

Abstract
Most fluorescent nucleoside analogues are quenched when base stacked and some maintain their brightness, but there has been little progress toward developing nucleoside analogues that markedly increase their fluorescence upon duplex formation. Here, we report on the design and synthesis of a new tricyclic cytidine analogue, 8-diethylamino-tC (8-DEA-tC), that responds to DNA duplex formation with up to a 20-fold increase in fluorescent quantum yield as compared with the free nucleoside, depending on neighboring bases. This turn-on response to duplex formation is the greatest of any reported nucleoside analogue that can participate in Watson–Crick base pairing. Measurements of the quantum yield of 8-DEA-tC mispaired with adenosine and, separately, opposite an abasic site show that there is almost no fluorescence increase without the formation of correct Watson–Crick hydrogen bonds. Kinetic isotope effects from the use of deuterated buffer show that the duplex protects 8-DEA-tC against quenching by excited state proton transfer. These results, supported by DFT calculations, suggest a rationale for the observed photophysical properties that is dependent on duplex integrity and the electronic structure of the analogue.
The recognition in 1969 of 2-aminopurine’s potential as a fluorescent adenine surrogate has driven a desire to expand the capabilities of fluorescent nucleosides.1–16 Recent highlights include Tor’s isomorphic RNA alphabet,17,18 Wilhelmsson’s internucleobase FRET pair,12,19 Sasaki’s fluorescent sensors for nucleobase damage,20 and a number of examples of nucleobase surrogates that can report on events in the major groove, such as protein binding.21,22 Still, significant unmet needs persist. For example, most fluorescent nucleobase analogues are quenched when base stacked, emit only at wavelengths <525 nm, and many perturb duplex structure.23,24 Here, we provide another unmet capability: a nucleoside analogue that is virtually nonfluorescent as a free nucleoside but becomes much brighter when base stacked. These properties have great value in turn-on fluorescence sensing applications for both enzymatic DNA synthesis and strand hybridization.
Little progress has yet been made on nucleoside analogues with substantial fluorescent turn-on responses to incorporation in duplex DNA.25–27 Probably the closest is Luedtke’s recently reported DMAT nucleoside, which exhibits up to 7-fold increase in quantum yield when base-paired with A, but this analogue is fluorescent as a free nucleoside (Φem = 0.03) and the mechanism of its fluorescence increase was not studied in detail.28 On the basis of our experience with tricyclic cytidines (tC molecules),29–31 we hypothesized that electronic modification of tC to favor a charge-separated excited state might be the key to unlocking a fluorescence turn-on response to duplex formation. Here, we report on a novel tricyclic cytidine analogue 8-DEA-tC 1 (Figure 1) that exhibits up to a 20-fold fluorescence enhancement in duplex DNA as compared with its almost nonfluorescent free nucleoside (Φem = 0.006). We provide the first mechanistic relationships between structure and this type of fluorescence turn-on response.
Figure 1.
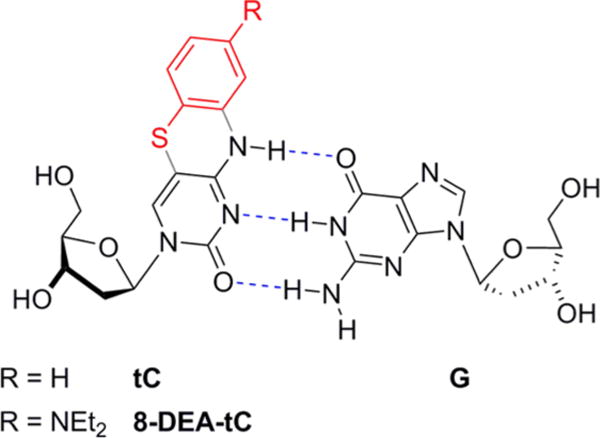
8-DEA-tC and parent tC nucleosides with Watson–Crick base pairing with G.
The synthesis of 8-DEA-tC 1 was developed based on previous syntheses of tC derivatives (Scheme 1).29 We began by doubly alkylating 5-amino-2-methylbenzothiazole 2 with bromoethane. Next, we performed a nucleophilic opening of the thiazole ring using hydrazine to afford 4-diethylamino-2-aminothiophenol, which oxidized under air to give the disulfide 3 (64% over two steps). Then, the disulfide was reduced using triethylphosphine followed by a substitution reaction with 5-bromouracil in one pot to yield thioether 4 (36%). Heating this compound at 120 °C in a mixture of concentrated HCl and butanol effected ring closure to afford the 8-DEA-tC nucleobase 5 (97%). Activation of 5 using BSA to give the TMS ether and ribosylation in the same pot using Hoffer’s chlorosugar 6 resulted in a 1:1 mixture of the α and β anomers in a combined yield of 86%. Isolation of the β anomer was facilitated by the removal of the toluoyl groups to give the 8-DEA-tC nucleoside 1. Standard procedures were used for dimethoxytrityl protection and phosphoramidite preparation to ready the nucleoside for solid-phase DNA synthesis (Supporting Information).
Scheme 1.
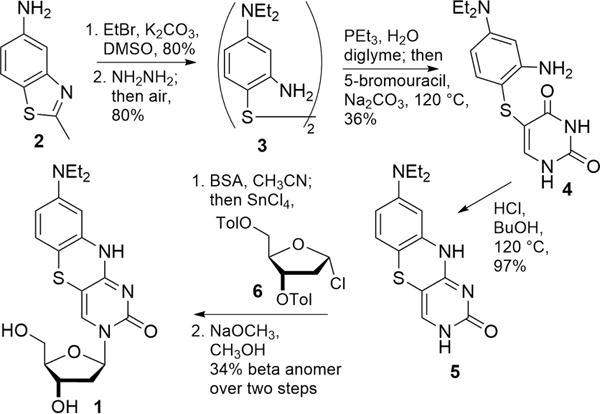
Synthesis of 8-DEA-tC Nucleoside 1
Photophysical measurements of the 8-DEA-tC nucleoside revealed ε = 2700 M−1 cm−1, λmax,abs = 395 nm, λmax,em = 493 nm and Φem = 0.006 in 1× PBS buffer. Because protic solvents often quench organic fluorophores, we made similar measurements in 1,4-dioxane and found that λmax,abs = 389 nm, λmax,em = 524 nm, and Φem = 0.06, a 10-fold increase. This increase is larger than that of the parent tC for the same solvent change (4-fold) but smaller than what we have observed in a past study of the 8-methoxy-tC nucleoside (30-fold).29
To test the properties of 8-DEA-tC in single-stranded and duplex oligonucleotides, the nucleoside phosphoramidite was used in solid-phase DNA synthesis to prepare 9 decameric oligos. Complementary sequences and sequences with an adenosine mismatch or dSpacer (1′,2′-dideoxyribose) as a stable abasic site surrogate were also prepared. We named the sequences with two-letter codes that identify the nucleobases 5′ and 3′ to 8-DEA-tC, respectively (Table 1). To study the impact of the 8-DEA-tC modification on tertiary structure, all sequences were annealed to their complements, and analyzed using circular dichroism (Supporting Information). All spectra are consistent with B form helices, indicating that 8-DEA-tC does not significantly perturb the tertiary structure.
Table 1.
Properties of Single- and Double-Stranded Oligonucleotides Containing 8-DEA-tC in 1× PBS buffer pH 7.4a
| Sequence Nameb | Sequence | ss Φem | ds Φem | ds λmax,abs/nm (ss) | ds λmax,em/nm (ss) | Tm (°C) | ΔTm (°C)c |
|---|---|---|---|---|---|---|---|
| GA | 5′-CGC-AGX-ATC-G-3′ | 0.020 | 0.050 | 422 (425) | 499 (499) | 50.7 | −5.1 |
| CT | 5′-CGC-ACX-TTC-G-3′ | 0.020 | 0.025 | 414 (413) | 492 (495) | 54.7 | +2.0 |
| GC | 5′-CGC-AGX-CTC-G-3′ | 0.032 | 0.12 | 348 (425) | 500 (499) | 49.1 | −14.5 |
| CA | 5′-CGC-ACX-ATC-G-3′ | 0.014 | 0.017 | 410 (420) | 494 (493) | 57.4 | +2.1 |
| GG | 5′-CGC-AGX-GTC-G-3′ | 0.027 | 0.056 | 400 (415) | 499 (498) | 58.2 | −7.0 |
| CC | 5′-CGC-ACX-CTC-G-3′ | 0.013 | 0.024 | 413 (421) | 496 (496) | 60.2 | +6.7 |
| TA | 5′-CGC-ATX-ATC-G-3′ | 0.014 | 0.026 | 420 (417) | 495 (495) | 48.7 | −1.6 |
| TT | 5′-CGC-ATX-TTC-G-3′ | 0.025 | 0.041 | 416 (413) | 495 (495) | 48.5 | +0.2 |
| AA | 5′-CGC-AAX-ATC-G-3′ | 0.008 | 0.042 | 410 (415) | 498 (497) | 48.4 | −2.6 |
| AA mismatchd | 5′-CGC-AAX-ATC-G-3′ | 0.008 | 0.015 | 417 (415) | 501 (497) | 33.1 | +9.6 |
| AA AP sitee | 5′-CGC-AAX-ATC-G-3′ | 0.008 | 0.007 | 421 (415) | 466 (497) | 36.9 | n.d.f |
| tC parent AAg | 5′-CGC-AAtC-ATC-G-3′ | 0.11 | 0.11 | 389 (390) | 501 (502) | 55.3 | +4.3 |
Detailed procedures for photophysical measurements are given in the Supporting Information. Quantum yield measurements were performed at least in duplicate and the reported numbers are averaged. Typical errors based on standard deviation are approximately ±15%. ds = double-stranded; ss = single-stranded.
Sequences named for neighboring bases.
Tm for natural DNA duplex subtracted from Tm for the 8-DEA-tC-containing duplex.
AA sequenced annealed to 5′-CGA-TAT-TGC-G-3′ (8-DEA-tC opposite A).
AA sequenced annealed to 5′-CGA-T(dSpacer)T-TGC-G-3′ (8-DEA-tC opposite the dSpacer surrogate for an abasic site, AP).
Temperature-dependent CD changes were nonsigmoidal for the natural DNA strand and Tm could not be determined.
Sequence AA made with parent tC in place of 8-DEA-tC.
Quantum yields of fluorescence emission were determined using the comparative method of Williams et al. and a fluorescence standard of quinine sulfate in 0.1 M H2SO4 (Table 1 and Figure 2).32 The single-stranded oligonucleotides have quantum yields of fluorescence emission ranging from Φem = 0.01–0.03, all brighter than the free 8-DEA-tC nucleoside by up to a factor of 5. Addition of the complementary sequences and duplex formation results in further fluorescence increases in all sequences of up to 4-fold, giving a maximum Φem = 0.12 for sequence GC. This quantum yield is 20-fold greater than that of the free 8-DEA-tC nucleoside, the largest such increase reported to date for a fluorescent nucleoside analogue. Correct base pairing is essential to the increased Φem in the duplex. Mispairing 8-DEA-tC with A resulted in only a modest, less than 2-fold increase in Φem, and placing 8-DEA-tC opposite an abasic site gave a Φem effectively the same as for the free nucleoside The three highest quantum yields observed for duplex oligonucleotides containing 8-DEA-tC all have guanine as the 5′-neighboring base. The brightest sequence, GC, is noteworthy for three reasons. First, it is known that intercalated ethidium has a greater Φem in poly(dG-dC) than in natural DNA sequences, paralleling our observations for 8-DEA-tC.33 Second, the λmax,abs is significantly blue-shifted as compared with any other sequence. Third, the ΔTm measurements show a striking inverse correlation to the percent quantum yield increase from free nucleoside to duplex (Figure 3). Electronic interactions between 8-DEA-tC and neighboring bases are therefore strongly tied to the fluorescence turn-on effect. Although the CD spectra indicate that overall B form tertiary structure is maintained, the changes in λmax,abs and ΔTm hint that there may be a localized perturbation in base stacking structure in the GC duplex.
Figure 2.
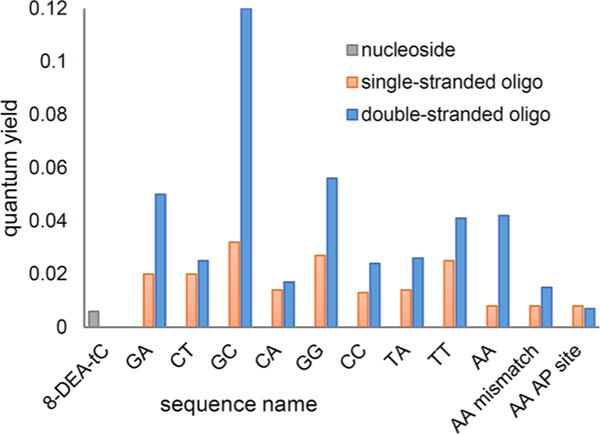
Quantum yields of the 8-DEA-tC nucleoside in single-stranded and double-stranded oligonucleotide sequences (sequence names are defined in Table 1).
Figure 3.
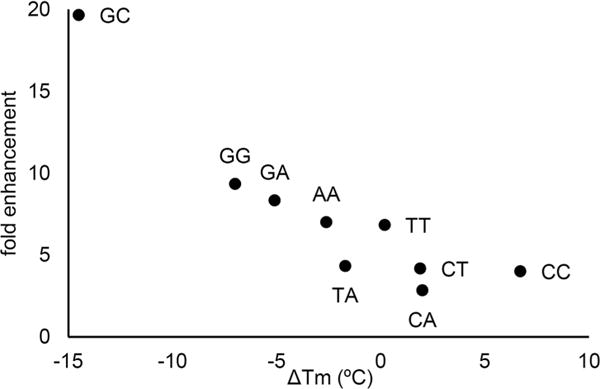
Correlation between the fold enhancement of Φem from 8-DEA-tC nucleoside to double-stranded DNA and the ΔTm for each sequence (Table 1).
We next sought to determine which aspects of the 8-DEA-tC nucleobase structure explain the photophysical properties. First, we note that SYBR Green I, ethidium bromide, and Luedtke’s DMAT nucleoside all have pronounced push–pull electronic motifs that can stabilize a charge-separated S1 state, and such a motif is not present in the parent tC. To test whether the diethylamino group in 8-DEA-tC enhances such character, we examined the appearance and energies of the HOMO and LUMO orbitals calculated by DFT (B3LYP/cc-pVDZ) at the optimized geometries (Figure 4; computational details are in the Supporting Information). These calculations show that the HOMO and LUMO of parent tC are distributed across the arene. In contrast, the HOMO of 8-DEA-tC is much more polarized toward the diethylaminobenzene ring and the LUMO toward the pyrimidine ring, indicating a push–pull character. The electronic modification imparted by the diethylamino group therefore makes this nucleoside’s π system more electronically similar to SYBR Green I, ethidium bromide, and DMAT.
Figure 4.
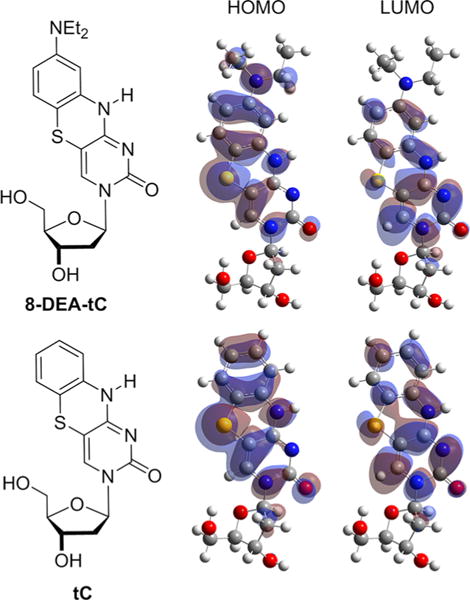
Molecular orbital calculations comparing HOMO and LUMO orbitals of the tC and 8-DEA-tC nucleosides.
Next, we sought to examine four potential quenching mechanisms for 8-DEA-tC that might be attenuated in the duplex and that could explain the fluorescence enhancement. These mechanisms are solvent quenching, chloride quenching, a molecular rotor effect, and excited-state proton transfer. As described above, 8-DEA-tC is approximately twice as sensitive to quenching by protic solvent as the parent tC, which is not sufficient to explain a 20-fold fluorescence increase. Our CD data show that the B form of DNA is intact when 8-DEA-tC is present opposite an abasic site, but there is no fluorescence increase as compared with the free nucleoside. Desolvation of the 8-DEA-tC nucleoside when base stacked therefore does not explain the fluorescence turn-on.
To test the ability of the duplex to attenuate chloride quenching of 8-DEA-tC, we performed a Stern–Volmer analysis using the 8-DEA-tC nucleoside and the TT duplex oligonucleotide (Supporting Information). To our surprise, the 8-DEA-tC nucleoside is more fluorescent with increasing chloride concentration, likely owing to salt-induced changes in solvation.34 In contrast, chloride has a modest quenching effect on 8-DEA-tC when present in the matched TT duplex. These results rule out the possibility that protection against chloride quenching contributes to the fluorescence turn-on effect.
Next, we hypothesized that the C–N bond appending the diethylamino group to tC may provide a molecular rotor effect, enabling nonemissive relaxation coupled to conformational change at the excited state. Although molecular modeling suggests that the diethylamino group would be relatively free to rotate in a duplex oligonucleotide (Supporting Information), water dynamics in the DNA major groove are known to slowed by up to 50-fold as compared with bulk water. For this reason, we hypothesized that slowed water dynamics in the major groove of the duplex could cause an increase in the fluorescence of 8-DEA-tC. To test this hypothesis, we compared the solvent viscosity sensitivity of fluorescence of the 8-DEA-tC nucleoside with parent tC and 9-(2,2-dicyanovinyl)julolidine (DCVJ, a frequently used reference compound for viscosity effects on fluorescence) using mixtures of glycerol and methanol (Supporting Information).35,36 We found that 8-DEA-tC, unlike parent tC, has fluorescence intensity that is positively correlated to viscosity, but the response of DCVJ is 3-fold greater. Other nucleoside analogue molecular rotors investigated by Tor are more sensitive to viscosity than 8-DEA-tC, but they lose Φem when base stacked.35 Moreover, when 8-DEA-tC is present opposite an abasic site in a duplex that maintains B form, there is no fluorescence increase. 8-DEA-tC has some molecular rotor character, but the fluorescence increase we observed in duplex DNA cannot be attributed to restricting the C–N bond rotation.
To test the influence of the duplex on excited-state proton transfer, we measured Φem in deuterated 1× PBS buffer, where a kinetic isotope effect slows proton transfer. In deuterated buffer, the 8-DEA-tC nucleoside is twice as bright (Φem = 0.012), but the quantum yield for the TT duplex hardly changes (Φem = 0.045). This result shows that the duplex protects 8-DEA-tC from excited state proton transfer, clearly a significant factor contributing to the fluorescence turn-on.
We last tested the ability of 8-DEA-tC to distinguish a single nucleotide by a visible fluorescence response (Figure 5). 0.26 mM solutions of GC oligo in 0.5× PBS buffer were prepared with the GC oligo alone, a duplex with the matching complement, and a duplex with an 8-DEA-tC mismatch. Visual inspection of the samples irradiated with a hand-held UV lamp clearly shows that the perfectly matched duplex is indicated by a large increase in fluorescence.
Figure 5.

Visual discrimination of single nucleotide polymorphism by 8-DEA-tC. Samples were prepared in 0.5× PBS buffer and illuminated by a hand-held UV lamp. Left to right: 8-DEA-tC ss GC oligo (Table 1), GC oligo annealed to its matched complement, GC oligo annealed with an 8-DEA-tC:A mismatch.
We have designed and synthesized a novel tricyclic cytidine analogue, 8-DEA-tC, that is almost nonfluorescent as a nucleoside but exhibits up to a 20-fold increase in Φem when base stacked in duplex DNA. This is the first nucleoside analogue to match the performance of ethidium bromide at fluorescence turn-on detection of DNA duplex formation, but it offers the distinct advantage of sequence-specificity. Studies of 8-DEA-tC mismatched with adenosine or positioned across from an abasic site reveal that correct Watson–Crick base pairing is essential to the fluorescence turn-on response, at least in part because base pairing protects 8-DEA-tC from quenching by excited state proton transfer involving the solvent. We expect that 8-DEA-tC will find application as a fluorescent turn-on probe for base pairing and, when converted to the triphosphate, a probe for enzymatic DNA synthesis. These applications are the subject of ongoing investigations in our laboratory.
Supplementary Material
Acknowledgments
We thank Prof. Andrew Cooksy (San Diego State University) for help with the DFT calculations, Prof. Nathan Luedtke (University of Zurich) for helpful discussions, and Brittney Rodgers for early studies in this project area. Financial support from San Diego State University, NIH IMSD support for K.L.T. (5R25GM058906), and prior NIH support for this research (GM093943 to B.W.P.) is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b10410.
Synthetic procedures and characterization data for novel compounds, UV/vis and fluorescence spectra and tabulated data, CD spectra, and computational details (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Tasara T, Angerer B, Damond M, Winter H, Dörhöfer S, Hübscher U, Amacker M. Nucleic Acids Res. 2003;31:2636–2646. doi: 10.1093/nar/gkg371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stengel G, Gill JP, Sandin P, Wilhelmsson LM, Albinsson B, Norden B, Millar D. Biochemistry. 2007;46:12289–12297. doi: 10.1021/bi700755m. [DOI] [PubMed] [Google Scholar]
- 3.Börjesson K, Sandin P, Wilhelmsson LM. Biophys Chem. 2009;139:24–28. doi: 10.1016/j.bpc.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 4.Neef AB, Luedtke NW. Proc Natl Acad Sci U S A. 2011;108:20404–20409. doi: 10.1073/pnas.1101126108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu W, Shin D, Tor Y, Cooperman BS. ACS Chem Biol. 2013;8:2017–2023. doi: 10.1021/cb400256h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin D, Lönn P, Dowdy SF, Tor Y. Chem Commun (Cambridge, U K) 2014;51:1662–1665. doi: 10.1039/c4cc08809c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dziuba D, Pohl R, Hocek M. Chem Commun. 2015;51:4880–4882. doi: 10.1039/c5cc00530b. [DOI] [PubMed] [Google Scholar]
- 8.Tanpure AA, Srivatsan SG. Nucleic Acids Res. 2015;43:e149. doi: 10.1093/nar/gkv743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mata G, Luedtke NWJ. Am Chem Soc. 2015;137:699–707. doi: 10.1021/ja508741u. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Zhang Y, Zhang H, Xuan X, Xie M, Xia S, Qu G, Guo H. Anal Chem. 2016;88:5554–5560. doi: 10.1021/acs.analchem.6b01395. [DOI] [PubMed] [Google Scholar]
- 11.Dziuba D, Jurkiewicz P, Cebecauer M, Hof M, Hocek M. Angew Chem, Int Ed. 2016;55:174–178. doi: 10.1002/anie.201507922. [DOI] [PubMed] [Google Scholar]
- 12.Dumat B, Larsen AF, Wilhelmsson LM. Nucleic Acids Res. 2016;44:e101. doi: 10.1093/nar/gkw114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward DC, Reich E, Stryer L. J Biol Chem. 1969;244:1228–1237. [PubMed] [Google Scholar]
- 14.Cekan P, Sigurdsson ST. Chem Commun (Cambridge, U K) 2008;(29):3393–3395. doi: 10.1039/b801833b. [DOI] [PubMed] [Google Scholar]
- 15.Saito Y, Suzuki A, Okada Y, Yamasaka Y, Nemoto N, Saito I. Chem Commun (Cambridge, U K) 2013;49:5684–5686. doi: 10.1039/c3cc42605j. [DOI] [PubMed] [Google Scholar]
- 16.Tainaka K, Tanaka K, Ikeda S, Nishiza KI, Unzai T, Fujiwara Y, Saito I, Okamoto A. J Am Chem Soc. 2007;129:4776–4784. doi: 10.1021/ja069156a. [DOI] [PubMed] [Google Scholar]
- 17.Shin D, Sinkeldam RW, Tor Y. J Am Chem Soc. 2011;133:14912–14915. doi: 10.1021/ja206095a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rovira AR, Fin A, Tor Y. J Am Chem Soc. 2015;137:14602–14605. doi: 10.1021/jacs.5b10420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Börjesson K, Preus S, El-Sagheer AH, Brown T, Albinsson B, Wilhelmsson LM. J Am Chem Soc. 2009;131:4288–4293. doi: 10.1021/ja806944w. [DOI] [PubMed] [Google Scholar]
- 20.Koga Y, Fuchi Y, Nakagawa O, Sasaki S. Tetrahedron. 2011;67:6746–6752. [Google Scholar]
- 21.Cservenyi TZ, Van Riesen AJ, Berger FD, Desoky A, Manderville RA. ACS Chem Biol. 2016;11:2576–2582. doi: 10.1021/acschembio.6b00437. [DOI] [PubMed] [Google Scholar]
- 22.Dziuba D, Pospíšil P, Matyašovský J, Brynda J, Nachtigallová D, Rulíšek L, Pohl R, Hof M, Hocek M. Chem Sci. 2016;7:5775–5785. doi: 10.1039/c6sc02548j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sinkeldam RW, Greco NJ, Tor Y. Chem Rev. 2010;110:2579–2619. doi: 10.1021/cr900301e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilhelmsson LMQ. Rev Biophys. 2010;43:159–183. doi: 10.1017/S0033583510000090. [DOI] [PubMed] [Google Scholar]
- 25.Xie Y, Maxson T, Tor Y. Org Biomol Chem. 2010;8:5053–5055. doi: 10.1039/c0ob00413h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dumas A, Luedtke NW. ChemBioChem. 2011;12:2044–2051. doi: 10.1002/cbic.201100214. [DOI] [PubMed] [Google Scholar]
- 27.Hudson RHE, Ghorbani-Choghamarani A. Org Biomol Chem. 2007;5:1845–1848. doi: 10.1039/b705805e. [DOI] [PubMed] [Google Scholar]
- 28.Mata G, Schmidt OP, Luedtke NW. Chem Commun. 2016;52:4718–4721. doi: 10.1039/c5cc09552b. [DOI] [PubMed] [Google Scholar]
- 29.Rodgers BJ, Elsharif NA, Vashisht N, Mingus MM, Mulvahill MA, Stengel G, Kuchta RD, Purse BW. Chem – Eur J. 2014;20:2010–2015. doi: 10.1002/chem.201303410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stengel G, Urban M, Purse BW, Kuchta RD. Anal Chem. 2010;82:1082–1089. doi: 10.1021/ac902456n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stengel G, Urban M, Purse BW, Kuchta RD. Anal Chem. 2009;81:9079–9085. doi: 10.1021/ac9017555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams ATR, Winfield SA, Miller JN. Analyst. 1983;108:1067–1071. [Google Scholar]
- 33.Pohl FM, Jovin TM, Baehr W, Holbrook JJ. Proc Natl Acad Sci U S A. 1972;69:3805–3809. doi: 10.1073/pnas.69.12.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joung JF, Kim S, Park S. J Phys Chem B. 2015;119:15509–15515. doi: 10.1021/acs.jpcb.5b09905. [DOI] [PubMed] [Google Scholar]
- 35.Sinkeldam RW, Wheat AJ, Boyaci H, Tor Y. ChemPhysChem. 2011;12:567–570. doi: 10.1002/cphc.201001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sutharsan J, Lichlyter D, Wright NE, Dakanali M, Haidekker MA, Theodorakis EA. Tetrahedron. 2010;66:2582–2588. doi: 10.1016/j.tet.2010.01.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.