

Abstract
Cervical small cell neuroendocrine tumors (CSCNETs) are rare, aggressive neuroendocrine tumors (NETs). Reliable diagnostic and prognostic CSCNET markers are lacking, making diagnosis and prognosis prediction difficult, and treatment strategies limited. Here we provide mutation profiles for five tumor-normal paired CSCNETs using whole exome sequencing (WES). We expanded our assessment of frequently mutated genes to include publicly available data from 55 small intestine neuroendocrine tumors, 10 pancreatic neuroendocrine tumors, 42 small cell lung cancers, six NET cell lines, and 188 cervical cancers, along with our five CSCNETs. We identified 1,968 somatic mutations, including 1,710 missense, 106 nonsense, 144 splice site, 4 lncRNA, 3 nonstop, and 1 start codon mutation. We assigned functions to the 114 most frequently mutated genes based on gene ontology. ATRX, ERBB4, and genes in the Akt/mTOR pathway were most frequently mutated. Positive cytoplasmic ERBB4 immunohistochemical staining was detected in all CSCNET tumors tested, but not in adjacent normal tissues. To our knowledge, this study is the first to utilize WES in matched CSCNET and normal tissues to identify somatic mutations. Further studies will improve our understanding of how ATRX and ERBB4 mutations and AKT/mTOR signaling promote CSCNET tumorigenesis, and may be leveraged in novel anti-cancer treatment strategies.
Keywords: cervical small cell neuroendocrine tumor, ATRX, ERBB4, AKT/mTOR, whole exome sequencing
INTRODUCTION
Neuroendocrine tumors (NETs) are a heterogeneous group of neoplasms derived from the diffuse neuroendocrine system. Cervical small cell NET (CSCNET) is an extremely rare neuroendocrine carcinoma of the cervix, accounting for up to 2% of all cervical carcinomas [1–3]. Based on the Surveillance Epidemiology and End Results (SEER) database, the mean annual CSCNET incidence in the United States from 1977 to 2003 was 0.06 cases per 100,000 women [4]. CSCNETs are morphologically and clinically categorized as poorly differentiated, high grade NETs, while typical and atypical carcinoids are categorized as well differentiated, low/intermediate grade cervical NETs. CSCNETs are clinically aggressive, with unfavorable outcomes and limited treatment strategies. As a result of early metastases to lymph nodes and distant organs, along with vascular invasion, CSCNET is usually not confined to the cervix at the time of diagnosis [5–7]. Disease diagnosis and prognosis prediction are limited by a lack of reliable standardized diagnostic or prognostic markers.
In this study, we performed whole exome sequencing (WES) using five CSCNETs and paired normal tissues to identify key mutations and disrupted pathways. Even with a limited sample number due to disease rarity, we identified frequent mutations in ATRX and ERBB4, and in the Akt/mTOR signaling pathway. Our findings suggest the possibility of a new anti-cancer treatment strategy using ERBB4-Akt/mTOR inhibitors in CSCNET patients.
RESULTS
CSCNET mutation profiles
Immunohistochemical (IHC) staining with chromogranin A and synaptophysin confirmed neuroendocrine differentiation of 16 CSCNETs (Figure 1A, Table 1). Of these 16 samples, the somatic mutation signatures from five tumor-adjacent normal paired CSCNETs were profiled via WES with 136.83X mean sequencing coverage across the targeted bases (Supplementary Table 1), and minimum read depths of 14 in tumors and 8 in normal tissues. We identified 1,968 somatic mutations, including 1,710 missense, 106 nonsense, 144 splice site, 4 lncRNA, 3 nonstop, and 1 start codon mutation, and compared our results with Cosmic data (http://cancer.sanger.ac.uk/cosmic/gene/; accessed 09202016) (Supplementary Table 2). Asian-specific variations were removed using normal lymphocyte DNA datasets from the 1000 Genome Project ASN. Germline and common polymorphisms were filtered via comparison to matching adjacent normal tissue samples and sequences in the Single Nucleotide Polymorphism database (dbSNP). CSCNET mutation spectrums were characterized by a predominance of C>T/G>A transitions, as observed in other solid tumors (Supplementary Figure 1) [8]. A total of 463 mutation sites were identified from 114 recurrently mutated genes (≥50%) in more than three samples (Figure 1B, Supplementary Table 3).
Figure 1. Representative histopathological analyses and somatic mutation signatures from five CSCNETs.
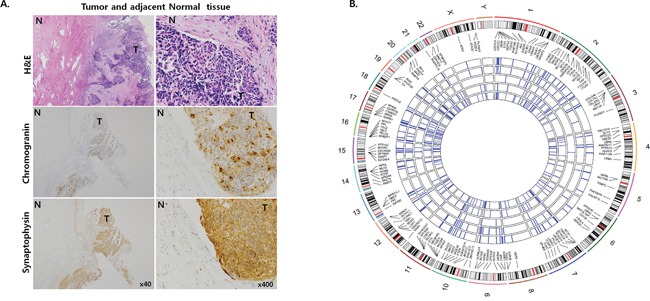
Representative (case 10) H&E and IHC staining for chromogranin and synaptophysin in tumor and adjacent normal tissues A. Tumor tissue stains positive for chromogranin and synaptophysin. Magnifications: x40 (left) and x400 (right). T: tumor tissue; N: adjacent normal tissue. Circos plots showing 114 recurrently mutated genes within 463 missense mutations found in ≥3 CSCNETs B. Five inner circles represent five CSCNETs each and blue lines represent nonsynonymous mutations in each sample. Outer circle represents chromosome with cytoband. Colors in the cytoband indicate: centromere (red), high gene density (black), low gene density (white), and gene empty loci (sky blue).
Table 1. Clinicopathological characteristics of 16 CSCNETs.
| Case | Reviewed Diagnosis | Age(year) | Tumor size(cm) | FIGOstage | Treatment | Metastasis | LVI | LNmetastasis | Follow up(months) | Clinicaloutcome | IHC | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ki-67(%) | CgA | Syn | ERBB4 | |||||||||||
| 1 | smcc | 51 | 1.4×2.0 | IB1 | OP | brain | No | No | 34 | Died | 5 | + | + | +++ |
| 2* | smcc | 45 | 2.5×1.0 | IB1 | OP | No | Yes | No | 9 | Alive | 57.9 | + | + | +++ |
| 3 | smcc | 39 | 2.5×2.5 | IV | No OP | Vertebra | Yes | No | 9 | Died | 90 | - | + | + |
| 4 | smcc | 40 | 3.5×3.2 | III | No OP | No | Yes | Yes | 101 | Alive | 63.5 | - | + | + |
| 5 | smcc | 51 | 2×1.8 | IB1 | OP, CTx | No | Yes | Yes | 166 | Alive | 1 | - | + | + |
| 6 | smcc,AIS withintestinal metaplasia | 51 | 1.2×0.6 | IB1 | OP, CTx | No | Yes | No | 160 | Alive | 47.5 | + | + | + |
| 7 | smcc | 28 | 1.0×0.36 | IB1 | OP, CTx | No | No | Yes | 152 | Alive | 0 | + | + | ++ |
| 8 | smcc | 37 | 2.3×1.5 | IB1 | OP, CTx | Lung | Yes | Yes | 11 | Died | 1.6 | + | + | ++ |
| 9 | smcc | 47 | 5.3×2.7 | IB2 | OP, CTx | No | Yes | Yes | 59 | Alive | 72.5 | + | + | +++ |
| 10* | smcc | 46 | 1.2×0.8 | IB1 | OP, CTx,RTx | No | Yes | No | 12 | Alive | 50.1 | + | + | +++ |
| 11* | smcc | 35 | 1.5×1.5 | IB1 | OP, CTx | No | Yes | No | 30 | Died | 79.6 | + | + | +++ |
| 12 | smcc+rosette | 45 | 2.0×2.2 | IB1 | OP | Lung, pancreas, bone | Yes | No | 60 | Alive | 41.3 | + | + | ++ |
| 13* | smcc+rosette | 48 | 4.5×1.6 | IIA | OP | No | Yes | Yes | 100 | Alive | 35 | - | + | ++ |
| 14 | smcc | 47 | 1.5×1.5 | IB1 | OP | no | Yes | No | 60 | Alive | 67.8 | + | + | ++ |
| 15 | smcc+rosette | 47 | 1×1.5 | IV | No OP,CTx, RTx | Bone, liver | Yes | Yes | 29 | Died | 60 | + | + | ++ |
| 16* | smcc | 33 | 2.1×1.0 | IV | No OP,CTx, RTx | Lung | Yes | No | 17 | Alive | 1 | + | + | ++ |
Abbreviations: *Normal and tumor tissues analyzed via WES; CNET: small cell neuroendocrine tumor; FIGO: Fédération Internationale de Gynécologieetd’ Obstétrique; LVI: lymphovascular invasion; LN: lymph node; IHC: immunohistochemistry; CgA: chromogranin A; Syn: synaptophysin; OP: radical hysterectomy; CTx: chemotherapy; RTx: radiotherapy.
Gene set analysis (GSA) was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID). We assigned functions to the 114 frequently mutated genes based on gene ontology [9]. Genes with functions in small GTPase mediated signal transduction, forebrain development, and protein kinase B/AKT signaling were enriched (P<0.001; Supplementary Table 4). Signal transduction pathways facilitated by small GTPases, like those in the RAS/Rho family, play important roles in multiple cellular and developmental processes, including differentiation, cell division, cytoskeletal dynamics, and cell survival. Deregulation of small GTPase-mediated transduction by various mechanisms is associated with multiple cancers [10]. Association with pathways involved in forebrain development may explain the origin of CSCNETs, which derive from a neuroendocrine cell that performs neuroendocrine integration. Protein kinase B signaling, also known as AKT signaling, promotes cell survival and growth in response to extracellular signals. This pathway, along with mTOR signaling, has been linked to a range of cancers, including cervical cancer and NETs [11–16].
In our study, genes with recurrent mutations were detected in five CSCNETs, and other NETs were analyzed by combining mutation data from 118 samples representing 10 pancreatic neuroendocrine tumors (panNETs) [17], 42 small cell lung cancers (SCLCs) [18], 55 small intestine neuroendocrine tumors (SINETs) [19], six NET cell lines [20] and our five CSCNETs (Supplementary Table 5A–5E). We selected a recurrent mutation rate of ≥5% in other NETs and ≥50% (in ≥3 samples) in our CSCNETs. Two genes (PLEC, OBSCN) were mutated in CSCNET, SCLC and NET cell lines, one gene (TSC2) was mutated in CSCNET, panNET and NET cell lines, five genes (PLEC, VPS13A, OBSCN, KIAA1109 and PCNX) were mutated in CSCNET and SCLC, and six genes (KANK1, TSC2, NF1, DYNC1H1, PTEN and TTC21B) were mutated in CSCNET and panNET. However, no single gene was mutated in every NET (Supplementary Figure 2).
Recurrent ATRX mutation in CSCNETs
Mutation profiles have been described in panNET, SINET, and SCLC using genome wide mutation analysis [17–21]. Mutations in ATRX, previously identified in SINET [19] and panNET with incidences of 17.65–30.23% [17, 21], were found in four CSCNET samples in this study (Supplementary Table 6). These mutations include p.R250X (case 12), p.N281S (case 16), p.G1042R (case 2) and p.R2387G (case 10), which were not identified in other published NETs [17, 19, 21] or Cosmic data (http://cancer.sanger.ac.uk/cosmic/gene/; accessed 09202016) (Supplementary Table 2). We predicted the functional impact of each annotated mutation site in silico to estimate domain activity (mutationassessor.org) (Supplementary Figure 3, Supplementary Table 6 and Supplementary Table 7) [22]. Two annotated mutation sites (p.R250X and p.N281S) were located in the ATRX-DNMT3-DNMT3L (ADD) domain. p.R250X produced a stop codon and p.N281S had a low predicted functional impact. The two other annotated mutation sites, p.G1042R and p.R2387G, had neutral predicted functional impacts. However, R2387G is in a highly conserved region.
Recurrent ERBB4 mutation and expression in CSCNETs
ERBB4 is a member of the epidermal growth factor receptor (EGFR) subfamily. When bound by ligands like neuregulin, ERBB4 activates numerous downstream pathways, including Ras/MAPK/ERK and PI3K/AKT signaling (via mTOR), to regulate both normal cellular processes and cancer development and progression [23–25]. We identified five ERBB4 mutations, three of which (p.P6619S, p.P981S and p.P996S) were annotated in Cosmic (http://cancer.sanger.ac.uk/cosmic/gene/; accessed 09202016) (Supplementary Table 2) and in four samples. We predicted functional impact for each annotated mutation site in silico (mutationassessor.org) (Figure 2A, Supplementary Table 6 and Supplementary Table 7) [22]. Three annotated mutation sites were located in functional domains, but were predicted to have low or neutral impacts. One of these mutations (p.T743P: case 16) was located in the protein binding site kinase domain, and two (p.Q558R: case 10, and p.P619S: case 2) were located in the growth factor (GF)-receptor IV domain site. Two additional mutations were located in the intracellular domain (p.P981S: case 13, and p.P996S: case 2) and were predicted to have moderate functional impacts. ERBB4 protein expression was examined by IHC staining in all 16 CSCNETs, including the 11 samples in which WES was not performed. All 16 tumor samples exhibited greater positive cytoplasmic ERBB4 staining as compared to adjacent normal tissues (Figure 2B, Table 1). Five samples, including cases 2 and 10, with ERBB4 mutations, and case 11, without ERBB4 mutation, showed the strongest expression. Seven cases, including cases 13 and 16, with ERBB4 mutations, exhibited modest expression. The remaining four cases showed minimal expression. We could not correlate ERBB4 mutation status with protein level, because mutation analysis in 11 samples could not be performed due to sample quality and/or quantity. However, higher ERBB4 expression correlated with lower OS by Kaplan-Meier analysis, although this correlation was not statistically significant, likely due to small sample size (Supplementary Figure 4).
Figure 2. EBRR4 mutation and protein expression.
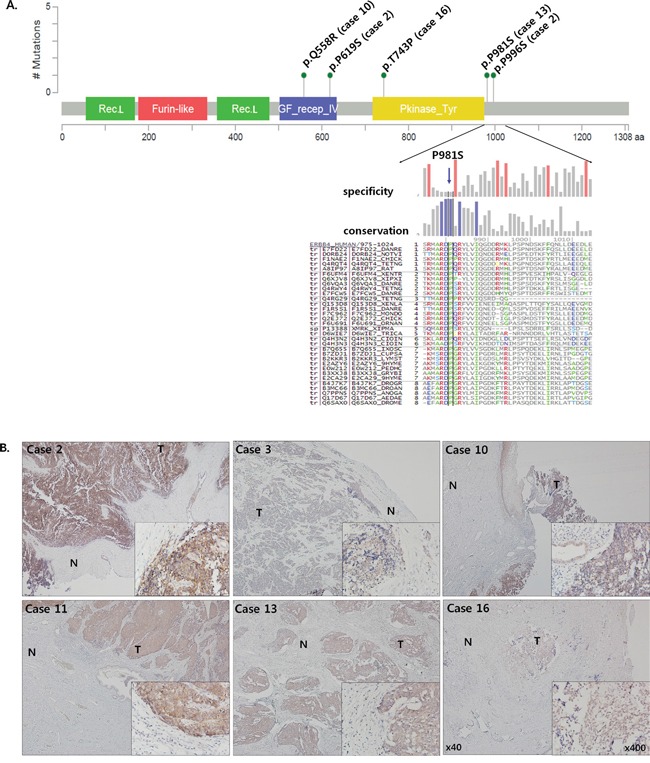
Functional domains containing five ERBB4 mutation sites from four CSCNETs A. Predictions for each mutation site indicated that p.P981S is highly conserved across species (mutationassessor.org). Representative IHC staining for ERBB4 B. Tumors, but not adjacent normal tissues, were positive for cytoplasmic ERBB4 expression. Cases 2, 10, and 11 exhibited the strongest expression, cases 13 and 16 showed medium expression, and case 3 showed the lowest expression. Original magnifications are x40 (big image) and x400 (small image). T: tumor tissue; N: adjacent normal tissue
Recurrent Akt/mTOR signaling pathway gene mutation in CSCNETs
The Akt/mTOR signaling pathway is a recurrent driver pathway promoting NETs [13, 14, 16]. GSA indicated enrichment of genes in the mTOR (GO:0032006) and Akt (GO:0051896) signaling pathway in CSCNETs in our study (P<0.01). The dynamic mutation profiles of Akt/mTOR signaling pathway genes downstream of the receptor tyrosine kinase (RTK) were assessed across several NETs, including panNET, SINET, SCLC, and CSCNETs. ERBB4, NF1, PTEN, RICTOR, and TSC1/2 were recurrently mutated in our CSCNETs (Figure 3, Supplementary Table 6).
Figure 3. Recurrently mutated RTK-Akt/mTOR pathway genes found in CSNCETs, other NETs, and CT.
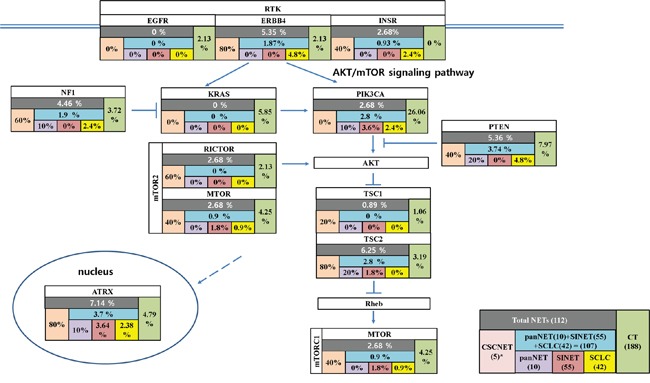
(): number of samples. *Indicated gene mutation rate was high, likely due to low sample number.
Cervical tumor (CT) signatures in CSCNET mutations
CT genomic studies have identified recurrent somatic driver mutations in PIK3CA, KRAS, TP53, ERBB2 and MAPK1 [26, 27] that were not found in our CSCNETs. However, the recurrent somatic TSC2, NF1 and PTEN mutations identified in our CSCNETs were also identified in NETs, but not in CTs (Figure 4, Supplementary Table 8). Although mutations in some genes, including in FLNC, MUC5B, PCLO, PLEC, OBSCN and VPS13A, were found in our CSCNETs and in CTs or NETs, they are not likely to drive CT or NET development or progression, because their recurrence rates are very low compared to those of predicted driver genes. This confirms that the CSCNET origin and genetic background are likely to be different from those of CT, and more similar to those of NETs.
Figure 4. Venn diagram of frequently mutated gene(s) shared across CSCNETs, NETs and CT.
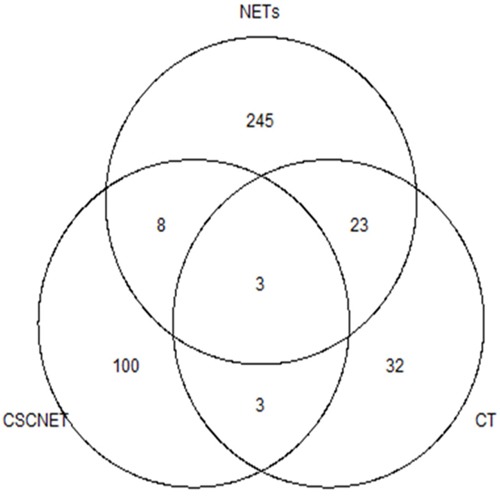
Recurrently mutated genes were selected at ≥5% in CTs and other NETs, and ≥50% (≥3 samples) in our CSCNETs. Mutated genes are listed in Supplementary Table 8A.
DISCUSSION
This study provides the first report of the CSCNET mutation landscape as determined by WES. A limitation of our study is the small number (five) of CSCNET cases, due to extremely low disease incidence. We identified recurrent mutations in ATRX, EBRR4, and in AKT/mTOR signaling pathway genes, such as NF1, PTEN, RICTOR and TSC2 in our CSCNETs. We found that the CSCNET mutation profile was more similar to that of other NETs as compared to CTs, indicating that genomic mutation may be correlated with tumor cell origin.
The ATRX mutation was highlighted in panNET, and identified in SINET, SCLC, and CT. In our CSCNETs, four out of five samples analyzed contained an ATRX mutation, although more samples must be tested to affirm this incidence rate. ATRX transcriptionally regulates genes activated during interphase and chromosomal segregation in mitosis [28]. Clinical implications of ATRX mutations are contradictory, and mutations were associated with better prognosis in Caucasian patients, but poorer prognosis in Chinese patients via panNET [17, 21]. These findings suggest that ATRX mutations may be frequent in NETs and underscore the need for understanding the common pathogenesis of NETs. The effects of ATRX mutations in CSCNET must still be explored.
ERBB4 mutations (found in 4/5 CSCNETs analyzed) and ERBB4 protein levels (positive in 11/11 CSCNETs) were examined. Due to small sample sizes, we could not precisely correlate ERBB4 mutation site and predicted functional impact with ERBB4 protein levels. However, IHC staining showed that cytoplasmic ERBB4 expression only occurred in tumor tissues and not in adjacent normal tissues. Somatic ERBB4 mutations have been reported in various cancers [29–31], and studies attempting to assess the impacts of these mutations have suggested both oncogenic and tumor suppressor functions for ERBB4 [22, 23, 30–33]. One model suggests that signaling downstream of wild type ERBB4 is enriched in the JAK/STAT pathway, whereas mutant ERBB4 signals more commonly via the PI3K/AKT pathway, constituting a cellular shift from a program of differentiation to that of proliferation [23, 24]. This suggests that ERBB4 gain or loss of function mutations, and other mechanisms up- or downregulating ERBB4, may affect downstream PI3K/AKT and mTOR signaling, which appear to be the key pathways activated in our CSCNETs. Members of the EGFR subfamily are frequently mutated oncogenes, and many are amenable therapeutic targets in various cancers, including non-small cell lung cancer and breast cancer [22]. ERBB4 may be an effective therapeutic target in CSCNETs, and further studies are necessary to better understand the clinical implications of ERBB4 expression changes.
Along with ATRX and ERRB4 mutations, high mutation incidences in AKT/mTOR signaling pathway components, such as NF1, PTEN, RICTOR, and TSC2, were observed in our CSCNETs. NF1, PTEN and TSC1/2 are tumor suppressors that inhibit PI3K/AKT signaling through the TSC complex, which is a critical negative regulator of mTORC1 [12, 34]. NF1, PTEN and TCS1/2 mutations have been reported both in familial syndromes, such as type 1 neurofibromatosis, Cowden syndrome, and tuberous sclerosis complex, and in sporadic NETs [35]. RICTORs are key partnering proteins that modulate mTOR, and RICTOR mutations have been identified in SCLC [18]. AKT activation through the mTOR-RICTOR complex was reported in various tumor types [16, 36–38]. Aberrant mTOR signaling, through altered mTOR pathway component expression or activation, has been associated with prognosis in panNETs, SINET, gastric NETs, and SCLC [14, 39, 40–43]. Targeting mTOR signaling has emerged as an effective therapeutic strategy for the management of advanced NETs [15, 16, 43]. Our results suggest new options for CSCNET treatment, similar to strategies for treating other NETs, which include AKT/mTOR pathway inhibitors.
To our knowledge, this study is the first to utilize WES in matched CSCNET and normal tissues to identify somatic mutations. We identified two frequently mutated genes, ATRX and ERBB4, and implicated AKT/mTOR signaling in CSCNET tumorigenesis. Future studies will help us better understand how these mutations and signaling pathways promote CSCNET development and progression, and how they may be applied in new anti-cancer treatment strategies.
MATERIALS AND METHODS
Tumor sample collection
This study included 16 formalin fixed paraffin embedded (FFPE) CSCNET tissue blocks diagnosed and collected between 1997 and 2012 in South Korea. All 16 hematoxylin and eosin (H&E)-stained slides were reviewed by pathologists to confirm histological diagnoses and classifications, which were performed according to the cervical NET criteria set in the 1997 workshop of the College of American Pathologists and the National Cancer Institute [1]. Clinicopathological data and Ki-67 indexes are described in Table 1. We were unable to collect information regarding HIV infection status. Mean patient age was 43.2 years (range 27–52) and eleven cases (68.9%) showed >50% Ki-67 index. Approval for this study was obtained from the institutional Ethics Committee of Korea University (IRB No. KU-IRB-14-88-A-1) and a waiver of informed consent was granted.
Immunohistochemical (IHC) analysis
Five of 16 tumor samples, for which paired adjacent normal tissues were available, were used to perform WES. Five FFPE paired tumor and adjacent normal tissue blocks were cut into 4μm sections, which were deparaffinized in xylene and hydrated by immersion in a graded ethanol series. Antigen retrieval was performed in a microwave by placing sections in epitope retrieval solution (0.01 M citrate buffer, pH 6.0, or 10 mM ethylenediaminetetraacetic acid, pH 8.0) for 20 min, and endogenous peroxidase was blocked via immersion in 0.3% hydrogen peroxide for 10 min. IHC staining was performed using the Dako Auto stainer plus Universal Staining System (Dako Cytomation, Carpinteria, CA, USA) with a Chem Mate DAKO En Vision detection kit (Dako Cytomation). Antibodies against chromogranin A (DAKO, Glostrup, Denmark), synaptophysin (Ventana, Roche, Tucson, USA), Ki-67 (monoclonal, MIB-1 clone, 1:50, DAKO), and ERBB4 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used.
DNA isolation
FFPE tissue blocks were cut into 10μm sections on slide glass. Tumor and normal tissues were visualized and separately scraped with a razor blade into 1.5 mL tubes. Scraped sections were deparaffinized twice for 5 min in xylene, rehydrated in series of 100%, 96%, and 70% ethanol for 30 sec each, stained with hematoxylin for 30 sec, rinsed with water, and incubated overnight in 1 M NaSCN at 37°C to remove crosslinks. After sample pre-treatment, DNA was isolated using the QIAamp DNA extraction kit (Qiagen, USA) according to the manufacturer's instructions. DNA samples were quantified using the Qubit® dsDNA HS Assay Kit (Life Technologies, Cat. no. Q32851). A total of 100 ng isolated DNA was treated with uracil-DNA glycoslyase (UDG, New England BioLabs, Inc.) according to the manufacturer's instructions and stored at 4°C before use in the subsequent target amplification reactions.
Ion ampliSeqTM exome library preparation
An Ion AmpliSeqTM Exome library was constructed using the Ion AmpliSeq™ Exome Kit (Life Technologies, Part #4487084 Rev. B.0) as per the manufacturer's protocol. Briefly, 100 ng of UDG-treated DNA was amplified for multiplex PCR with each of 12 primer-pools. The amplicons, which were partially digested primer sequences, were ligated to the Ion Torrent adapters P1 and the Ion XpressTM Barcode using DNA ligase. Adapter-ligated products were then purified using AMPure XP reagent (Beckman Coulter, Brea, CA, USA), and PCR-amplified for five cycles. The resulting library was purified using AMPure XP reagent (Beckman Coulter). 200–350 base read libraries were selected for the Pippin Prep™ instrument (Sage Science) using 2% agarose gel cassettes (Sage Science). The size-selected library was purified using AMPure XP reagent (Beckman Coulter), and library concentration and size were determined using the Agilent 2100 BioAnalyzer and the Agilent BioAnalyzer DNA High-Sensitivity LabChip (Agilent Technologies).
Ion proton sequencing
Sample emulsion PCR, emulsion breaking, and enrichment were performed using the Ion PITM Template OT2 200 Kit v3 (Life Technologies, Part #4488318 Rev. B.0) according to the manufacturer's instructions. Equal molar ratios of multiple barcoded libraries were combined for one Ion PITM v2 chip. Two pooled Ion AmpliSeqTMExome libraries were loaded onto a single Ion PITM v2 chip. Five pooled Ion AmpliSeq™ Transcriptome libraries were loaded onto a single Ion PITM v2 chip. Subsequent emulsion PCR and enrichment of the pooled library sequencing beads was performed using the Ion OneTouchTM system (Life Technologies) according to the manufacturer's protocol within approximately seven hours. Finally, sequencing (520 flows) was performed with the Ion PITM v2 chip using the Ion PITM Sequencing 200 Kit v3 (Life Technologies, Part #4488315 Rev. B.0) on the Ion ProtonTM sequencer (Life Technologies).
Processing of whole-exome sequencing data
After the FastQC step, trimmed FASTQ reads were aligned to human reference genome version 19 (hg19) using the TMAP mapping program with default parameters [44]. The results were sorted and compressed in BAM format using Picard SortSam. In accordance with manufacturer's instructions, duplicates were not removed from AmpliSeq data [45]. Local realignment around indels and recalibration were performed by GATK [46]. Recalibrated BAM files were used to call variants with MuTect software (version 1.1.4) using default parameters [47]. Nonsynonymous mutations within each sample were visualized by circos plot using the RCircos library (http://www.ncbi.nlm.nih.gov/pubmed/23937229). The RCircos plot produced a human chromosome ideogram heatmap with five sample tracks for mutations. We used the chromosome ideogram tables from the UCSC genome browser (https://genome.ucsc.edu/). The chromosome ideogram is shown in high-resolution to plot gene locations. Tools in the Database for Annotation, Visualization, and Integrated Discovery (DAVID) were used to assess gene ontology enrichment and to identify biological pathway categories associated with somatically mutated genes [9]. Sequence data for CSCNET samples used in this study were deposited in the European Nucleotide Archive under accession number, PRJEB12274.
Recurrent mutations in CTs and NETs
The mutated genes in the NETs dataset are from a recent WES study of 55 SINETs [19], 10 panNETs [17], 42 SCLCs [18], six NET cell lines [20] and our five CSCNETs. The mutated CT dataset is from 188 samples from The Cancer Genome Atlas (TCGA). Publicly available somatic variant calls in mutation annotation format (MAF) files were used in this study. All MAF files were downloaded from TCGA cBioPortal or supplementary tables from each study. TCGA Pan-cancer analysis provided the distributions of mutation frequencies, types, and contexts across 12 tumor types [48]. In the TCGA study, only genes mutated in at least 5% of tumors were analyzed. With this cutoff percentage, mutated genes were selected with ≥5% recurrent rates from other NET and CT studies. We used ≥50% (≥3 samples) recurrent mutation as a cutoff for our CSCNETs. We also added significantly reported genes from each study. Selected gene mutations were nonsynonymous. Venn diagram analysis shows co-occurrence among NETs, and mutated gene overlap between CT, NETs, and CSCNETs.
SUPPLEMENTARY MATERIALS FIGURES AND TABLES
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This research was supported by the Collaborative Genome Program for Fostering New Post-Genome Industry through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Science ICT and Future Planning (NRF-2014M3C9A3064693), by the Basic Science Research Program Grant (NRF-2014R1A2A2A01003566) from the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education, Science and Technology (MEST), Republic of Korea, and Republic of Korea, and Future Planning and Bio-Synergy Research Project (NRF-2014M3A9C4066487) of the Ministry of Science, ICT and Future Planning through the National Research Foundation.
REFERENCES
- 1.Albores-Saavedra J, Gersell D, Gilks CB, Henson DE, Lindberg G, Santiago H, Scully RE, Silva E, Sobin LH, Tavassoli FJ, Travis WD, Woodruff JM. Terminology of endocrine tumors of the uterine cervix: results of a workshop sponsored by the College of American Pathologists and the National Cancer Institute. Arch Pathol Lab Med. 1997;121:34–39. [PubMed] [Google Scholar]
- 2.Van Nagell JR, Jr, Donaldson ES, Wood EG, Maruyama Y, Utley J. Small cell cancer of the uterine cervix. Cancer. 1977;40:2243–2249. doi: 10.1002/1097-0142(197711)40:5<2243::aid-cncr2820400534>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 3.Scully RE, Aguirre P, DeLellis RA. Argyrophilia, serotonin, and peptide hormones in the female genital tract and its tumors. Int J Gynecol Pathol. 1984;3:51–70. doi: 10.1097/00004347-198403010-00005. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Macdonald OK, Gaffney DK. Incidence, mortality, and prognostic factors of small cell carcinoma of the cervix. Obstet Gynecol. 2008;111:1394–1402. doi: 10.1097/AOG.0b013e318173570b. [DOI] [PubMed] [Google Scholar]
- 5.Cohen JG, Kapp DS, Shin JY, Urban R, Sherman AE, Chen LM, Osann K, Chan JK. Small cell carcinoma of the cervix: treatment and survival outcomes of 188 patients. Am J Obstet Gynecol. 2010;203 doi: 10.1016/j.ajog.2010.04.019. 347.e341-346. [DOI] [PubMed] [Google Scholar]
- 6.Chan JK, Loizzi V, Burger RA, Rutgers J, Monk BJ. Prognostic factors in neuroendocrine small cell cervical carcinoma: a multivariate analysis. Cancer. 2003;97:568–574. doi: 10.1002/cncr.11086. [DOI] [PubMed] [Google Scholar]
- 7.Straughn JM, Jr, Richter HE, Conner MG, Meleth S, Barnes MN. Predictors of outcome in small cell carcinoma of the cervix--a case series. Gynecol Oncol. 2001;83:216–220. doi: 10.1006/gyno.2001.6385. [DOI] [PubMed] [Google Scholar]
- 8.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O'Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarz JK, Payton JE, Rashmi R, Xiang T, Jia Y, Huettner P, Rogers BE, Yang Q, Watson M, Rader JS, Grigsby PW. Pathway-specific analysis of gene expression data identifies the PI3K/Akt pathway as a novel therapeutic target in cervical cancer. Clin Cancer Res. 2012;18:1464–1471. doi: 10.1158/1078-0432.CCR-11-2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Populo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci. 2012;13:1886–1918. doi: 10.3390/ijms13021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Briest F, Grabowski P. PI3K-AKT-mTOR-signaling and beyond: the complex network in gastroenteropancreatic neuroendocrine neoplasms. Theranostics. 2014;4:336–365. doi: 10.7150/thno.7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan J, Kulke M. Targeting the mTOR signaling pathway in neuroendocrine tumors. Curr Treat Options Oncol. 2014;15:365–379. doi: 10.1007/s11864-014-0294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polivka J, Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142:164–175. doi: 10.1016/j.pharmthera.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 16.Qian ZR, Ter-Minassian M, Chan JA, Imamura Y, Hooshmand SM, Kuchiba A, Morikawa T, Brais LK, Daskalova A, Heafield R, Lin X, Christiani DC, Fuchs CS, Ogino S, Kulke MH. Prognostic significance of MTOR pathway component expression in neuroendocrine tumors. J Clin Oncol. 2013;31:3418–3425. doi: 10.1200/JCO.2012.46.6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, Velculescu VE, Diaz LA, Jr, Vogelstein B, Kinzler KW, Hruban RH, Papadopoulos N. DAXX/ATRX MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, Bhatt D, Stinson J, Gnad F, Haverty PM, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, Banck MS, Kanwar R, Kulkarni AA, Karpathakis A, Manzo V, Contractor T, Philips J, Nickerson E, Pho N, Hooshmand SM, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet. 2013;45:1483–1486. doi: 10.1038/ng.2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boora GK, Kanwar R, Kulkarni AA, Pleticha J, Ames M, Schroth G, Beutler AS, Banck MS. Exome-level comparison of primary well-differentiated neuroendocrine tumors and their cell lines. Cancer Genet. 2015;208:374–381. doi: 10.1016/j.cancergen.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Yuan F, Shi M, Ji J, Shi H, Zhou C, Yu Y, Liu B, Zhu Z, Zhang J. KRAS and DAXX/ATRX gene mutations are correlated with the clinicopathological features, advanced diseases, and poor prognosis in Chinese patients with pancreatic neuroendocrine tumors. Int J Biol Sci. 2014;10:957–965. doi: 10.7150/ijbs.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roskoski R., Jr The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 24.Telesco SE, Vadigepalli R, Radhakrishnan R. Molecular modeling of ErbB4/HER4 kinase in the context of the HER4 signaling network helps rationalize the effects of clinically identified HER4 somatic mutations on the cell phenotype. Biotechnol J. 2013;8:1452–1464. doi: 10.1002/biot.201300022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tvorogov D, Sundvall M, Kurppa K, Hollmen M, Repo S, Johnson MS, Elenius K. Somatic mutations of ErbB4: selective loss-of-function phenotype affecting signal transduction pathways in cancer. J Biol Chem. 2009;284:5582–5591. doi: 10.1074/jbc.M805438200. [DOI] [PubMed] [Google Scholar]
- 26.Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio L, Cibulskis K, Bertelsen B, Romero-Cordoba S, Trevino V, Vazquez-Santillan K, Guadarrama AS, Wright AA, Rosenberg MW, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–375. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spaans VM, Trietsch MD, Peters AA, Osse M, N Ter Haar, Fleuren GJ, Jordanova ES. Precise Classification of Cervical Carcinomas Combined with Somatic Mutation Profiling Contributes to Predicting Disease Outcome. PLoS One. 2015;10:e0133670. doi: 10.1371/journal.pone.0133670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bérubé NG, Smeenk CA, Picketts DJ. Cell cycle-dependent phosphorylation of the ATRX protein correlates with changes in nuclear matrix and chromatin association. Hum Mol Genet. 2000;9:539–547. doi: 10.1093/hmg/9.4.539. [DOI] [PubMed] [Google Scholar]
- 29.Soung YH, Lee JW, Kim SY, Wang YP, Jo KH, Moon SW, Park WS, Nam SW, Lee JY, Yoo NJ, Lee SH. Somatic mutations of the ERBB4 kinase domain in human cancers. Int J Cancer. 2006;118:1426–1429. doi: 10.1002/ijc.21507. [DOI] [PubMed] [Google Scholar]
- 30.Kurppa KJ, Denessiouk K, Johnson MS, Elenius K. Activating ERBB4 mutations in non-small cell lung cancer. Oncogene. 2016;35:1283–1291. doi: 10.1038/onc.2015.185. [DOI] [PubMed] [Google Scholar]
- 31.Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA, Samuels Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41:1127–1132. doi: 10.1038/ng.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sundvall M, Korhonen A, Paatero I, Gaudio E, Melino G, Croce CM, Aqeilan RI, Elenius K. Isoform-specific monoubiquitination, endocytosis, and degradation of alternatively spliced ErbB4 isoforms. Proc Natl Acad Sci U S A. 2008;105:4162–4167. doi: 10.1073/pnas.0708333105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams EE, Trout LJ, Gallo RM, Pitfield SE, Bryant I, Penington DJ, Riese DJ., 2nd A constitutively active ErbB4 mutant inhibits drug-resistant colony formation by the DU-145 and PC-3 human prostate tumor cell lines. Cancer Lett. 2003;192:67–74. doi: 10.1016/s0304-3835(02)00690-0. [DOI] [PubMed] [Google Scholar]
- 34.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7:183–188. doi: 10.1007/s11523-012-0226-9. [DOI] [PubMed] [Google Scholar]
- 36.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 38.Moraitis D, Karanikou M, Liakou C, Dimas K, Tzimas G, Tseleni-Balafouta S, Patsouris E, Rassidakis GZ, Kouvaraki MA. SIN1, a critical component of the mTOR-Rictor complex, is overexpressed and associated with AKT activation in medullary and aggressive papillary thyroid carcinomas. Surgery. 2014;156:1542–1548. doi: 10.1016/j.surg.2014.08.095. discussion 1548-1549. [DOI] [PubMed] [Google Scholar]
- 39.Hay N, Qian ZR, Ter-Minassian M, Chan JA, Imamura Y, Hooshmand SM, Kuchiba A, Morikawa T, Brais LK, Daskalova A, Heafield R, Lin X, Christiani DC, Fuchs CS, Ogino S, Kulke MH. Prognostic significance of MTOR pathway component expression in neuroendocrine tumors. J Clin Oncol. 2013;31:3418–3425. doi: 10.1200/JCO.2012.46.6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komori Y, Yada K, Ohta M, Uchida H, Iwashita Y, Fukuzawa K, Kashima K, Yokoyama S, Inomata M, Kitano S. Mammalian target of rapamycin signaling activation patterns in pancreatic neuroendocrine tumors. J Hepatobiliary Pancreat Sci. 2014;21:288–295. doi: 10.1002/jhbp.26. [DOI] [PubMed] [Google Scholar]
- 41.Righi L, Volante M, Rapa I, Tavaglione V, Inzani F, Pelosi G, Papotti M. Mammalian target of rapamycin signaling activation patterns in neuroendocrine tumors of the lung. Endocr Relat Cancer. 2010;17:977–987. doi: 10.1677/ERC-10-0157. [DOI] [PubMed] [Google Scholar]
- 42.Ruza A, Griniak K, Phillip L, Felder S, Pape FU, Manfred D. Immunohistochemical Analysis of mTOR Pathway Expression in Gastric Neuroendocrine Tumors. J Clin Exp Pathol. 2014;4:1000173. [Google Scholar]
- 43.Wolin EM. PI3K/Akt/mTOR pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett. 2013;335:1–8. doi: 10.1016/j.canlet.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 44.Cartwright DA, Troggio M, Velasco R, Gutin A. Genetic mapping in the presence of genotyping errors. Genetics. 2007;176:2521–2527. doi: 10.1534/genetics.106.063982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samorodnitsky E, Jewell BM, Hagopian R, Miya J, Wing MR, Lyon E, Damodaran S, Bhatt D, Reeser JW, Datta J, Roychowdhury S. Evaluation of Hybridization Capture Versus Amplicon-Based Methods for Whole-Exome Sequencing. Hum Mutat. 2015;36:903–914. doi: 10.1002/humu.22825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.