

Abstract
Apart from their essential role in generating ATP, mitochondria also act as local calcium (Ca2+) buffers to tightly regulate intracellular Ca2+ concentration. To do this, mitochondria utilize the electrochemical potential across their inner membrane (ΔΨm) to sequester Ca2+. The influx of Ca2+ into the mitochondria stimulates three rate-limiting dehydrogenases of the citric acid cycle, increasing electron transfer through the oxidative phosphorylation (OXPHOS) complexes. This stimulation maintains ΔΨm, which is temporarily dissipated as the positive calcium ions cross the mitochondrial inner membrane into the mitochondrial matrix.
We describe here a method for simultaneously measuring mitochondria Ca2+ uptake and ΔΨm in live cells using confocal microscopy. By permeabilizing the cells, mitochondrial Ca2+ can be measured using the fluorescent Ca2+ indicator Fluo-4, AM, with measurement of ΔΨm using the fluorescent dye tetramethylrhodamine, methyl ester, perchlorate (TMRM). The benefit of this system is that there is very little spectral overlap between the fluorescent dyes, allowing accurate measurement of mitochondrial Ca2+ and ΔΨm simultaneously. Using the sequential addition of Ca2+ aliquots, mitochondrial Ca2+ uptake can be monitored, and the concentration at which Ca2+ induces mitochondrial membrane permeability transition and the loss of ΔΨm determined.
Keywords: Cellular Biology, Issue 119, Mitochondria, membrane potential, calcium, fluorescent staining, live cells, confocal microscopy
Introduction
Mitochondria play an important role in regulating intracellular Ca2+ concentration by acting as local Ca2+ buffers1. Ca2+ enters the mitochondria via the Ca2+ uniporter, a process driven by the electrochemical gradient that exists across the mitochondrial inner membrane (ΔΨm)2. Once inside the mitochondrial matrix, Ca2+ can activate oxidative phosphorylation by stimulating three rate-limiting dehydrogenases of the citric acid cycle3. This stimulation maintains ΔΨm, which is temporarily dissipated as the positive calcium ions cross the mitochondrial inner membrane into the mitochondrial matrix. If the Ca2+ concentration within the mitochondria becomes very high, mitochondrial permeability transition can be initiated, resulting in the dissipation of ΔΨm, the cessation of oxidative phosphorylation and the induction of cell death signaling pathways4.
The important role that mitochondria play in the spatial buffering of cellular calcium makes the accurate monitoring of mitochondrial calcium critical. Various methods have been established to monitor mitochondrial calcium, including the use of rhodamine based dyes. One such dye, Rhod-2, AM, is quite effective at partitioning to the mitochondria to measure mitochondrial Ca2+ levels5,6. However, care must be used as some dye will accumulate in other organelles, such as liposomes, or remain in the cell cytosol. Nevertheless, downstream analyses can be employed to distinguish these signals from those from the mitochondria7.
Another technique to monitor mitochondrial calcium utilizes fluorescent reporter constructs8. The benefit of these genetically encoded probes is that they can be specifically targeted to the mitochondria by using endogenous N-terminal peptides, for example the N-terminal targeting signal of human COX subunit VIII. This system has been employed to generate a mitochondrial-targeted aequorin probe which has proved extremely useful for investigating mitochondrial calcium signaling9. The main drawback of these genetically encoded probes is that they need to be introduced into the cells by transient expression (which is not feasible for certain cell types and can produce variable results) or by creating stable expression systems (which is time consuming).
To circumvent the problems outlined above, we have developed a new protocol to measure mitochondrial Ca2+ and ΔΨm simultaneously. This protocol is based on a previously described method that adds exogenous calcium to permeabilized cells10. Our protocol has three main advantages over other methods: firstly, we use Fluo-4, AM and TMRM to monitor mitochondrial Ca2+ and ΔΨm, two dyes that have very distinct spectral properties; secondly, the cells are permeabilized so that the Fluo-4 signal is only detecting mitochondrial Ca2+ and not Ca2+ localized to other organelles or the cell cytosol; and thirdly, the use of Fluo-4 to detect mitochondrial Ca2+ allows for fast and simple cell staining, negating any cell transfection or transformation issues that exist if using genetically encoded probes.
Protocol
1. Preparation of Cells
Grow cells on 10 cm cell culture dishes or 75 cm2 flasks in culture media [10 ml Dulbecco's Modified Eagles Medium (DMEM) supplemented with 5% (v/v) fetal bovine serum (FBS) and 1x penicillin/streptomycin (p/s)] at 37 °C/5% CO2.
To harvest cells, remove media by aspiration, then wash with 5 ml 1x phosphate buffered saline (1x PBS). Remove 1x PBS by aspiration, then add 1.5 ml 0.25% (w/v) Trypsin/0.25% (w/v) ethylenediaminetetraacetic acid (EDTA) and incubate at 37 °C/5% CO2 for 2 min. Tap dish or flask gently to remove cells, then resuspend in 5 ml culture media.
Count cells by adding 12 µl of resuspended cells onto a hemocytometer.
Plate cells in dishes or chambered coverslips for confocal imaging. NOTE: These will have a suitable plastic or glass bottom that has been designed for use with inverted microscopes.
Plate cells so that ~80% confluency is achieved on the day of imaging. For example, approximately 2 x 104 143B osteosarcoma cells can be plated into one well of an 8-well chamber slide one day before imaging.
Incubate cells overnight at 37 °C/5% CO2 to allow cells to attach and recover.
2. Buffers for TMRM and Fluo-4 Imaging
Prepare Record Solution (RS) containing 156 mM NaCl, 3 mM KCl, 2 mM MgSO4, 1.25 mM KH2PO4, 10 mM D-glucose, 2 mM CaCl2 and 10 mM HEPES pH 7.35. Store RS in aliquots at -20 ºC.
Prepare Intracellular Medium (IM) containing 6 mM NaCl, 130 mM KCl, 7.8 mM MgCl2, 1 mM KH2PO4, 0.4 mM CaCl2, 2 mM EGTA, 10 mM HEDTA, 2 mM malate, 2 mM glutamate, 2 mM ADP and 20 mM HEPES pH 7.1. 200 mM stock solutions of malate, glutamate and ADP can be prepared separately, aliquoted, and stored at -20 °C. These stocks can be added to the IM just before use.
Ca2+ free Hank's buffered salt solution (HBSS) can be obtained pre-prepared from commercial suppliers. Add ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA) to a final concentration of 500 µM.
Prepare a stock solution of 10 mM tetramethylrhodamine, methyl ester, perchlorate (TMRM) in 100% methanol. From this stock, make a working stock of 2 µM in distilled H2O.
Prepare a stock solution of 10 mM Verapamil in 100% ethanol.
Prepare a 1 mg/ml (w/v) stock solution of Fluo-4 acetoxymethyl ester (Fluo-4, AM) in dimethyl sulfoxide (DMSO). NOTE: Fluo-4, AM is a calcium indicator that exhibits increased fluorescence upon binding Ca2+. Fluo-4 is an analog of fluo-3, with the two chlorine substituents replaced by fluorines. This results in increased fluorescence excitation at 488 nm and higher fluorescence signal levels. The AM ester group results in an uncharged Fluo-4 molecule that can permeate cell membranes. Once inside the cell, the AM ester is cleaved by nonspecific esterases, resulting in a charged form of Fluo-4 that is trapped within the cell.
Prepare a stock solution of 1 mM carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) in 100% ethanol.
Prepare a stock solution of 25 mg/ml (w/v) digitonin in distilled H2O.
Prepare a stock solution of 100 µM thapsigargin in DMSO.
Prepare a stock solution of 40 mM calcium chloride (CaCl2) in distilled H2O.
3. Staining of Cells with TMRM and Fluo-4, AM
Prepare RS staining solution with 20 nM TMRM, 5 µg/ml (w/v) Fluo-4, AM and 0.005% surfactant such as Pluronic F-127 in RS. NOTE: This surfactant is a nonionic polyol that facilitates the solubilization of Fluo-4, AM. Note: 10 µM Verapamil can also be added to inhibit TMRM export by the plasma membrane multidrug transporter if it is expressed in the cell type being analyzed.
Remove culture media from cells by pipette and wash with 100 µl 1x PBS.
Remove 1x PBS by pipette and incubate cells in 250 µl RS staining solution for 45 min at room temperature. NOTE: Once the Fluo-4, AM enters the cell, the AM ester is cleaved by cellular esterases. Incubating at room temperature inhibits esterase activity, allowing Fluo-4, AM to enter the mitochondria.
Remove RS staining solution by pipette and wash cells in 100 µl Ca2+ free HBSS to remove excess Fluo-4, AM and TMRM.
Prepare IM imaging solution with 25 µg/ml (w/v) digitonin, 200 nM TMRM and 1 µM thapsigargin in IM. NOTE: The concentration of digitonin may need to be adjusted to ensure that permeabilization of the mitochondrial inner membrane does not occur. This can be determined empirically by assessing the intensity of the TMRM signal at different concentrations of digitonin.
Remove the Ca2+ free HBSS by pipette and add 300 µl of IM imaging solution to the cells. Leave cells to equilibrate at room temperature for 10 min. Cells are now ready for imaging with CaCl2 additions.
4. Cell Imaging
Place dish or chambered coverslip with cells in IM imaging solution onto an inverted laser scanning confocal microscope.
Use setting on the microscope lasers for the excitation and emission spectra of TMRM and Fluo-4. For example, use 543 nm He-Ne and 473 nm argon laser lines to excite TMRM and Fluo-4 respectively. Use a low laser power of approximately 5% to minimize any photo-damage to the cells.
Set microscope to scan images every 25 sec. Scan cells for 10 min to establish baseline readings for the TMRM and Fluo-4 signals. NOTE: The Fluo-4 signal may be weak or undetectable at resting calcium concentrations.
Add 3 μl of 40 mM CaCl2 stock solution directly to the cells in 300 μl of IM imaging solution (a 1:100 dilution) and mix gently with a pipette. Pause the image scanning while adding and mixing the CaCl2 if necessary. NOTE: The final free Ca2+ ion concentration [Ca2+] in the IM imaging solution can be calculated using software such as Maxchelator WEBMAXC EXTENDED (http://www.stanford.edu/%7Ecpatton/maxc.html) or Chelator11. A detailed description of how to calculate the final free Ca2+ ion concentration [Ca2+] is described in section 6 below.
Continue image scanning for approximately 4 min, then repeat CaCl2 additions every 4 min until the desired TMRM or Fluo-4 signals are reached, usually around 8 to 10 additions.
Using a pipette, add a 1:100 dilution of 1 mM FCCP (final concentration of 10 μM) directly to the cells to dissipate ΔΨm. Image cells for a further 5 min. The experiment is now finished.
5. Image Analysis
Once imaging is complete, determine the intensity of the Fluo-4 and TMRM signals using image analysis software, for example ImageJ software with the Bio-Formats plugin (download 'bioformats_package.jar' from http://downloads.openmicroscopy.org/bio-formats/ and save it in the 'C:\Program Files\ImageJ\plugins' folder).
Open the imaging file (for example an .oif file) in ImageJ. Click on the selection tool and select a region of interest (ROI) that contains mitochondria. Use the initial TMRM signal to select the ROI.
Open 'Analyze>Tools>ROI Manager', click 'Add' to include the selected ROI for intensity measurement. Multiple ROIs can be selected on a single image for measurement. Repeat previous steps until all ROIs have been added to the ROI Manager.
Click 'More>Multi Measure’, make sure that ‘Measure all slices’ is selected and that ‘One Row Per Slice’ is deselected, then click ‘OK’ to obtain a table of mean fluorescent intensity of the TMRM and Fluo-4 signals for each ROI at each time point.
Use data from all ROIs to calculate the average intensities and standard deviation for the TMRM and Fluo-4 signals.
6. Calculating the Final Free Ca2+ Ion Concentration [Ca2+]
The free Ca2+ ion concentration [Ca2+] in the IM imaging solution can be determined using software such as Maxchelator WEBMAXC EXTENDED (http://www.stanford.edu/%7Ecpatton/maxc.html) or Chelator11. For example, set the variables in WEBMAXC EXTENDED as follows: Temperature = 24 °C, pH = 7.1, Ionic strength = 0.162, EGTA = 0.002 M, HEDTA = 0.01 M, Ca2+ = 0.0004 M and Mg2+ = 0.0078 M. This results in a final free Ca2+ ion concentration [Ca2+] of 62 nM. Note that these programs cannot take into account every variable, such as reagent purity, accuracy of measurements and pH determination. The most accurate method for determining the free Ca2+ concentration is to use a calibrated Ca2+ selective electrode.
Representative Results
We have used this protocol to examine the effects of an MT-ND5 mutation on the ability of 143B cell mitochondria to buffer increases in calcium12. In the example shown here, control 143B cells were loaded with TMRM and Fluo-4, AM before permeabilization with digitonin. After 5 min of imaging, eight sequential additions of a 1:100 dilution of 40 mM exogenous CaCl2 were made, with the final free Ca2+ ion concentration [Ca2+] calculated.
Following each CaCl2 addition, the TMRM signal decreases as the influx of Ca2+ ions into the mitochondria dissipates the mitochondrial membrane potential ΔΨm (Figure 1). ΔΨm then recovers as respiration is increased to maintain ΔΨm. This occurs four times after each CaCl2 addition. Following the fifth addition, the mitochondrial Ca2+ ion concentration reaches a critical level, at which point mitochondrial permeability transition occurs and ΔΨm dissipates rapidly. A small ΔΨm is evident following permeability transition, as the addition of 10 µM FCCP still induces a collapse of ΔΨm (Figure 1).
The mitochondrial calcium concentration (Fluo-4 signal) begins to increase following the second CaCl2 addition when the free [Ca2+] in the media reaches 0.32 μM (Figure 2). Each subsequent CaCl2 addition caused a progressive increase in mitochondrial calcium.
Images of the simultaneous measurements of ΔΨm (TMRM, red signal) and mitochondrial calcium (Fluo-4, green signal) are shown (Figure 3).
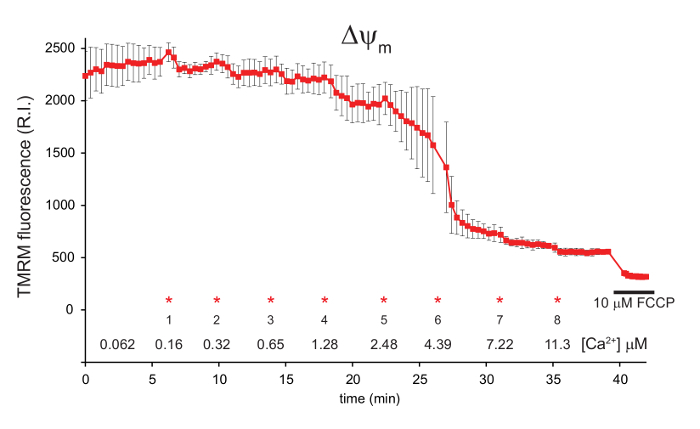 Figure 1: Measurement of mitochondrial ΔΨm in digitonin permeabilized 143B cells using TMRM. 143B cells were preloaded with Fluo-4, AM and TMRM before permeabilization with digitonin. A 1:100 dilution of 40 mM exogenous CaCl2 was added in sequential aliquots. The final free [Ca2+] in the media is indicated for each addition (*). FCCP was added to a final concentration of 10 µM after approximately 40 min. ΔΨm is shown in red, represented by the relative intensity (R.I.) of the TMRM signal. Data is mean ± s.d. n = 3. Figure is adapted with permission from reference12. Please click here to view a larger version of this figure.
Figure 1: Measurement of mitochondrial ΔΨm in digitonin permeabilized 143B cells using TMRM. 143B cells were preloaded with Fluo-4, AM and TMRM before permeabilization with digitonin. A 1:100 dilution of 40 mM exogenous CaCl2 was added in sequential aliquots. The final free [Ca2+] in the media is indicated for each addition (*). FCCP was added to a final concentration of 10 µM after approximately 40 min. ΔΨm is shown in red, represented by the relative intensity (R.I.) of the TMRM signal. Data is mean ± s.d. n = 3. Figure is adapted with permission from reference12. Please click here to view a larger version of this figure.
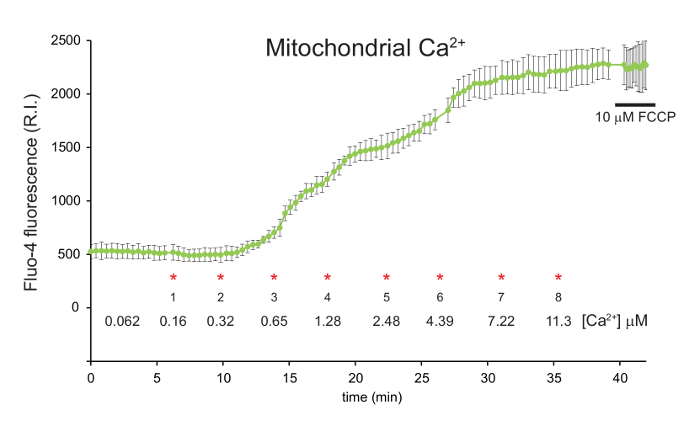 Figure 2: Measurement of mitochondrial calcium in digitonin permeabilized 143B cells using Fluo-4, AM. 143B cells were preloaded with Fluo-4, AM and TMRM before permeabilization with digitonin. A 1:100 dilution of 40 mM exogenous CaCl2 was added in sequential aliquots. The final free [Ca2+] in the media is indicated for each addition (*). FCCP was added to a final concentration of 10 µM after approximately 40 min. Mitochondrial calcium is shown in green, represented by the relative intensity (R.I.) of the Fluo-4 signal. Data is mean ± s.d. n = 3. Figure is adapted with permission from reference12. Please click here to view a larger version of this figure.
Figure 2: Measurement of mitochondrial calcium in digitonin permeabilized 143B cells using Fluo-4, AM. 143B cells were preloaded with Fluo-4, AM and TMRM before permeabilization with digitonin. A 1:100 dilution of 40 mM exogenous CaCl2 was added in sequential aliquots. The final free [Ca2+] in the media is indicated for each addition (*). FCCP was added to a final concentration of 10 µM after approximately 40 min. Mitochondrial calcium is shown in green, represented by the relative intensity (R.I.) of the Fluo-4 signal. Data is mean ± s.d. n = 3. Figure is adapted with permission from reference12. Please click here to view a larger version of this figure.
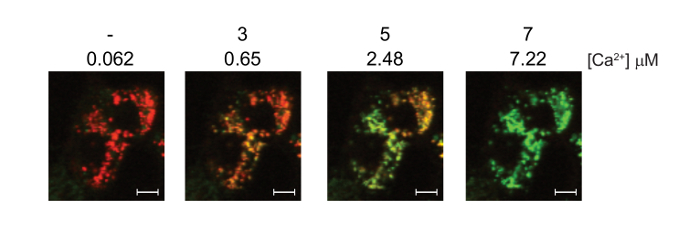 Figure 3: Simultaneous measurement of mitochondrial calcium and ΔΨm in digitonin permeabilized 143B cells using Fluo-4, AM and TMRM. 143B cells were preloaded with Fluo-4, AM and TMRM before permeabilization with digitonin. A 1:100 dilution of 40 mM exogenous CaCl2 was added in sequential aliquots. The final free [Ca2+] in the media is indicated for each addition. Representative confocal images showing the simultaneous staining of ΔΨm (TMRM, red) and mitochondrial calcium (Fluo-4, green). Note that the Fluo-4 signal is hard to detect at the resting calcium concentration of 0.062 μM. White scale bars = 10 μm. Figure is adapted with permission from reference12. Please click here to view a larger version of this figure.
Figure 3: Simultaneous measurement of mitochondrial calcium and ΔΨm in digitonin permeabilized 143B cells using Fluo-4, AM and TMRM. 143B cells were preloaded with Fluo-4, AM and TMRM before permeabilization with digitonin. A 1:100 dilution of 40 mM exogenous CaCl2 was added in sequential aliquots. The final free [Ca2+] in the media is indicated for each addition. Representative confocal images showing the simultaneous staining of ΔΨm (TMRM, red) and mitochondrial calcium (Fluo-4, green). Note that the Fluo-4 signal is hard to detect at the resting calcium concentration of 0.062 μM. White scale bars = 10 μm. Figure is adapted with permission from reference12. Please click here to view a larger version of this figure.
Discussion
Calcium plays a critical role in many cell processes, including muscle contraction, neuronal signaling and cell proliferation13. Increases in cell calcium concentrations are often associated with energy demand, with calcium able to directly stimulate mitochondrial oxidative phosphorylation to raise ATP generation3. It is therefore essential that we have the ability to effectively monitor mitochondrial calcium accumulation and to be able to compare how this function is affected by both genetic factors and pharmacological agents.
Critical steps within the protocol
This protocol describes the ability to monitor mitochondria calcium accumulation and ΔΨm simultaneously with Fluo-4, AM and TMRM. During the staining process it is critical to incubate the cells at room temperature to optimize the effectiveness of Fluo-4, AM in detecting mitochondrial calcium. Incubation at room temperature reduces the activity of cytosolic esterases, allowing the uncharged Fluo-4, AM to cross the mitochondrial membranes and enter the mitochondrial matrix. Conversely, incubation at 37 °C enhances esterase activity in the cytosol, cleaving off the AM ester to leave a charged Fluo-4 dye that would not be able to cross the mitochondrial membranes effectively.
Once cell staining has been performed, the cells are immersed in IM imaging solution. This solution has a higher TMRM concentration compared to the initial RS staining solution. Now that the cells are permeabilized, the TMRM no longer equilibrates across the plasma membrane. Therefore, the TMRM concentration is raised in the IM imaging solution to achieve the correct equilibration across the mitochondrial inner membrane. It is also critical at this stage to include thapsigargin in the IM imaging solution. Thapsigargin blocks the endoplasmic reticulum (ER) Ca2+ATPase, ensuring that the Fluo-4 signal is not detecting ER calcium.
Modifications and troubleshooting
Some cell types can express the multidrug resistance transporter (also known as MDR1 or P-glycoprotein) on their plasma membranes. This protein is known to export fluorescent indicators, including AM ester derivatives, from the cytosol out of the cell14. If the transporter is expressed, both Fluo-4, AM and TMRM may be exported from the cell cytosol, resulting in sub-optimal staining. To block this effect, Verapamil can be added to the RS staining solution to block the multidrug resistance transporter, inhibiting Fluo-4, AM and TMRM export during the staining process.
By permeabilizing the cells to be analyzed, Fluo-4, AM can be used to detect mitochondrial Ca2+, with no competing signals from other organelles or the cytosol. We routinely use the non-ionic detergent digitonin for permeabilization at a concentration of 25 µg/ml. This concentration may need to be adjusted to ensure that solubilization of the mitochondrial membranes does not occur. This can be determined empirically by assessing the intensity of the TMRM signal at different concentrations of digitonin. If the concentration of digitonin is too high, the mitochondrial membrane will be partially solubilized, reducing the TMRM signal.
Limitations of the technique
One of the main benefits of this protocol is that by using Fluo-4, AM to detect mitochondrial calcium, TMRM can be used to simultaneously measure ΔΨm, with negligible spectral overlap between the two dyes. To ensure the specificity of Fluo-4 for mitochondrial calcium, the cells being examined need to be permeabilized to eliminate the cytosolic Fluo-4 signal. This permeabilization is one limitation of this technique. While the IM imaging solution closely matches the ionic strength of the cell cytosol, it can't completely replicate all of its constituents. This may affect the results if specific cytosolic components are required in the experimental system being examined. Furthermore, as the cells are permeabilized, the protocol may not be suitable for longer experiments greater than 1 hr.
Significance of the technique with respect to existing/alternative methods
Various fluorescent dyes have been developed for assessing mitochondrial calcium8. These dyes are simple to use and can provide useful data in a short space of time. While these dyes accumulate predominantly in the mitochondria, some can also accumulate in other organelles, including liposomes, or remain in the cell cytosol. Therefore, care must be taken to ensure that these other signals do not influence the mitochondrial calcium signal.
In this protocol, mitochondrial Ca2+ is measured in permeabilized cells in the presence of thapsigargin. The advantage of this methodology is that the cytosolic and ER calcium signals are eliminated. Furthermore, permeabilization allows the use of Fluo-4, AM to detect mitochondrial Ca2+. Fluo-4, AM has very little spectral overlap with TMRM, meaning that mitochondrial Ca2+ and ΔΨm can be measure simultaneously using these two dyes.
Mitochondrial Ca2+ can also be measured using genetically encoded fluorescent reporters8. These reporters can be targeted to the mitochondria, resulting in specific mitochondrial Ca2+ signals. Genetically encoded probes need to be introduced into the cells being studied, which in some cases can take considerable time and effort. Conversely, our protocol uses the dyes Fluo-4, AM and TMRM, allowing for fast and simple cell staining to measure mitochondrial Ca2+ and ΔΨm simultaneously.
Future applications or directions after mastering this technique
Mitochondria act as local calcium buffers to regulate intracellular calcium concentrations and the energy requirements of the cell. When this process is disrupted, excessive mitochondrial calcium uptake can induce mitochondrial permeability transition, resulting in the collapse of ΔΨm and the release of pro-apoptotic molecules that trigger cell death induction4.
We present a fast and straight-forward protocol for the examination of mitochondrial calcium and its relationship to ΔΨm and the induction of mitochondrial permeability transition. This can be used to investigate how these mitochondrial parameters influence pathogenesis in a wide range of human diseases, including diabetes15 and age-related neurodegeneration conditions such as Alzheimer's and Parkinson's Disease16. Furthermore, this protocol can be used to examine how mitochondrial calcium and ΔΨm are affected by genetic defects or environmental toxins, and also how these affects can be modulated by therapies or drugs which target the mitochondria. These types of future experiments can provide important new insights into the role that mitochondrial calcium plays in both human health and disease.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Dr Kirstin Elgass and Dr Sarah Creed from Monash Micro Imaging for technical assistance, and the Wellcome Trust and Medical Research Council UK for financial support. MMcK is supported the Australian Research Council Future Fellowship Scheme (FT120100459), the William Buckland Foundation, The Australian Mitochondrial Disease Foundation (AMDF), The Hudson Institute of Medical Research and Monash University. This work was supported by the Victorian Government Operational Infrastructure Support Scheme.
References
- Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology. 2008;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- Jacobson J, Duchen MR. Interplay between mitochondria and cellular calcium signalling. Mol. Cell. Biochem. 2004;256-257:209–218. doi: 10.1023/b:mcbi.0000009869.29827.df. [DOI] [PubMed] [Google Scholar]
- Bhosale G, Sharpe JA, Sundier SY, Duchen MR. Calcium signaling as a mediator of cell energy demand and a trigger to cell. Ann. N. Y. Acad. Sci. 2015;1350:107–116. doi: 10.1111/nyas.12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch. 2012;464:111–121. doi: 10.1007/s00424-012-1112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond RM, Mix TC, Tuft RA, Walsh JV, Fay FS. Mitochondrial Ca2+ homeostasis during Ca2+ influx and Ca2+ release in gastric myocytes from Bufo marinus. J. Physiol. 2000;522:375–390. doi: 10.1111/j.1469-7793.2000.t01-2-00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Davidson SM, Duchen MR. Imaging mitochondrial calcium signalling with fluorescent probes and single or two photon confocal microscopy. Methods Mol. Biol. 2012;810:219–234. doi: 10.1007/978-1-61779-382-0_14. [DOI] [PubMed] [Google Scholar]
- Pozzan T, Rudolf R. Measurements of mitochondrial calcium in vivo. Biochim. Biophys. Acta. 2009;1787:1317–1323. doi: 10.1016/j.bbabio.2008.11.012. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–327. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- Pitter JG, Maechler P, Wollheim CB, Spat A. Mitochondria respond to Ca2+ already in the submicromolar range: correlation with redox state. Cell Calcium. 2002;31:97–104. doi: 10.1054/ceca.2001.0264. [DOI] [PubMed] [Google Scholar]
- Schoenmakers TJ, Visser GJ, Flik G, Theuvenet AP. CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. BioTechniques. 1992;12:870–879. [PubMed] [Google Scholar]
- McKenzie M, Duchen MR. Impaired Cellular Bioenergetics Causes Mitochondrial Calcium Handling Defects in MT-ND5 Mutant Cybrids. PLoS One. 2016;11:e0154371. doi: 10.1371/journal.pone.0154371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Homolya L, Hollo Z, Germann UA, Pastan I, Gottesman MM, Sarkadi B. Fluorescent cellular indicators are extruded by the multidrug resistance protein. J. Biol. Chem. 1993;268:21493–21496. [PubMed] [Google Scholar]
- Fujimoto K, Chen Y, Polonsky KS, Dorn GW 2nd. Targeting cyclophilin D and the mitochondrial permeability transition enhances beta-cell survival and prevents diabetes in Pdx1 deficiency. Proc. Natl. Acad. Sci. U.S.A. 2010;107:10214–10219. doi: 10.1073/pnas.0914209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VK, Carlson EA, Yan SS. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta. 2014;1842:1267–1272. doi: 10.1016/j.bbadis.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]