

Abstract
Childhood acute lymphoblastic leukemia (cALL) is the most common pediatric cancer and, despite an 85% cure rate, still represents a major cause of disease-related death in children. Recent studies have implicated long non-coding RNAs (lncRNAs) in cALL etiology, progression, and treatment response. However, barring some exceptions little is known about the functional impact of lncRNAs on cancer biology, which limits their potential as potential therapeutic targets. We wanted to investigate the functional role of lncRNAs identified as specifically overexpressed in pre-B cALL by whole-transcriptome sequencing. Here we report five lncRNAs specifically upregulated in pre-B cALL that had significant impacts on cancer hallmark traits such as cell proliferation, migration, apoptosis, and treatment response. In particular, silencing of the RP11-137H2.4 lncRNA effectively restored normal glucocorticoid (GC) response in a GC-resistant pre-B cALL cell line and specifically modulated expression of members of both the NRAS/BRAF/NF-?B MAPK cascade and cell cycle pathways. Since GC form the cornerstone of cALL chemotherapy and resistance in cALL confers a dismal prognosis, characterizing RP11-137H2.4sexact role and function in this process will be critical to the development of new therapeutic approaches to overcome GC resistance in children treated for cALL.
Keywords: long non-coding RNA, acute lymphoblastic leukemia, glucocorticoids, RNASeq, treatment resistance
INTRODUCTION
Childhood acute lymphoblastic leukemia (cALL) is the most common cancer among children under 14 years of age. Despite remarkable improvements in survival, with 5 year event-free survival rates of up to 85%, non-responding children with ALL still represent one of the most frequent cause of death from cancer in pediatrics.[1] Childhood ALL is a complex disease comprising multiple subtypes with distinctive somatic genetic alterations, including aneuploidy, chromosomal rearrangements, and point mutations.[1] These genetic alterations contribute to leukemogenesis by altering key regulatory processes, subverting normal proliferation control, blocking differentiation, and promoting resistance to death signals.[2] Despite this understanding of the molecular basis of this disease, accurate patient risk stratification is an ongoing challenge in cALL treatment and the development of innovative therapies. Several studies have described expression signatures for classifying molecularly-defined cALL subtypes and improving outcome prediction.[3–10] These studies focused on the analysis of protein-coding transcripts, probably because most of their translated proteins are important signaling molecules. A new class of non-coding RNAs, designated as long non-coding RNAs (lncRNAs) have been described recently. LncRNAs are expressed in most cell types and at most stages of development and play regulatory roles in various biological processes, including cell pluripotency and tumorigenesis.[11–15] LncRNAs can exert their effects through many cellular processes, such as spatial conformation of chromosomes, chromatin and DNA modifications, RNA transcription, pre-mRNA splicing, mRNA degradation, and mRNA translation.[13, 14] A well-known example of this is the Hox transcript antisense intergenic lncRNA, HOTAIR, which cooperates with the polycomb repressive complex to modulate gene expression. HOTAIR is deregulated in a spectrum of cancers, and its overexpression is associated with poor prognosis in breast, [16] liver, [17] colorectal, [18] gastrointestinal, [19] and pancreatic [20] cancers, and is proposed to increase tumor invasiveness and metastasis.[16] Recent expression studies performed on pre-B cALL samples have shown that lncRNA expression profiles can accurately classify disease subtypes and are correlated with outcome, [3, 21] (Lajoie et al., submitted). These studies suggest that lncRNAs might be utilized as diagnostic and prognostic markers in leukemia, but additional studies are needed to understand their clinical usefulness. Exploring the mechanisms underlying lncRNA functions is critical to recognizing their contribution to biological processes involved in cALL.[22]
In this report, we further investigated five lncRNAs that were shown to be specifically upregulated in pre-B cALLs (Lajoie et al. submitted; see Material and Methods for details). Silencing the expression of any one of these five lncRNAs in pre-B cALL cells had a significant impact on at least one of hallmark cancer traits (apoptosis, cell proliferation, migration, and treatment response). One of these lncRNAs, RP11-137H2.4, had a considerable impact on apoptosis, proliferation, and cell migration, or effectively restored glucocorticoid (GC) sensitivity in GC-resistant pre-B cALL cells. Glucocorticoids are the cornerstone of cALL therapy, inducing G1 arrest and apoptosis in leukemic blasts, and resistance to these agents significantly worsens prognosis. In addition, we showed that decreased expression of RP11-137H2.4 modulates the NRAS/BRAF/NF-κB MAPK pathway that is then translated into a strong transcriptional modulation of E2F targets and NF-κB/Jun-Fos pathway members upon exposure to GCs. In summary, this study showed that the deregulation of a single lncRNA might interfere with apoptotic and GC responses. These observations strongly suggest that lncRNAs could have novel and unexplored therapeutic potential, at least in childhood pre-B ALL.
RESULTS
LncRNAs specifically overexpressed in pre-B cALL modulate cell proliferation and treatment response
In a previous study, we identified 799 lncRNAs that were specifically overexpressed in primary pre-B cALL samples, as compared to CD10+CD19+ pre-B cells isolated from human cord blood (Lajoie et al. submitted; see Material and Methods above for details about cohort and gene expression analyses) (Figure S1). We validated that five such lncRNA transcripts overexpressed in pre-B cALL samples (RP11-137H2.4, RP11-68I18.10, AC156455.1, KB-208E9.1, and CTA-331P3.1) were also overexpressed in the Reh and NALM-6 pre-B cALL cell lines (Figure S2). We then assessed their impact on cancer traits, such as cell proliferation and apoptosis, through siRNA-mediated loss-of-function assays in NALM-6 (average silencing of 61.4%; see Figure S3). Two lncRNAs, RP11-137H2.4 and RP11-68I18.10, caused a significant reduction in proliferation (20-24%, P ≤ 0.05; see Figure 1) when silenced, while knockdown of AC156455.1, KB-208E9.1, or CTA-331P3.1 had no effect on proliferation (data not shown). Furthermore, RP11-137H2.4, AC156455.1, KB-208E9.1, or CTA-331P3.1 knockdown significantly increased apoptosis (3-15%, P ≤ 0.05) when treated with either camptothecin (CPT), doxorubicin (DOX), or prednisolone (Figure 2), while silencing RP11-68I18.10 had no effect on apoptosis (data not shown). Although the magnitudes of the effects on apoptosis are relatively modest (yet statistically significant), they are the product of the deregulation of single lncRNAs in pre-B cALL. These results thus show that individual lncRNAs can have large impacts on cancer phenotypes.
Figure 1. Silencing of lncRNAs upregulated in cALL reduces cell proliferation.
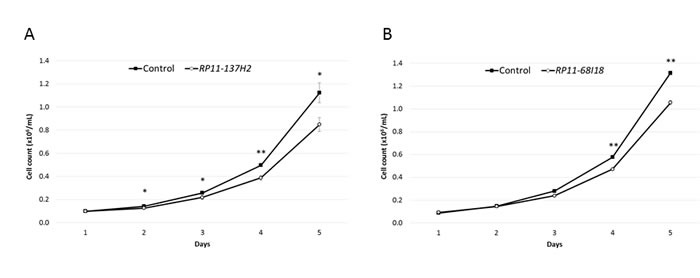
A. Reduction of cell proliferation in NALM-6 cells transfected with dsiRNA against RP11-137H2.4, measured by counting cells in triplicate each day for 5 days. B. Reduction of cell proliferation in NALM-6 cells transfected with siRNA against RP11-68I18.10, measured as in (A). Control cells were transfected with negative control siRNA/dsiRNA (see Methods). Comparisons were made using a two-tailed T-test. *: P ≤ 0.05; **: P ≤ 0.01.
Figure 2. Silencing of lncRNAs upregulated in cALL increases apoptosis in response to cytotoxic treatment.
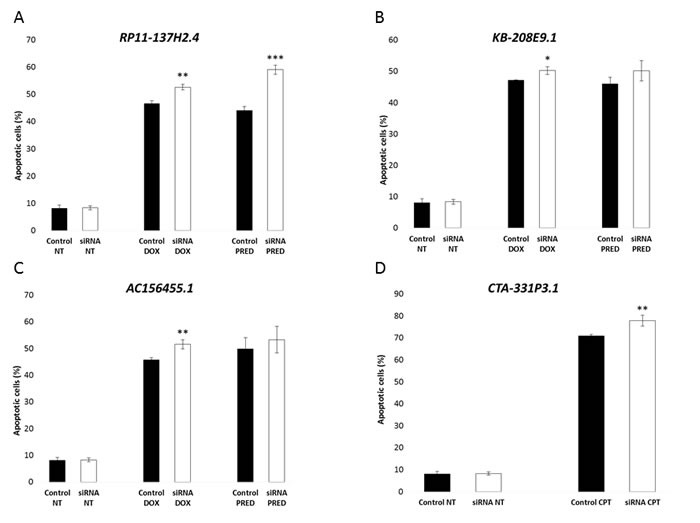
A. Increased apoptosis with doxorubicin and prednisolone treatment measured by AnnexinV / propidium iodide staining. NALM-6 cells transfected with dsiRNA against RP11-137H2.4 were treated with 150 nM doxorubicin for 24 hours or with 750 µM prednisolone for 20 hours. DMSO was used as vehicle control for prednisolone. B. Increased apoptosis with doxorubicin and prednisolone treatment measured by AnnexinV / propidium iodide staining. NALM-6 cells transfected with dsiRNA against KB-208E9.1 were treated with 150 nM doxorubicin for 24 hours or with 750 µM prednisolone for 20 hours. DMSO was used as vehicle control for prednisolone. C. Increased apoptosis with doxorubicin and prednisolone treatment measured by AnnexinV / propidium iodide staining. NALM-6 cells transfected with dsiRNAs against AC156455.1 were treated with 150 nM doxorubicin for 24 hours or with 750 µM prednisolone for 20 hours. DMSO was used as vehicle control for prednisolone. D. Increased apoptosis with camptothecin treatment measured by AnnexinV / propidium iodide staining. NALM-6 cells transfected with siRNA against CTA-331P3.1 were treated with 500 nM camptothecin for 24 hours. Control cells were transfected with negative control siRNA/dsiRNA (see Methods). Comparisons were made using a two-tailed T-test. *: P ≤ 0.05; **: P ≤ 0.01; ***: P ≤ 0.001.
RP11-137H2.4 knockdown inhibits cell proliferation and migration and restores glucocorticoid sensitivity
The lncRNA RP11-137H2.4 had the most pronounced impact upon siRNA-mediated silencing in NALM-6 cells, with an observed 24% reduction of proliferation at 5 days (Figure 1A) and increased apoptosis by 6% and 15% after exposure to DOX or prednisolone, respectively (Figure 2A). We repeated these experiments in a second pre-B cALL cell line, Reh that overexpresses this lncRNA (Figure S2a). Since, contrary to NALM-6, the Reh cell line is hard to transfect we stably transduced Reh with a shRNA specifically targeting RP11-137H2.4. We then validated the silencing (Figure S4) and repeated the phenotyping analysis (Figure 3). The sensitivity of this stable cell line was exacerbated for CPT and prednisolone, with 18% and 26% cell death after treatment, respectively (Figure 3B). This is particularly interesting since Reh does not express functional NR3C1 GC receptor (GR) protein (confirmed in our lab, data not shown) and is therefore resistant to GCs.[23] Furthermore, a significant decrease (27%; P ≤ 0.005) in cell migration was also observed (Figure 3C). Since RP11-137H2.4 has only one documented isoform (Figure S5), rescue experiments performed by introducing a vector constitutively overexpressing RP11-137H2.4 in cells expressing this shRNA (Figure S4) could be performed. These resulted in increased cell proliferation, partial rescue of CPT- and prednisolone-induced apoptosis, and partial rescue of migration, indicating that these effects were specific to RP11-137H2.4 silencing (Figure 3). These results strongly suggest that RP11-137H2.4 silencing specifically restores GC sensitivity in a NR3C1-independent manner in the Reh and, possibly, NALM-6 cell lines.
Figure 3.
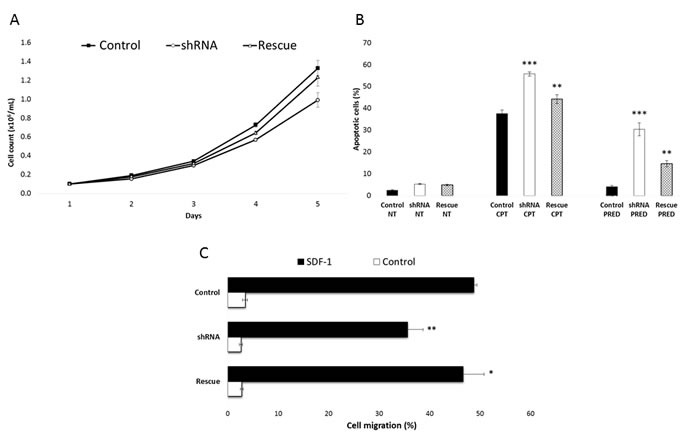
RP11-137H2.4 silencing restores normal glucocorticoid apoptotic response. A. Reduction of cell proliferation in Reh cells stably transduced with shRNAs against RP11-137H2.4 (“shRNA”) and partial rescue of the phenotype by overexpressing the lncRNA in a cell line where RP11-137H2.4 is silenced (“Rescue”, see Methods), measured by counting cells in triplicate each day for 5 days. B. Increased apoptosis with camptothecin or prednisolone treatment and partial rescue of the phenotype by overexpressing the lncRNA measured by AnnexinV / propidium iodide staining. Reh cells stably transduced with shRNAs against RP11-137H2.4 were treated with 10 µM camptothecin for 6 hours or 750 µM prednisolone for 24 hours. DMSO was used as vehicle control for prednisolone. C. Reduction of cell migration in Reh cells stably transduced with shRNAs against RP11-137H2.4 and partial rescue of the phenotype by overexpressing the lncRNA, measured by counting cells in triplicate, using SDF-1 as a chemoattractant. Control cells were stably transduced with scrambled shRNA. Comparisons were made using a two-tailed T-test. *: P ≤ 0.05; **: P ≤ 0.01; ***: P ≤ 0.001.
RP11-137H2.4 silencing modulates the MAPK and cell cycle pathways to restore prednisolone sensitivity
In light of these results, we further investigated how RP11-137H2.4 silencing affected global transcriptional profiles upon prednisolone exposure. We first identified genes significantly deregulated RP11-137H2.4 silencing upon prednisolone exposure by sequencing the whole transcriptome of cells expressing RP11-137H2.4 shRNA exposed to prednisolone and comparing it to cells expressing RP11-137H2.4 shRNA exposed to vehicle (DMSO). Gene ontology (GO) analyses indicated that E2F target genes, DNA damage response, and p53 pathway and TNFα/NF-κB signaling genes were significantly deregulated by RP11-137H2.4 silencing upon prednisolone exposure (Figure 4A, right). Interestingly, genes in cell cycle-related categories were specifically downregulated by RP11-137H2.4 silencing, while those corresponding to TNFα/NF-κB signaling were upregulated (Figure 4A, left). Closer examination of the cell cycle and MAPK extracellular signal transduction cascade pathways confirmed that they were indeed down- and up-regulated, respectively, by RP11-137H2.4 silencing upon prednisolone exposure (Figure 4B-4C). Furthermore, we observed a significant overlap (p ≤ 0.005, exact hypergeometric probability) between genes significantly deregulated by RP11-137H2.4 silencing upon prednisolone exposure in our dataset (1131 transcripts at FDR ≤ 0.005) and GC-induced genes whose promoters contain glucocorticoid-response elements (GREs) (Reddy et al.) (Table 1).[24] Indeed, 10/61 of the genes in the latter list were differentially expressed in our experiments and were consistently expressed in the same direction (up- or down-regulated) in both datasets, with the exception of CKB, suggesting that GC response genes are modulated specifically by RP11-137H2.4 silencing upon prednisolone exposure. These data strongly suggest that RP11-137H2.4 expression in pre-B cALL can abrogate normal GC response by modulating the expression of members of MAPK cascade and cell cycle pathways in a NR3C1-independent fashion.
Figure 4. Gene ontology (GO) analysis of.
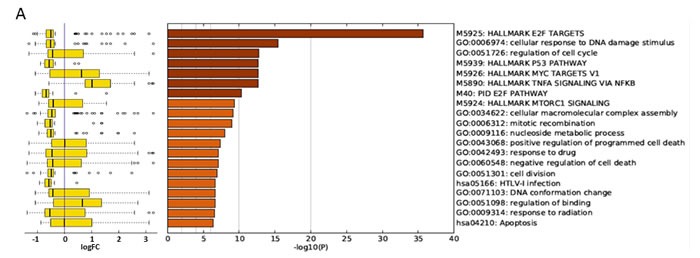
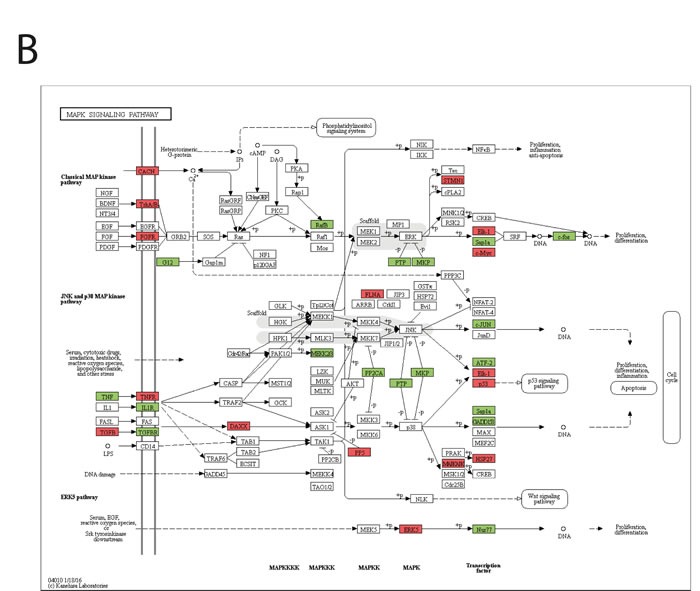
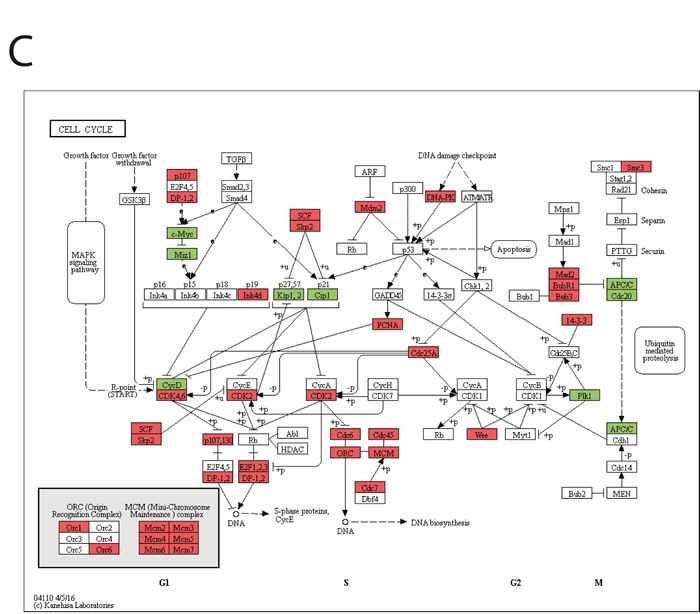
RP11-137H2 silencing-specific gene expression deregulation upon prednisolone exposure. Whole-transcriptome deep sequencing was performed on Reh cell lines expressing either scrambled or RP11-137H2.4-specific shRNA. Cells were exposed for 24 h to either vehicle (DMSO) or 750 µM prednisolone. A. Deregulated genes’ enriched GO categories are reported with significance (-log10(P)) (right) along with a box-plot of log2 expression fold changes for the corresponding GO categories (left). Edges of the box are the first and third quartiles, while the band inside the box is the median. Whiskers represent minimum and maximum values. B. Genes in the MAPK cascade deregulated in Reh cells following RP11-137H2.4 silencing and prednisolone treatment (red is downregulated, green is upregulated). C. Genes in the cell cycle pathway deregulated in Reh cells following RP11-137H2.4 silencing and prednisolone treatment (red is downregulated, green is upregulated).
Table 1. GC-modulated genes common to both Reddy et al. and our datasets.
| Gene Name | Ensembl ID | Log2 Fold-change (Reddy et al.) | Log2 Fold-change (Ouimet et al.) | FDR(Ouimet et al.) |
|---|---|---|---|---|
| ZFP36 | ENSG00000128016 | 2.29 | 0.99 | 1.95E-06 |
| BIRC3 | ENSG00000023445 | 1.87 | 2.48 | 8.54E-11 |
| ERN1 | ENSG00000178607 | 1.11 | 0.46 | 3.53E-03 |
| ALOX5AP | ENSG00000132965 | 1.02 | 0.74 | 2.58E-03 |
| CEBPB | ENSG00000172216 | 0.98 | 0.59 | 1.57E-03 |
| FOSL2 | ENSG00000075426 | 0.80 | 0.66 | 3.79E-03 |
| CKB | ENSG00000166165 | 0.71 | −0.69 | 1.52E-04 |
| RHOB | ENSG00000143878 | 0.46 | 1.24 | 2.72E-16 |
| KLF5 | ENSG00000102554 | 0.44 | 1.23 | 1.21E-03 |
| ID3 | ENSG00000117318 | −0.46 | −0.44 | 2.37E-05 |
Interestingly, RP11-137H2.4 silencing in vehicle-treated (DMSO) conditions, as compared with control cells expressing a scrambled shRNA, also seemed to significantly affect gene expression profiles (Figure S6). Indeed, GO analyses revealed significant enrichment of deregulated genes in several GO categories related to external signal transduction, cell differentiation, adhesion, and morphogenesis (Figure S5a, right). Although no clear overall direction was observed in gene expression modulation for these GO categories (Figure S5a, left), the expression of multiple key members of the MAPK extracellular signal transduction cascade and cell cycle pathways was significantly altered (Figure S5b-c).
Taken together, these data indicate that pre-B cALL-specific lncRNA overexpression can induce malignant behaviors, such as increased cell proliferation, resistance to apoptosis, and increased cell migration. Finally, our best candidate, RP11-137H2.4, seems to be implicated in GC resistance in pre-B cALL patients.
DISCUSSION
To date, childhood leukemia research has mainly focused on the deregulated expression of protein-coding genes that could be used as diagnostic and prognostic biomarkers. In this study, we have shown that five lncRNAs specifically overexpressed in pre-B cALL can significantly impact cancer traits, such as cell proliferation and migration, apoptosis, and treatment response. Furthermore, while the magnitude of the observed effects was relatively modest (while still statistically significant), they are caused by the partial silencing of single lncRNAs. The importance of this cannot be understated: modulating the expression of individual, non-coding transcripts can significantly impact cancer hallmark phenotypes. Moreover, we demonstrated that RP11-137H2.4 expression in cALL is linked to GC treatment resistance, and that its silencing is sufficient to restore a NR3C1-independent cellular response to these compounds, leading to GC-induced apoptosis. Furthermore, stably overexpressing RP11-137H2.4 in Reh cells already expressing the shRNA specific to it resulted in a partial rescue of these phenotypes, further demonstrating the RP11-137H2.4 specificity of the effects we observed.
Deregulation of lncRNAs has been linked to several complex human diseases, including cancer.[15, 25] For instance, MALAT1 was found to be highly expressed and associated with metastasis and poor prognosis in many cancer types [26] including non-small cell lung carcinoma [27, 28] and hepatocellular carcinoma.[29] The up-regulation of several other lncRNAs, such as HOTAIR and MVIH, and the downregulation of H19 have been associated with poor prognosis in cancers.[30] The up-regulation of lncRNA PCA3 (DD3) has proven to be a reliable biomarker for early detection of prostate cancer.[31] To date, few lncRNAs have been directly linked to leukemogenesis. In childhood pre-B ALL, two studies showed that expression of lncRNAs correlated with cytogenetic abnormalities, disease subtypes, and survival of B-ALL patients.[21] (Lajoie et al. submitted) The B-ALL-associated long RNA-2 (BALR-2) has been shown to be specifically upregulated in MLL-rearranged ALL.[21] BALR-2 was identified as a modulator of the response to GC treatment.[21]
These results are very interesting considering that GCs are key chemotherapeutic agents in pre-B cALL therapy. GCs are involved in many biological processes, such as metabolism, development, differentiation, immunity, reproduction, and neural activity. The diverse actions of GC have led to their use as therapeutic agents in the treatment of many diseases, including cancer. Their effect on lymphoid cells is dramatic, and includes the induction of G1 cell cycle arrest and apoptosis.[32] Furthermore, relapsed cALL patients acquire prednisolone resistance disproportional to other anti-leukemic agents. Indeed, resistance to GCs is a hallmark of relapsed cALL, since relapsed leukemia blasts are significantly more resistant to prednisolone and dexamethasone, and is a strong predictor of negative outcome at diagnosis.[32–35]
Several members of key signalling pathways were overrepresented in the transcripts deregulated by RP11-137H2.4 silencing. Indeed, we found that NRAS, BRAF, NF-κB, and several other genes in the MAPK cascade were downregulated following RP11-137H2.4 silencing, whereas members of the activating protein-1 (AP-1) complex, JUN and FOS, were upregulated. Genes involved in cell cycle control were also affected. Prednisolone treatment showed a strong, RP11-137H2.4 silencing-specific repression of E2F target genes involved in cell cycle control and a modulation of MAPK cascade gene expression, as expected in GC-sensitive cells. Importantly, RP11-137H2.4 silencing restores GC sensitivity in the Reh prednisolone-resistant pre-B cALL cell line, despite it having both copies of the NR3C1 GC receptor gene inactivated (one allele is deleted while the other carries a nonsense mutation (p.Gln528*), which we confirmed independently; data not shown).[36] Prednisolone, a GC, diffuses passively into the cell, where it binds the GC receptor (NR3C1, also known as GR), causing it to dimerize. Dimerized GR acts as a transcription factor by either binding to positive or negative glucocorticoid response elements in the DNA, or by binding to other transcription factors, such as NF-kB and AP-1.[37] The AP-1 family of transcription factors consists of multiple Jun and Fos members and integrates growth signals at the transcriptional level. AP-1 factors regulate the expression of CCND1 and E2F, which in turn regulates E2F-downstream genes, leading to the modulation of cellular proliferation, differentiation, apoptosis, oncogene-induced transformation, and cancer cell invasion.[38] While further studies are required to characterize the molecular mechanisms involved, our results strongly suggest that RP11-137H2.4 expression abrogates normal GC response in pre-B cALL samples by modulating the expression of MAPK cascade genes. These results are strikingly similar to those of Fernando et al. where they demonstrated that knocking down another lncRNA specifically overexpressed in pre-B cALL, BALR-2. They found that knockdown of BALR-2 restored prednisolone sensitivity through modulation of the GC receptor signaling pathway, specifically by upregulating JUN, FOS, SGK1, and SERPINE1. We also see these genes upregulated by prednisolone, but not DMSO, in our system (Tables S2 and S3, respectively).[21]
In summary, we found that specific lncRNAs play important roles in pre-B cALL progression and treatment response. In particular, one lncRNA, RP11-137H2.4, seems to play a role into GC resistance in treatment of cALL. Characterizing its exact role and function in this process will be critical to the development of new therapeutic approaches to overcome GC resistance in children treated for cALL.
MATERIALS AND METHODS
Childhood ALL sample cohort and transcriptome profiling
Our study cohort consisted of 56 pre-B cALL patients (28 females and 28 males) with a mean age at diagnosis of 6.1±3.6 years. All subjects were French-Canadians of European descent diagnosed in the Division of Hematology-Oncology at the Sainte-Justine Hospital (Montreal, Canada) and part of the Quebec childhood ALL cohort (QcALL).[39] CD10+CD19+ cells isolated from human cord blood were used as controls. Briefly, after being isolated using a Ficoll-Paque gradient fragmentation, PBMCs were positively selected using MACS Separation with CD19 MicroBeads (Miltenyi Biotec). Cell sorting was performed on the CD19+ cells using CD19-PE and CD10-FITC antibodies (Miltenyi Biotec). Purity was >90 %. Total RNA was extracted from white blood cell pellets obtained from bone marrow and peripheral blood at diagnosis using the mirVana Isolation kit (Ambion) according to manufacturer's protocol. Following a DNAse treatment to remove possible contamination by genomic DNA, ribosomal RNAs were removed using the RiboMinus Eukaryote kit (Invitrogen). cDNA libraries were prepared using the SOLiD Total RNA-seq kit based on manufacturer's protocol and sequenced on the Life Technologies SOLiD 4/5500 System (paired-end: 50×35bp and 75×35bp). Reads were aligned to the human genome (hg19) using the Lifescope Genomic Analysis Software (Applied Biosystems; Whole Transcriptome Analysis pipeline, default parameters). Expression levels by gene were determined with the HTseq-count software[40] using gene models from Ensembl75 combined with (non-overlapping) lncRNA transcripts provided in Casero et al.[41] The identification of differentially expressed transcripts relative to HCB controls was done using the Generalized Linear Model implemented in the edgeR package.[42] The Sainte-Justine Institutional Review Board approved the research protocol, and informed consent was obtained from all participating individuals and/or their parents.
Cell culture
Reh (Human B cell precursor leukemia; ATCC CRL-1567) and NALM-6 (Human B cell precursor leukemia; DSMZ ACC-128) cells were cultured in RPMI 1640 medium (Wisent) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Wisent). Reh_shRNA control and Reh_shRNA-RP11-137H2.4 cells were cultured in RPMI 1640 medium (Wisent) supplemented with 10% heat-inactivated FBS and 1µg/mL puromycin (Wisent). Reh_shRNA-RP11-137H2.4_RP11-137H2.4 cells (“Rescue” cell line) were cultured in the same media as Reh_shRNA RP11-137H2.4, but supplemented with 5µg/mL blasticidin (Wisent). Human embryonic kidney 293T cells (HEK293T; ATCC CRL-3216) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Wisent) supplemented with 10% heat-inactivated FBS. All culture media were supplemented with 1% penicillin/streptomycin (Wisent), and all cell lines were cultured in a 37 °C incubator.
RT-qPCR
Total RNA was extracted from cells using RNeasy mini Kit (Qiagen). Total RNA was retro-transcribed into cDNA using the QuantiTect Reverse Transcription Kit (Qiagen), and qPCR amplifications (triplicates) were performed on the ABI Prism 7000 Sequence Detection System (Thermo Fisher Scientific) using SYBR Green PCR Master Mix (Applied Biosystems). Primer sequences used are listed in Table S1. The cycling parameters were 95 °C for 10 min, 40 cycles (95°C for 15 sec, 60°C for 1 min) followed by a denaturation curve at 60°C. GAPDH was used as reference gene. Expression values were calculated as 2-(∆∆CT), as per Livak and Schmittgen.[43]
siRNA-mediated silencing
Custom Silencer Select siRNA (Ambion by Life Technologies) and Custom dsiRNA Duplex (Integrated DNA Technologies) were used to silence the lncRNA. Silencer Select negative control No.1 siRNA (Ambion) and dsiRNA NC1 (Integrated DNA Technologies) were used as negative control siRNA/dsiRNA. Lonza's Nucleofector™ Technology was used for the transfection of siRNA/dsiRNA into the NALM-6 cells using the Nucleofector kit T (Lonza) and the program C-005; silencing was measured 24 hours later by RT-qPCR.
shRNA and overexpression constructs
The structure region of RP11-137H2.4 (Ensembl ENSG00000226659) was used for cDNA synthesis (IDT), which was subcloned into the pLenti vector with blasticidin resistance using Gateway technology. The integrity of the clone selected was confirmed by sequencing. shRNA target sequences for RP11-137H2.4 were cloned into the psi-LVRU6P vector with puromycin resistance (GeneCopoeia). A scrambled shRNA for psi-LVRU6P (GeneCopoeia) was used as control.
Vector particle production and transduction
For preparation of lentiviral particles, we used a co-transfection procedure. Briefly, 293T cells were co-transfected with 12 µg of the pREV vector, 15.6 µg of the pVSVG vector, 30 µg of the pMDL vector, and 18 µg of the pLenti vector containing the gene of interest or the shRNA of the gene of interest using Lipofectamine 2000 (Invitrogen) to produce lentiviral particles. Forty-eight hours later, vector particles were collected from culture supernatants, filtered through 0.22 µm pore-size nitrocellulose membranes, concentrated, and aliquoted before being frozen at -80 °C until use. The amount of viral particles produced was determined by the HIV-1 p24 antigen capture assay (ABL Inc.). Transductions of Reh cells were performed using 1 × 106 cells and 600 ng of lentiviral particles in the presence of 8 µg/mL polybrene (Sigma-Aldrich). Forty-eight hours after infection, cells were placed in medium containing antibiotics for selection.
Proliferation, apoptosis, and drug response assays
To measure proliferation, 150µL of cells at 0.1 × 106 cells/mL were plated in 96-well plates in triplicate for five days. Each day, cells were counted in triplicate using a cell counter (Beckman Coulter). For drug response assays, 2mL of cells were diluted to 0.5 × 105 cells/mL in 12-well plates in their culture medium and treated with the corresponding agent, camptothecin (Tocris Bioscience), doxorubicin (Sigma-Aldrich), prednisolone (Sigma-Aldrich) to promote DNA damage-induced apoptosis, then harvested to assay for apoptosis. DMSO (vehicle) was used as control for prednisolone treatment, while other drugs were diluted in water. Cell apoptosis was measured using the Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Stained cells were analyzed by flow cytometry using the BD FACSCantoTM II cell analyser and the BD FACSDiva software (BD Biosciences) according to manufacturer's guidelines.
Cell migration assay
To measure migration, 30 µL of media containing 30 ng/mL of the chemoattractant SDF-1 (ProSpec) or not (controls) were filled in triplicate into microplate wells of a 96-well cell migration system (ChemoTx #101-5) with 5 µm pore size (Neuro Probe). 25µL of cells at 8 × 106 cells/mL were dropped on the filter of each well, and the filled instrument was incubated at 37 °C in humidified air with 5% CO2 for 1.5 hours. After incubation, 25µL of migrated cells in the microplate were counted using a cell counter (Beckman Coulter).
Whole transcriptome sequencing
Total RNA was extracted from white blood cell pellets obtained from bone marrow and peripheral blood at diagnosis using the mirVana Isolation kit (Ambion) according to the manufacturer's protocol. Following a DNase treatment to remove possible contamination by genomic DNA, ribosomal RNAs were removed using the RiboMinus Eukaryote kit (Invitrogen). cDNA libraries were prepared using the Illumina TruSeq Stranded Total RNA Library Prep Kit based on manufacturer's protocol and sequenced on an Illumina HiSeq 2500 (2 × 100 bp paired-end).
Sequencing data were analyzed using the GATK best practice pipeline.[44] Expression levels by gene were determined with the HTseq-count software[40] using gene models from Ensembl75 combined with (non-overlapping) lncRNA transcripts provided in Casero et al.[41] Transcript differential expression was assessed using Deseq2.[45]
Gene expression and ontology analyses
Differential gene expression analyses were performed using Deseq2.[45] First, genes regulated by the shRNA in DMSO were retrieved using the design “~ group”, where “group” corresponds to the presence/absence of the shRNA or scrambled control. Positive gene expression log2 fold-changes in that design correspond to genes being overexpressed in the presence of the shRNA in DMSO. Second, genes having different expression log2 fold-changes in prednisolone vs. DMSO due to the presence of the shRNA were retrieved using the design “~ treatment + shRNA + treatment:shRNA”, where “treatment” corresponds to prednisolone/DMSO and “shRNA” corresponds to presence of the shRNA or scrambled control. Positive gene expression log2 fold-changes in that design correspond to genes having a positive shRNA-specific log2 fold-change difference in prednisolone vs. DMSO. The top 500 differentially expressed genes (p-adjusted ≤ 0.05) of the above two designs were used as input into Metascape (http://metascape.org/; Aug 2016) for the Gene Ontology (GO) analyses. Boxplots of gene expression log2 fold-changes were generated based on genes assigned to the enriched GO terms. Significantly differentially expressed genes were mapped on the KEGG MAPK signaling and cell cycle pathways.[46]
SUPPLEMENTARY MATERIALS TABLES FIGURES
Acknowledgments and Funding
This study was supported by research funds provided by the Canadian Cancer Society Research Institute (CCSRI). DS holds the François-Karl-Viau Research Chair in Pediatric Oncogenomics.
Abbreviations
- cALL
childhood acute lymphoblastic leukemia
- lncRNA
long non-coding RNA
- GC
glucocorticoid
- siRNA / dsiRNA
short interfering RNA / double-stranded short interfering RNA
- shRNA
short hairpin RNA
- CPT
camptothecin
- DOX
doxorubicin
- GO
gene ontology
- QcALL
Quebec childhood ALL cohort
- RT-qPCR
quantitative reverse transcription polymerase chain reaction
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest.
Authorship contributions
MO designed and performed all experiments, analyzed the data, and wrote the first draft. SD participated in experimental design, and co-wrote the manuscript. ML, MC, and PSO performed bioinformatics analyses. RG assisted MO and provided critical feedback for the manuscript. CR assisted MO and generated RNA sequencing libraries. DS is the principal investigator of the research and participated in the design and interpretation of the results as well as in the writing of the paper.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
- 1.Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med. 2015;373:1541–52. doi: 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- 2.Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350:1535–48. doi: 10.1056/NEJMra023001. [DOI] [PubMed] [Google Scholar]
- 3.Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, Behm FG, Raimondi SC, Relling MV, Patel A, Cheng C, Campana D, Wilkins D, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–43. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 4.Ross ME, Zhou X, Song G, Shurtleff SA, Girtman K, Williams WK, Liu HC, Mahfouz R, Raimondi SC, Lenny N, Patel A, Downing JR. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102:2951–9. doi: 10.1182/blood-2003-01-0338. [DOI] [PubMed] [Google Scholar]
- 5.Andersson A, Eden P, Lindgren D, Nilsson J, Lassen C, Heldrup J, Fontes M, Borg A, Mitelman F, Johansson B, Hoglund M, Fioretos T. Gene expression profiling of leukemic cell lines reveals conserved molecular signatures among subtypes with specific genetic aberrations. Leukemia. 2005;19:1042–50. doi: 10.1038/sj.leu.2403749. [DOI] [PubMed] [Google Scholar]
- 6.Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, Chen IM, Atlas SR, Kang H, Ar K, Wilson CS, Wharton W, Murphy M, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874–84. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhojwani D, Kang H, Menezes RX, Yang W, Sather H, Moskowitz NP, Min DJ, Potter JW, Harvey R, Hunger SP, Seibel N, Raetz EA, Pieters R, et al. Gene expression signatures predictive of early response and outcome in high-risk childhood acute lymphoblastic leukemia: A Children's Oncology Group Study [corrected] J Clin Oncol. 2008;26:4376–84. doi: 10.1200/JCO.2007.14.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silveira VS, Scrideli CA, Moreno DA, Yunes JA, Queiroz RG, Toledo SC, Lee ML, Petrilli AS, Brandalise SR, Tone LG. Gene expression pattern contributing to prognostic factors in childhood acute lymphoblastic leukemia. Leuk Lymphoma. 2013;54:310–4. doi: 10.3109/10428194.2012.710330. [DOI] [PubMed] [Google Scholar]
- 9.Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, Borowitz MJ, Camitta BM, Carroll AJ, Devidas M, Pullen DJ, Payne-Turner D, Tasian SK, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood. 2012;119:3512–22. doi: 10.1182/blood-2011-11-394221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, De Vos J, Hernandez JM, Hofmann WK, Mills KI, Gilkes A, Chiaretti S, Shurtleff SA, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–37. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goff LA, Rinn JL. Linking RNA biology to lncRNAs. Genome Res. 2015;25:1456–65. doi: 10.1101/gr.191122.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15:423–37. doi: 10.1038/nrg3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morlando M, Ballarino M, Fatica A. Long Non-Coding RNAs: New Players in Hematopoiesis and Leukemia. Front Med (Lausanne) 2015;2:23. doi: 10.3389/fmed.2015.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, Poliakov A, Cao X, Dhanasekaran SM, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1:391–407. doi: 10.1158/2159-8290.cd-11-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Z, Zhou L, Wu LM, Lai MC, Xie HY, Zhang F, Zheng SS. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann Surg Oncol. 2011;18:1243–50. doi: 10.1245/s10434-011-1581-y. [DOI] [PubMed] [Google Scholar]
- 18.Kogo R, Shimamura T, Mimori K, Kawahara K, Imoto S, Sudo T, Tanaka F, Shibata K, Suzuki A, Komune S, Miyano S, Mori M. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011;71:6320–6. doi: 10.1158/0008-5472.CAN-11-1021. [DOI] [PubMed] [Google Scholar]
- 19.Niinuma T, Suzuki H, Nojima M, Nosho K, Yamamoto H, Takamaru H, Yamamoto E, Maruyama R, Nobuoka T, Miyazaki Y, Nishida T, Bamba T, Kanda T, et al. Upregulation of miR-196a and HOTAIR drive malignant character in gastrointestinal stromal tumors. Cancer Res. 2012;72:1126–36. doi: 10.1158/0008-5472.CAN-11-1803. [DOI] [PubMed] [Google Scholar]
- 20.Kim K, Jutooru I, Chadalapaka G, Johnson G, Frank J, Burghardt R, Kim S, Safe S. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene. 2013;32:1616–25. doi: 10.1038/onc.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernando TR, Rodriguez-Malave NI, Waters EV, Yan W, Casero D, Basso G, Pigazzi M, Rao DS. LncRNA Expression Discriminates Karyotype and Predicts Survival in B-Lymphoblastic Leukemia. Mol Cancer Res. 2015;13:839–51. doi: 10.1158/1541-7786.MCR-15-0006-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Wu P, Lin R, Rong L, Xue Y, Fang Y. LncRNA NALT interaction with NOTCH1 promoted cell proliferation in pediatric T cell acute lymphoblastic leukemia. Sci Rep. 2015;5:13749. doi: 10.1038/srep13749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bachmann PS, Gorman R, Papa RA, Bardell JE, Ford J, Kees UR, Marshall GM, Lock RB. Divergent mechanisms of glucocorticoid resistance in experimental models of pediatric acute lymphoblastic leukemia. Cancer Res. 2007;67:4482–90. doi: 10.1158/0008-5472.CAN-06-4244. [DOI] [PubMed] [Google Scholar]
- 24.Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, Myers RM. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19:2163–71. doi: 10.1101/gr.097022.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutschner T, Diederichs S. The Hallmarks of Cancer: A long non-coding RNA point of view. RNA Biol. 2012;9:703–19. doi: 10.4161/rna.20481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Zhang B, Wang T, Wang H. LncRNA MALAT1 overexpression is an unfavorable prognostic factor in human cancer: evidence from a meta-analysis. Int J Clin Exp Med. 2015;8:5499–505. [PMC free article] [PubMed] [Google Scholar]
- 27.Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, Thomas M, Berdel WE, Serve H, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–41. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt LH, Spieker T, Koschmieder S, Schaffers S, Humberg J, Jungen D, Bulk E, Hascher A, Wittmer D, Marra A, Hillejan L, Wiebe K, Berdel WE, et al. The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol. 2011;6:1984–92. doi: 10.1097/JTO.0b013e3182307eac. [DOI] [PubMed] [Google Scholar]
- 29.Lai MC, Yang Z, Zhou L, Zhu QQ, Xie HY, Zhang F, Wu LM, Chen LM, Zheng SS. Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med Oncol. 2012;29:1810–6. doi: 10.1007/s12032-011-0004-z. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Chen Z, Wang X, Huang Z, He Z, Chen Y. Long non-coding RNA: a new player in cancer. J Hematol Oncol. 2013;6:37. doi: 10.1186/1756-8722-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Kok JB, Verhaegh GW, Roelofs RW, Hessels D, Kiemeney LA, Aalders TW, Swinkels DW, Schalken JA. DD3(PCA3), a very sensitive and specific marker to detect prostate tumors. Cancer Res. 2002;62:2695–8. [PubMed] [Google Scholar]
- 32.Tissing WJ, Meijerink JP, den Boer ML, Pieters R. Molecular determinants of glucocorticoid sensitivity and resistance in acute lymphoblastic leukemia. Leukemia. 2003;17:17–25. doi: 10.1038/sj.leu.2402733. [DOI] [PubMed] [Google Scholar]
- 33.Pieters R, den Boer ML, Durian M, Janka G, Schmiegelow K, Kaspers GJ, van Wering ER, Veerman AJ. Relation between age, immunophenotype and in vitro drug resistance in 395 children with acute lymphoblastic leukemia—implications for treatment of infants. Leukemia. 1998;12:1344–8. doi: 10.1038/sj.leu.2401129. [DOI] [PubMed] [Google Scholar]
- 34.Schmiegelow K, Nyvold C, Seyfarth J, Pieters R, Rottier MM, Knabe N, Ryder LP, Madsen HO, Svejgaard A, Kaspers GJ. Post-induction residual leukemia in childhood acute lymphoblastic leukemia quantified by PCR correlates with in vitro prednisolone resistance. Leukemia. 2001;15:1066–71. doi: 10.1038/sj.leu.2402144. [DOI] [PubMed] [Google Scholar]
- 35.Klumper E, Pieters R, Veerman AJ, Huismans DR, Loonen AH, Hahlen K, Kaspers GJ, van Wering ER, Hartmann R, In Henze G. vitro cellular drug resistance in children with relapsed/refractory acute lymphoblastic leukemia. Blood. 1995;86:3861–8. [PubMed] [Google Scholar]
- 36.Grausenburger R, Bastelberger S, Eckert C, Kauer M, Stanulla M, Frech C, Bauer E, Stoiber D, von Stackelberg A, Attarbaschi A, Haas OA, Panzer-Grumayer R. Genetic alterations in glucocorticoid signaling pathway components are associated with adverse prognosis in children with relapsed ETV6/RUNX1-positive acute lymphoblastic leukemia. Leuk Lymphoma. 2016;57:1163–73. doi: 10.3109/10428194.2015.1088650. [DOI] [PubMed] [Google Scholar]
- 37.Kfir-Erenfeld S, Sionov RV, Spokoini R, Cohen O, Yefenof E. Protein kinase networks regulating glucocorticoid-induced apoptosis of hematopoietic cancer cells: fundamental aspects and practical considerations. Leuk Lymphoma. 2010;51:1968–2005. doi: 10.3109/10428194.2010.506570. [DOI] [PubMed] [Google Scholar]
- 38.Shen Q, Uray IP, Li Y, Krisko TI, Strecker TE, Kim HT, Brown PH. The AP-1 transcription factor regulates breast cancer cell growth via cyclins and E2F factors. Oncogene. 2008;27:366–77. doi: 10.1038/sj.onc.1210643. [DOI] [PubMed] [Google Scholar]
- 39.Healy J, Belanger H, Beaulieu P, Lariviere M, Labuda D, Sinnett D. Promoter SNPs in G1/S checkpoint regulators and their impact on the susceptibility to childhood leukemia. Blood. 2007;109:683–92. doi: 10.1182/blood-2006-02-003236. [DOI] [PubMed] [Google Scholar]
- 40.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2014. p. btu638. [DOI] [PMC free article] [PubMed]
- 41.Casero D, Sandoval S, Seet CS, Scholes J, Zhu Y, Ha VL, Luong A, Parekh C, Crooks GM. Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages. Nat Immunol. 2015;16:1282–91. doi: 10.1038/ni.3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta. C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 44.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, G Del Angel, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43(11 0):1–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.