrate).
- Add substrates using clean syringes as follows: 10 µL 2 M glutamate [final concentration (f.c) 0.01 M], 5 µL 0.8 M malate [f.c. 2 mM], 20 µL 0.5 M adenosine diphosphate (ADP) [f.c. 5 mM], 1 µL 1 mM rotenone [f.c. 0.5 µM], 20 µL 1M succinic acid [10 mM], 1 µL 5 mM antimycin A [f. c. 2.5 µM], 1 µL 0.8 M ascorbate [f.c. 0.4 mM], and 5 µL 0.2 M tetramethyl-p-phenylenediamine (TMPD). [f.c. 0.5 mM] When adding substrates to the chamber be careful not to introduce air bubbles into the system. NOTE: Detailed information on the functional effect induced by each substrate and inhibitor is described in a previously published protocol 7. Individual dose-response curves may need to be generated to determine optimal concentrations of the substrates and inhibitors in preparations/tissues/species not previously studied. Likewise, the protocol can be optimized for the innate metabolic tendencies of the tissue/species being studied, such as substrate preferences.
Once data collection is completed, gently aspirate the cerebellar homogenates and clean the chamber with ethanol and deionized water. Once clean, additional cerebellar homogenate samples can be run without additional calibration for as long as the P.T.F.E. membrane remains intact. After final completion, clean electrode with deionized water and ethanol and store with silica gel or desiccant. NOTE: Between same-day runs, fill the chamber with deionized water ensuring that the membrane does not dry out.
Using the data sheet created by the instrument software, export 40 min of respiration data beginning two minutes post substrate addition to a spreadsheet program.
Calculate the rate of respiration per substrate or inhibitor. Normalize data by dividing the total respiration by the respiration rate during the ascorbate-TMPD substrate addition. NOTE: The respiration rate can be further normalized to the protein content of the tissue which may be particularly desirable in neurodegeneration experiments in which the tissue content may vary. To do so, reserve 50 μL of the sample homogenate (from step 9.3) for use in a standard protein assay.
Representative Results
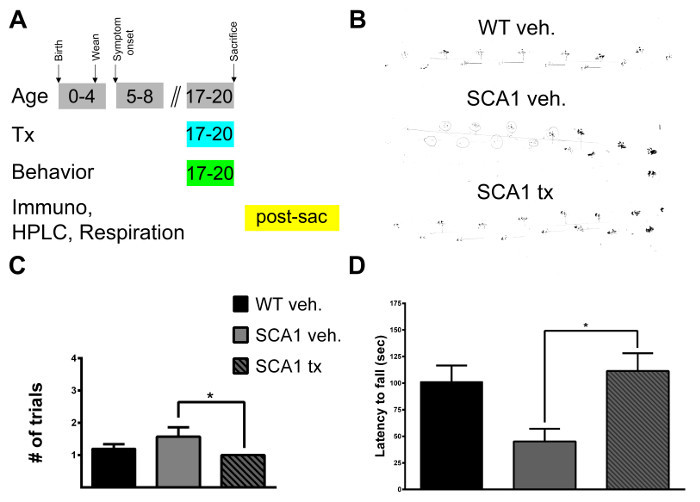
Through pharmacological targeting of cerebellar mitochondria with succinic acid we are able to prevent mitochondrial dysfunction in a mouse model of the cerebellar neurodegenerative disease SCA1. The canonical electron donor of succinate dehydrogenase, succinic acid, was dissolved in home cage drinking water of SCA1 mice for one month, with behavioral assessment beginning during the second week of treatment and neuropathological assessment following treatment (Figure 1A). Succinic acid treatment 22 improves performance of SCA1 mice as measured by the footprint assay, beam assay and accelerating rotarod assay. The behavioral improvements detected indicates enhanced motor coordination and motor learning (Figure 1B-D).
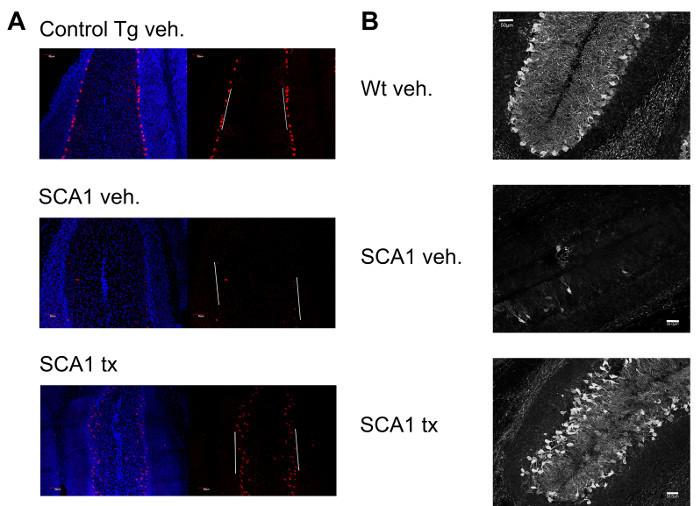
Rescue of behavioral performance was found in concert with a preservation of cerebellar tissue (Figure 2). Succinic acid treatment significantly preserved Purkinje cell bodies and increased molecular layer thickness (indicative of Purkinje cell dendritic arbor extension) in the cerebellar primary fissure in treated SCA1 mice compared to untreated SCA1 mice. Purkinje cell dendritic length and Purkinje cell body counts provide strong measures of overall cerebellar cytoarchitecture 5.
HPLC was performed on the cerebellar tissue from treated mice to verify that succinic acid dissolved in the cage drinking water successfully crossed the blood brain barrier and penetrated cerebellar tissue. A dose response curve varying the length of treatment further supports ad libitum treatment of succinic acid as a viable way of reaching cerebellar tissue (Figures 3A-B). Our respiration experiments are on-going and our results will be published in a research manuscript. Here we show that we can successfully record the rate of respiration through cerebellar oxidative phosphorylation complexes upon addition of varying electron transport chain substrates and inhibitors (Figure 3C). Ultimately, we will use the respiration assay to show oxidative phosphorylation deficits in SCA1 mouse cerebellum and the reversal of those deficits by succinic acid treatment.
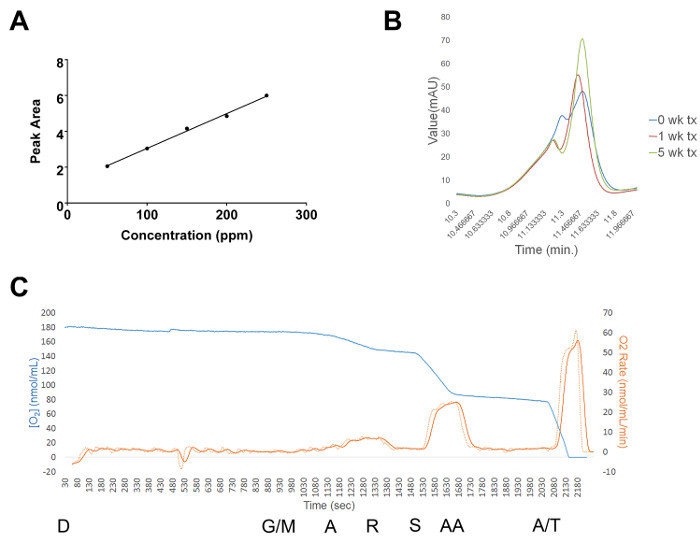
Figure 1:Treatment Scheme and Representative Data from the Footprint Assay, Beam Assay and Accelerating Rotarod.(A) Succinic acid was dissolved in home cage drinking water of SCA1 mice for four weeks beginning at 17 weeks of age. SCA1 mice begin to display the ataxia phenotype at 5 weeks of age. Not depicted in the schematic are three control cohorts: succinic acid-treated wildtype mice, vehicle-treated SCA1 mice and vehicle-treated wildtype mice. Behavioral assessments were conducted during the treatment period as follows: footprint assay during week 17, beam assay during week 18 and accelerating rotarod during week 19. At 20 weeks of age, mice were sacrificed and cerebellar tissue was harvested for immunopathology assays, HPLC assays and respiration assays. (B) Representative data from the footprint assay showing a vehicle-treated wildtype gait (top), vehicle-treated SCA1 gait (middle) and a succinic acid-treated SCA1 gait (bottom). (C) Beam analysis data showing improvement in beam performance (measured as number of trials for a successful run) of SCA1 mice with succinic acid treatment. (* p < 0.05). (D) Succinic acid treatment significantly improves accelerating rotarod performance of SCA1 22 mice as shown by increased latency to fall (* p < 0.05). Error bars in C-D reflect the standard error of the mean. Significance was determined by one-way ANOVA and multiple comparisons post-hoc analysis. Please click here to view a larger version of this figure.
Figure 2: Representative Data from the Purkinje Cell Counts and Molecular Layer Thickness Immunopathology Assays. (A) Representative results from the Purkinje cell counts assay showing a vehicle-treated control transgenic cerebellar primary fissure (top), vehicle-treated SCA1 cerebellar primary fissure (middle) and a succinic acid-treated SCA1 cerebellar primary fissure (bottom) stained for ATXN1-positive nuclei (red) and counterstained with DAPI (blue). White lines depict the 200 μm regions of tissue designated for cell counts. For this particular analysis, a control transgenic mouse over-expressing a non-pathogenic ATXN1 gene under control of a Purkinje cell-specific promoter was used instead of a wildtype mouse because the transgenic control features easily detectable levels of nuclear Purkinje ATXN1 and the wildtype mouse does not. The control transgenic mouse does not display an ataxic phenotype or cerebellar neurodegeneration. (B) Representative results from the molecular layer thickness assay showing a vehicle-treated wildtype cerebellar primary fissure (top), vehicle-treated SCA1 cerebellar primary fissure (middle) and a succinic acid-treated SCA1 cerebellar primary fissure (bottom) stained for calbindin, a marker of Purkinje cells (white). Two of the three cerebellar cell layers can be visualized with calbindin. The middle Purkinje cell layer (consisting of Purkinje cell bodies) is visible along with the outermost molecular layer (consisting of Purkinje cell dendrites). The molecular layer appears in the middle of the primary fissure due to the natural folds of the tissue. Please click here to view a larger version of this figure.
Figure 3:Representative Results from the HPLC and Respiration Assays. (A) Standard curve of known concentrations up to 250 ppm succinic acid diluted in acetate buffer and plotted against the area beneath the measured HPLC peak. The equation of the line was used to calculate unknown succinic acid concentrations from treated tissue samples. (B) Representative dose response succinic acid peaks from mouse cerebellar tissue following different times of treatment (either 0, 1 or 5 weeks). Under the conditions described, succinic acid typically eluted between 10 and 12 min. (C) Representative respiration run from vehicle-treated wildtype mouse cerebellar tissue. The blue line depicts the change in oxygen concentration, the dashed red line depicts the change in respiration rate and the solid red line depicts a smoothed version of the dashed line. D = addition of digitonin, G/M = addition of glutamate/malate, A = addition of ADP, R = addition of rotenone, S = addition of succinic acid, AA = addition of antimycin A and A/T = addition of Asc/TMPD. All substrates were added at the times depicted in the figure and at the concentrations described in the protocol (step 9.5). Please click here to view a larger version of this figure.
Discussion
If these methods are used as described, they are capable of detecting and alleviating oxidative phosphorylation-mediated mitochondrial dysfunction in cerebellar neurodegenerative disease mice models. The combined biochemical and behavioral assays are multifaceted methods for determining the extent of mitochondrial contribution to cerebellar neurodegenerative disease pathology. By treating mice with succinic acid to stimulate metabolism and boost mitochondrial function, we are able to show a rescue of cerebellar behavioral deficits and cerebellar degeneration.
The methodology presented here can be used to test a wide variety of water-soluble metabolically targeted compounds for potential treatment of cerebellar neurodegenerative disease. Succinic acid is readily soluble in water at 0.75 mg/mL which allows for simple delivery via cage drinking water and does not necessitate the creation of supplemented food pellets. Other water-soluble compounds can be easily substituted into the protocol. If using metabolically targeted compounds that are not water-soluble, our methodology can be readily adapted for treatment with supplemented food pellets 11, 12. When testing a new compound, it is critical to verify cerebellar penetration of the drug before commencing treatment via HPLC analysis or other suitable detection method.
The battery of pathological assessments was chosen due to their reported efficacy at detecting cerebellar neurodegenerative pathologies. The footprint assay, beam assay and rotarod assay are widely used assessments of cerebellar neurodegenerative disease including various SCAs, and Friedreich's ataxia 6, 23, 24. These particular behavioral assessments strongly correlate with cerebellar degeneration. However, other behavioral paradigms can be substituted or added as needed.
The labeling of Purkinje cells in SCA1 mice with ataxin-1 and calbindin is widely used to detect cerebellar degeneration in SCA1 models 5, 20, 21. Other neuronal-selective antibodies and tissue regions can be used to visualize the mitigation of neurodegeneration as needed.
Respiration analysis is a powerful tool for the specific detection of dysfunction or repair within individual mitochondrial complexes from cerebellar tissue. Care should be taken to minimize the time between harvesting the tissue and running the assay to achieve reproducible results. Also, it is important to note the homogeneity of the tissue being analyzed. Specifically, cerebellar tissue consists overwhelmingly of granule neurons which do not express the SCA1 transgene in our model. Therefore, a critical step of this protocol is to determine if whole cerebellar respiration is a viable method for detection of oxidative complex function. Respiration changes in the transgenic SCA1 mouse cerebellum may be subtle and may require increased numbers of subjects to achieve statistically significant results.
The combination of behavioral and neuropathological methods detailed in this protocol provide distinct quantification of SCA1 disease and can robustly detect improvement upon treatment with water-soluble compounds. The significance of this protocol is in its wide adaptability to various treatments and a diverse array of mouse models. Furthermore, many of the techniques used in this protocol do not require expensive equipment or complicated techniques and are therefore suitable for an undergraduate research setting. Future applications of this protocol will be used to assess the degree of treatment efficacy upon age-dependent delivery of the compound.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank Dr. Harry Orr at the University of Minnesota for his generous gift of transgenic mice. We would also like to thank the following Skidmore College alum for their work performing the preceding experiments: Monica Villegas, Porter Hall, Mitchell Spring, Nicholas Toker, Jenny Zhang, Chloe Larson and Cheyanne Slocum. Furthermore, we would like to thank Skidmore College for funding the development of these methods.
References
- Stucki DM, et al. Mitochondrial impairments contribute to Spinocerebellar ataxia type 1 progression and can be ameliorated by the mitochondria-targeted antioxidant MitoQ. Free Radic Biol Med. 2016;97:427–440. doi: 10.1016/j.freeradbiomed.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Breuer ME, Willems PH, Russel FG, Koopman WJ, Smeitink JA. Modeling mitochondrial dysfunctions in the brain, from mice to men. J Inherit Metab Dis. 2012;35(2):193–210. doi: 10.1007/s10545-011-9375-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer ME, et al. The role of mitochondrial OXPHOS dysfunction in the development of neurologic diseases. Neurobiol Dis. 2012. [DOI] [PubMed]
- Hroudová J, Singh N, Fizar Z. Mitochondrial dysfunctions in neurodegenerative diseases, relevance to Alzheimer's disease. Biomed Res Int. 2014;2014:175062. doi: 10.1155/2014/175062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burright EN, et al. SCA1 transgenic mice, a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82(6):937–948. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- Clark HB, et al. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17(19):7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov AV, et al. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3(6):965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- Deng-Bryant Y, Singh IN, Carrico KM, Hall ED. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab. 2008;28(6):1114–1126. doi: 10.1038/jcbfm.2008.10. [DOI] [PubMed] [Google Scholar]
- Vaishnav RA, Singh IN, Miller DM, Hall ED. Lipid peroxidation-derived reactive aldehydes directly and differentially impair spinal cord and brain mitochondrial function. J Neurotrauma. 2010;27(7):1311–1320. doi: 10.1089/neu.2009.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RT, Yang L, Browne S, Baik M, Beal MF. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci U S A. 1998;95(15):8892–8897. doi: 10.1073/pnas.95.15.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, et al. Neuroprotective effects of creatine in a transgenic mouse model of Huntington's disease. J Neurosci. 2000;20(12):4389–4397. doi: 10.1523/JNEUROSCI.20-12-04389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, et al. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci. 2002;22(5):1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2'dG. Neurology. 2006;66(2):250–252. doi: 10.1212/01.wnl.0000194318.74946.b6. [DOI] [PubMed] [Google Scholar]
- Yang L, et al. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson's and Huntington's diseases. J Neurochem. 2009;109(5):1427–1439. doi: 10.1111/j.1471-4159.2009.06074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Dai G, Li G, Yang ES. Coenzyme Q10 reduces beta-amyloid plaque in an APP/PS1 transgenic mouse model of Alzheimer's disease. J Mol Neurosci. 2010;41(1):110–113. doi: 10.1007/s12031-009-9297-1. [DOI] [PubMed] [Google Scholar]
- Sandhir R, Mehrotra A. Quercetin supplementation is effective in improving mitochondrial dysfunctions induced by 3-nitropropionic acid, implications in Huntington's disease. Biochim Biophys Acta. 2013;1832(3):421–430. doi: 10.1016/j.bbadis.2012.11.018. [DOI] [PubMed] [Google Scholar]
- Ergonul PG, Nergiz C. Determination of organic acids in olive fruit by HPLC. 'Czech Food Sci. 2010;28(3):202–205. [Google Scholar]
- Jones BJ, Roberts DJ. The quantiative measurement of motor inco-ordination in naive mice using an acelerating rotarod. J Pharm Pharmacol. 1968;20(4):302–304. doi: 10.1111/j.2042-7158.1968.tb09743.x. [DOI] [PubMed] [Google Scholar]
- Luong TN, Carlisle HJ, Southwell A, Patterson PH. Assessment of motor balance and coordination in mice using the balance beam. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]
- Servadio A, Koshy B, Armstrong D, Antalffy B, Orr HT, Zoghbi HY. Expression analysis of the ataxin-1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat Genet. 1995;10(1):94–98. doi: 10.1038/ng0595-94. [DOI] [PubMed] [Google Scholar]
- Klement IA, et al. Ataxin-1 nuclear localization and aggregation, role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95(1):41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- Serra HG, et al. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet. 2004;13(20):2535–2543. doi: 10.1093/hmg/ddh268. [DOI] [PubMed] [Google Scholar]
- Carter RJ, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19(8):3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjomani Virmouni S, et al. A novel GAA-repeat-expansion-based mouse model of Friedreich's ataxia. Dis Model Mech. 2015;8(3):225–235. doi: 10.1242/dmm.018952. [DOI] [PMC free article] [PubMed] [Google Scholar]