

Abstract
Scaffold-based delivery of bioactive molecules capable of directing stem cell differentiation is critical to the development of point-of-care cell therapy for orthopaedic repair. Dexamethasone-conjugated hyaluronic acid (HA-DXM) was synthesized and combined with hydrolytically degradable, photocrosslinkable PEG-bis-(2-acryloyloxy propanoate) (PEG-bis-AP) to form semi-IPNs. Dexamethasone (DX) release was limited in physiological buffer and substantially increased in the presence of encapsulated human mesenchymal stem cells (hMSCs) or exogenous hyaluronidase, confirming that release occurred primarily by a cell-mediated enzymatic mechanism. hMSCs encapsulated in PEG-bis-AP/HA-DXM semi-IPNs increased osteoblast-specific gene expression, alkaline phosphatase activity, and matrix mineralization, attaining levels that were not significantly different from positive controls consisting of hMSCs in PEG-bis-AP/native HA cultured with DX supplementation in the culture medium. These studies demonstrate that PEG-bis-AP/HA-DXM semi-IPNs can provide cell-mediated release of bioactive free DX that induces hMSC osteogenic differentiation. This approach offers an efficient system for local delivery of osteogenic molecules empowering point of care applications.
Keywords: Hyaluronic acid, dexamethasone, semi-IPNs, cell-mediated release, osteogenesis
Graphical abstract
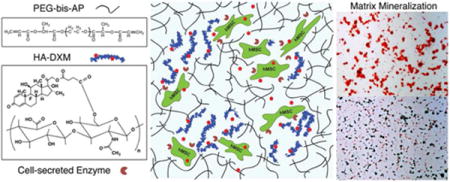
1. Introduction
Bone tissue engineering is a complex and dynamic field that utilizes biomimetic scaffolds, osteoinductive molecules, and osteogenic cells to achieve bone regeneration in pathological and large defects.1,2 Mesenchymal stem cells (MSCs) are a multipotent cell source with osteogenic potential that can be isolated from a variety of tissues. Culture-expanded MSCs in combination with biomaterial scaffolds have been shown to be an effective strategy for enhanced bone formation in many preclinical studies.3 However, clinical application of this approach is still controversial due to the need for two separate procedures for isolation and transplantation; the time and cost required for expansion under GMP guidelines, and the current FDA classification of such cells as medical products that creates a substantial regulatory barrier to clinical translation.4,5 In order to address this primary obstacle, intraoperative stem cell therapy strategies have been developed that include the selection and concentration of autologous progenitor cells by minimal processing within the operating room and their immediate re-implantation to patients at the time of care.6
Because intraoperative therapy removes the opportunity for priming/pre-differentiation that has been shown to enhance subsequent tissue formation post-transplantation,7–9 there is a critical need to simultaneously deliver bioactive stimuli to direct the osteogenic differentiation of MSCs in vivo. Currently, proteins/growth factors integrated into scaffolds such as BMPs, FGF, IGF, PDGF, TGF-βs, and VEGF are of special interest for bone regeneration because of their ability to stimulate the differentiation of osteoprogenitor cells to bone-specific lineages and promote angiogenesis.10 Specifically, BMPs have been extensively studied as osteoinductive agents in clinical bone repair and several polymeric products incorporating rhBMPs have received FDA approval for orthopedic surgical applications.11,12 However, the clinical use of recombinant BMPs still shows several risks, including swelling, vertebral osteolysis, ectopic bone formation, urogenital complications, and cost concerns.13–17 These challenges have motivated the development of alternative strategies for delivering osteogenic stimuli incorporated within biomaterials.18,19
Dexamethasone is a synthetic glucocorticoid, which has been shown to stimulate the proliferation and osteogenic lineage differentiation of MSCs in vitro, together with β-glycerophosphate and ascorbic acid, in defined concentrations.20,21 In order to maintain the required concentration of dexamethasone at a site of bone regeneration in situ, several investigators have developed polymeric microparticles or hydrogels for localized, sustained delivery. For example, Dawes et al. developed dexamethasone-loaded PLGA microparticles that provided sustained release over 30 days and stimulated osteoblast differentiation and matrix mineralization.22,23 Shi et al. fabricated composite scaffolds containing PLGA microspheres loaded with alendronate and dexamethasone that supported osteogenic differentiation of synovium-derived MSCs.24,25 Controlled release of osteogenic small molecules has also been achieved from hydrogels by covalent conjugation with degradable linkages.26–28 For example, Nuttelman et al. developed dexamethasone-releasing PEG-based hydrogels incorporating a heterobifunctional methacrylate-PEG-lactide-dexamethasone macromer.26 Photoencapsulated hMSCs exhibited increased core binding factor alpha 1 (Cbfa1) gene expression indicative of osteogenic differentiation. One limitation of current microparticle- and hydrogel-based delivery systems is that drug release rate is chemically controlled by the susceptibility of the ester bonds to hydrolysis and difficult to control in a predictable manner, because the hydrolysis of ester bonds is accelerated by water ingress and the formation of acidic degradation products.29
Recently, several groups have investigated strategies for immobilizing bioactive molecules to hydrogels through proteolytically-sensitive linkages in order to recapitulate the sequestration of native growth factors within the extracellular matrix and subsequent release during cell-mediated remodeling.30–33 Yang et al. reported the conjugation of dexamethasone to a cysteine-containing MMP-sensitive peptide that was co-polymerized into PEG-based thiol-ene networks with encapsulated MSCs.34 A dexamethasone derivative retaining part of the conjugated peptide was released in a cell-mediated manner and stimulated significant increases alkaline phosphatase activity and calcium deposition of encapsulated hMSCs. One limitation of this approach was that the dexamethasone derivatives exhibited modestly but significantly lower bioactivity than free dexamethasone when tested in 2D cultures at equal concentrations.
Previously, we have reported semi-interpenetrating polymer networks (semi-IPNs) composed of hydrolytically degradable PEG diacrylates (PEG-bis-AP) and native hyaluronic acid that support increased cell spreading/migration relative to fully synthetic networks through cell-mediated hyaluronidase activity.35–38 We hypothesized that dexamethasone covalently conjugated to HA through a double ester linkage (HA-DXM) and PEG-bis-AP could form semi- IPN structures that release free dexamethasone in response to cell-mediated remodeling and stimulate osteogenic differentiation of encapsulated MSCs (Figure 1). In this study, we synthesized HA-DXM and characterized DX release in response to chemical and enzymatic hydrolysis. We show that free DX is released from semi-IPNs by cell-mediated enzymatic degradation and supports osteogenic differentiation of encapsulated human MSCs comparable to semi-IPNs containing native HA cultured in the presence of conventional soluble dexamethasone supplementation.
Figure 1.
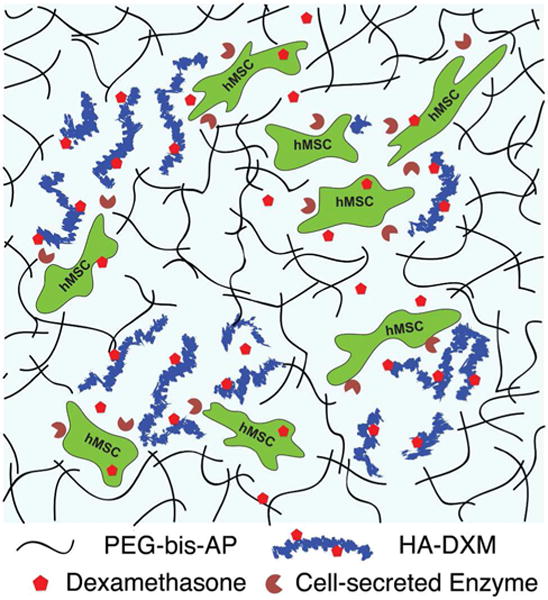
Photo-crosslinked semi-IPN hydrogels composed of PEG-bis-AP and HA-DXM conjugates with encapsulated human mesenchymal stem cells (hMSCs). Cell-mediated HA degradation enables cell spreading/migration and dexamethasone (DX) release.
2. Materials and Methods
2.1 Materials
Hyaluronic acid (HA, sodium salt, MW 1.5 MDa) and PEG diol (4 kDa) were purchased from LifeCore Biomedical (Chaska, MN) and Fluka (Buchs, Switzerland), respectively. 1,12-Carbonyldiimidazole (CDI), 2-chloropropionyl chloride, sodium acrylate, triethylamine (TEA), 4-methoxyphenol, anhydrous sodium sulfate, β-glycerophosphate, ascorbic acid 2-phosphate, dexamethasone (DX), 10 mM 4-nitrophenol solution, 1.5 M alkaline buffer solution, sodium bicarbonate were purchased from Sigma-Aldrich (St. Louis, MO). Tetrabutyl ammonium bromide (TBA-Br) and Dowex® 50WX8 200–400(H) were purchased from Alfa Aesar (Alfa Aesar, Ward Hill, MA). Dichloromethane (HPLC grade), anhydrous dimethylformamide (DMF), ethyl ether, and chloroform were obtained from Fisher Chemical (Fair Lawn, NJ). GRGDS peptide was purchased from Bachem (Torrance, CA), and acrylate-PEG-NHS (MW 3.4 kDa) was obtained from Jenkem (Beijing, China) and Laysan Bio Inc. (Arab, AL). Irgacure 2959 (2-hydroxy-1-[4-(hydroxyethoxy) phenyl]-2-methyl-1-propanone, I-2959) was donated by BASF (Florham Park, NJ). Human mesenchymal stem cells (hMSC, PT-2501) were purchased from Lonza (Walkersville, MD). Dulbecco’s Modified Eagle Medium (low-glucose) and bovine growth serum (hMSC culture grade) were purchased from Gibco (Grand Island, NY). All other reagents for cell culture were obtained from Mediatech (Herdon, VA).
2.2 Synthesis and Characterization
2.2.1 PEG-bis-AP Macromer
PEG-bis-(2-acryloyloxy propanoate) (PEG-bis-AP) was synthesized by a two-step reaction as previously described.37 Briefly, the PEG hydroxyl groups were first activated with 2-chloropropionyl chloride to form PEG-bis-(2-chloropropanoate), which was then reacted with sodium acrylate to obtain PEG-bis-AP. The structure of PEG-bis-AP and the degree of acrylation were determined from the 1H NMR (Brucker 300 MHz, DMSO-d6) spectra as previously described.
2.2.2 HA-DXM Conjugate
Dexamethasone-conjugated hyaluronic acid (HA-DXM) was prepared by two steps: Firstly, the hydroxyl groups of dexamethasone (DX) were reacted with succinic anhydride to obtain dexamethasone monosuccinate (DXM). Briefly, DX (250 mg, 0.637 mmole) was dissolved in 70 ml anhydrous acetonitrile and TEA (177.6 μl, 1.27 mmole) was added. Succinic anhydride (127.5 mg, 1.27 mmole) dissolved in 10 ml anhydrous acetonitrile was added drop wise into the dexamethasone solution and reacted for 7 days at room temperature. The reaction was monitored using thin layer chromatography (TLC, Whatman silica gel 60Å F254). After solvent removal by rotary evaporation, the residue was precipitated in ethanol/water (29:71 v/v), filtered, dried, and then stored at 4 °C until analysis. The chemical structure of DXM was characterized by 1H NMR (DMSO-d6) and ATR-FTIR. Secondly, the carboxyl end group of DXM was activated with carbonyldiimidazole (CDI) and then conjugated to the hydroxyl groups of hyaluronic acid in the presence of TEA to obtain HA-DXM. Briefly, sodium hyaluronate (250 mg) was dissolved in 50 ml distilled water, mixed with TBA-Br (5.0 g, 15.5 mmol) in the presence of activated Dowex® ion-exchange resin (3.0 g) for 3 hours, and then filtered and freeze-dried to obtain HA-tetrabutyl ammonium salt (HA-TBA).39,40 HA-TBA (50 mg, 0.0835 mmole) was then dissolved in 30 ml anhydrous DMSO. The terminal carboxyl group of DXM (50 mg, 0.10 mmole) was activated with carbonyldiimidazole (35.7 mg, 0.22 mmole) in the presence of TEA (33.5 μl, 0.24 mmole) for 2 hrs at room temperature and then added drop wise to the HA-TBA solution and stirred overnight at room temperature in the dark. The reaction was monitored using thin layer chromatography (TLC, Whatman silica gel 60Å F254). Upon completion, the reactant solution was dialyzed against DMSO for 24 hrs (MWCO 12–14 kDa) to remove unconjugated DXM. 10% v/v NaCl (2.0 M) was added into the dialysis membrane for the exchange of TBA+ by Na+ ions and then dialysis was continued for 3 days against distilled water to remove TBA+ and any residual traces of DMSO. After dialysis, the solution was filtered through a Millipore 0.45 μm membrane and HA-DXM recovered by lyophilization. The structure of HA-DXM was determined by 1H NMR using D2O:DMSO-d6 mixture (1:4 v/v) and FTIR spectrometer equipped with a Thermo-SpectraTech Foundation Series Endurance Diamond ATR (Thermo-Nicolet Magna 550). The degree of substitution (DS) of DX on HA was determined by high-performance liquid chromatography (Waters Breeze HPLC system) after alkaline hydrolysis as previously reported.41 Briefly, 5 mg/ml HA-DXM was incubated in 0.1 N NaOH for 1 hour at 60 °C. The samples were neutralized with 0.1 N HCl, centrifuged and then filtered by 0.2 μm membrane filter. 20 μl sample was injected in Waters HPLC system composed of: Waters 1525 binary HPLC pump, Waters 2707 autosampler, Waters 2998 photodiode array detector, and Waters Symmetry® C18 column (3.5 μm, 4.6 mm × 75 mm) (Milford, MA). Acetonitrile/water (35:65 v/v) was used as the mobile phase at a flow rate of 0.6 mL/min and the eluate was monitored by measuring the absorbance at 240 nm. The concentration of DX was calculated from a calibration curve constructed from the absorbance of DX standard solutions at 240 nm. The amount of DX conjugated to HA was expressed as mg of DX per 100 mg of HA-DXM.
2.3 Semi-IPN Photopolymerization
Human mesenchymal stem cells (hMSC) were cultured in 75 cm2 tissue culture flasks with low glucose DMEM medium supplemented with 10% v/v fetal bovine serum and 1% penicillin/streptomycin. Medium was changed every 2 days and cells were passaged at a 1:3 ratio for expansion. Encapsulation studies were performed using hMSCs between 5th and 6th passages after receipt from the supplier.
PEG-bis-AP macromer (30% w/v, sterile-filtered) and native HA and HA-DXM (2% w/v) were prepared in 0.1 M PBS (pH 7.4). A fibronectin-derived cell adhesion peptide (GRGDS) was conjugated to acrylate-PEG-NHS as previously described.42 10 mM acrylate–PEG–GRGDS was prepared as a stock solution in 0.1 M PBS and sterile filtered. To prepare semi-IPNs (PEG-bis-AP/HA or PEG-bis-AP/HA-DXM), 50 μl solutions containing PEG-bis-AP (6% w/v); HA or HA-DXM (0.36% w/v); 1 mM acrylate-PEG-GRGDS; 1.25 × 107 hMSCs/ml; and I-2959 (0.1% w/v) (all final concentrations) were prepared and photopolymerized between glass microscope slides separated by 1 mm Teflon spacers by exposure to low intensity UV (365 nm, 10 mW/cm2, Black-Ray B100-AP, Upland, CA) for 5 min per side.
2.4 Release of Dexamethasone from HA-DXM conjugate and semi-IPN in Vitro
DX release from soluble HA-DXM and HA-DXM entrapped within semi-IPNs was first evaluated in physiological buffer alone and with the addition of hydrolytic enzymes such as hyalulonidase (Hase) and esterase (Ease). HA-DXM and PEG-bis-AP/HA-DXM semi-IPNs were incubated in 0.1 M PBS (pH 7.4) solution, 0.1 M PBS containing Ease (5 U/ml), and 0.1 M PBS containing Ease and Hase (5 U/ml each) at 37 °C. One hundred microliter samples of each solution were removed at predetermined time intervals and stored at −20 °C until analysis.
In order to investigate cell-mediated DX release, PEG-bis-AP/HA-DXM semi-IPNs with and without encapsulated hMSCs were incubated in 12-well plates containing low glucose DMEM media with 10% FBS and 1% penicillin/streptomycin. To further characterize the specific role of Hase in DX release, acellular PEG-bis-AP/HA-DXM semi-IPNs were also incubated in the same medium with and without the addition of Hase (5 U/ml). At predetermined time points, the media containing released dexamethasone was completely removed and stored at −20 °C until analysis.
The release of free DX and DX derivatives from HA-DXM after incubation in PBS alone or PBS with Ease/Hase was evaluated by HPLC. The samples collected on day 7 were treated with methanol (4X volume) to precipitate enzymes and HA-DXM, centrifuged, and filtered through 0.2 μm syringe filter. DX, DXM, and the release samples were analyzed by HPLC as described above.
Free DX released from soluble HA-DXM or PEG-bis-AP/HA-DXM hydrogels in buffer and media was measured using an enzyme-linked immunosorbent assay (ELISA) (Dexamethasone, Neogen Corporation, Lexington, KY) according to the manufacturer’s instructions.
2.5 In Vitro Osteogenesis
2.5.1 hMSC Culture in Semi-IPN Hydrogel
After photopolymerization, semi-IPNs with encapsulated hMSCs were transferred to 12 well plates, incubated for 24 hours in growth medium without any osteogenic supplements, and then cultured for 21 days in low glucose DMEM containing 10% FBS, 1% penicillin/streptomycin, 10 mM β-glycerophosphate, and 50 μg/mL L-ascorbic acid-2-phosphate in the absence (−DX) or presence (+DX) of soluble dexamethasone supplementation (100 nM). Three sample groups were tested: Group 1 (negative control): PEG-bis-AP/HA semi-IPN (−DX), Group 2 (positive control): PEG-bis-AP/HA semi-IPN (+DX), and Group 3 (experimental): PEG-bis-AP/HA-DXM (-DX). Semi-IPNs (n = 4 per group) were collected at 7, 14, and 21 days for analysis of osteogenic differentiation.
2.5.2 Real time RT-PCR
At one day post-encapsulation before the addition of osteogenic supplements, one sample group (n = 3) of hMSCs within PEG-bis-AP/HA semi-IPNs was collected to serve as a reference for gene expression levels at later time points. Semi-IPN samples were minced in lysis buffer, homogenized, and centrifuged at 14,825 rcf for 15 min at 4 °C. The supernatant was collected and total RNA was purified by using RNeasy mini kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The quality and quantity of isolated total RNA was determined using a Take 3 microplate reader (Biotek Instruments, Winooski, VT). 1 μg of total RNA from each sample was used to synthesize cDNA according to the manufacturer’s instructions (Retroscript kit, Life Technologies). Real-time RT-PCR was performed with Quantitect SYBR green PCR Kit (Qiagen) using custom-designed sense and anti-sense primers (Supporting information, Table S1) in a Rotorgene 3000 thermal cycler (Corbett Research, Mortlake, NSW, Australia). Relative expression levels of target genes were quantified by the 2−ΔΔCt method, using β2-microglobulin (β2MG) as an internal standard43 and expressed as relative fold changes with respect to expression levels at day 1 post-encapsulation. Melt curve analysis was performed at the end of all reactions to verify formation of single products.
2.5.3 Alkaline Phosphatase (ALP) Activity
Semi-IPN samples were collected, rinsed twice with PBS, and encapsulated hMSCs were lysed in 0.1% Triton-X solution by three cycles of freezing and thawing. ALP activity was assayed using p-nitrophenyl phosphate (pNPP) as the substrate. Lysate samples (50 μl) were mixed with phosphatase substrate (100 μl, 2 mg/ml) in 0.75 M alkaline buffer solution and incubated at 37 °C for 15 min on a bench shaker. The reaction was stopped by the addition of 0.25 N NaOH (50 μl) per reaction mixture. ALP activity was quantified by absorbance measurements at 405 nm using a standard curve prepared by serial dilution of 4-nitrophenol stock solution of known concentration. ALP levels were normalized to the total protein content determined by BCA assay (Pierce, Rockford, IL).
2.5.4 Mineralized Matrix: Histology and Calcium Contents
The extent of mineralization by hMSCs encapsulated in different semi-IPNs was evaluated by histology (Alizarin red S and von Kossa staining) and calcium quantification assay. For histological analysis, the samples were fixed in 4% paraformaldehyde solution for 20 min, washed, transferred into 50% OCT compound for 1 h, and incubated in 80% OCT compound overnight. The samples were then embedded in 100% OCT compound, frozen in liquid nitrogen, and stored at −80 °C until sectioning. Frozen sections (6 μm) were stained with 2% Alizarin red S solution (pH 4.2; Acros Organics, NJ) for 10 min and washed with distilled water. For von Kossa staining, the sections were immersed in 5% silver nitrate solution under UV light for 1 hour, washed with distilled water several times, and then treated with 5% sodium thiosulfate solution for 1 min. Slides were counterstained with 0.5% Safranin-O solution. Sections were imaged using an optical microscope (Axiovert 200, Carl Zeiss, Hamburg, Germany) equipped with color CCD camera.
For calcium analysis, semi-IPN samples encapsulating hMSCs were hydrolyzed using 6 N Ultrex II pure hydrochloric acid (J.T. Baker, Philipsburg, NJ) for 8 hours in a boiling water bath. HCl was evaporated under nitrogen gas and the dried samples were then reconstituted in 1 ml of 0.01 N Ultrex II HCl. The calcium content of the samples was measured using Atomic absorption spectroscopy (Perkin-Elmer 3030 Atomic Absorption Spectrophotometer, Norwalk, CT) as previously reported.44,45
2.6 Statistical Analysis
Statistical comparisons were performed using independent group t-test for unpaired observations. One-way analysis of variance (ANOVA) with a post-hoc Bonferroni correction was used for statistical analysis with multiple comparisons. P values less than 0.05 were considered significant. All quantitative data are expressed as mean ± standard deviation. Significant differences are marked as * (p < 0.05), ** (p < 0.01), or ns (p > 0.05).
3. Results and Discussion
The goal of this study was to incorporate dexamethasone (DX) within PEG-bis-AP/HA semi-IPNs as a means to provide localized delivery of an osteoinductive small molecule to direct the in situ differentiation of MSCs transplanted intraoperatively for orthopaedic applications. However, the delivery of therapeutic small molecules from hydrogels is challenging due to initial burst release and rapid diffusion of hydrophilic molecules and limited solubility of hydrophobic drugs. HA-DXM conjugates incorporated within semi-IPNs offer a unique approach to increase DX solubility/loading, sequester DX within the network structure, and release it in response to cell-mediated enzymatic degradation.
3.1 Synthesis and Characterization of Hydrogel Precursors
HA-DXM conjugate was synthesized using a carbodiimide/active ester-mediated coupling reaction. Briefly, succinic anhydride was coupled to the hydroxyl group of dexamethasone, and then dexamethasone monosuccinate (DXM) was coupled to the hydroxyl groups of hyaluronic acid.
The structure of DXM and HA-DXM conjugate was verified by 1H NMR and ATR-FTIR. First, the structure of DXM was confirmed by 1H NMR (DMSO-d6). The peaks of 21-methylene protons (–CCH2O–) of dexamethasone were shifted from δ 4.46 and 4.69 to 4.83 and 5.02 ppm, respectively. Also, the peaks of methylene protons (–CCH2CH2C–) of succinic acid were observed at δ 2.59 and 2.62 ppm (data not shown). Second, the synthesis of HA-DXM was also confirmed by 1H NMR (D2O:DMSO-d6 mixture (1:4, v/v)) (Figure 2A). The peaks of C1, C2, and C4 protons (–CH) of DXM conjugated to HA were observed at δ 6.21, 7.32, and 6.02 ppm, respectively. The acetyl (–NHCOCH3) peak of HA at δ 1.80 ppm along with glucosidic H (10H) at δ 3.01–3.53 ppm were also clearly shown on NMR spectrum (Figure 2A). Figure 2B shows FT-IR spectra of HA and HA-DXM. HA exhibited its broad O–H stretch at 2800–3600 cm−1 and C=O stretching at 1609 cm−1, which corresponds to carboxylic groups in HA. The IR spectrum of HA-DXM showed a typical ester carbonyl peak at 1721 cm−1 along with peaks originating from HA and DXM. The amount of DX conjugated to HA was 3.01 % (w/w) as quantified by HPLC.
Figure 2.
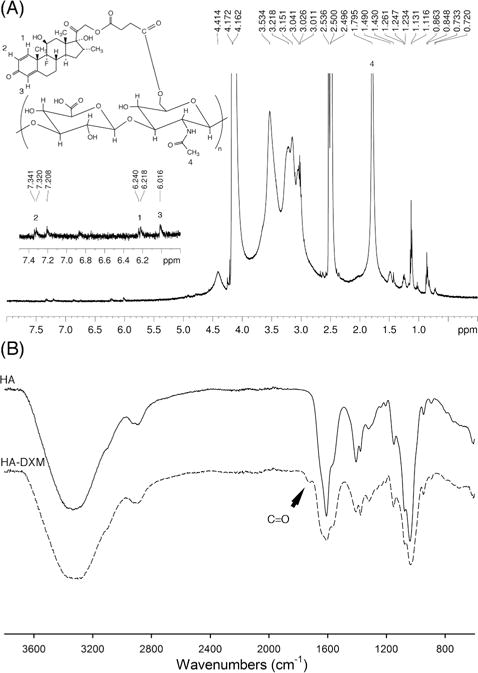
Characterization of dexamethasone-conjugated HA (HA-DXM) by 1H NMR and ATR-FTIR. (A) 1H NMR spectrum (D2O:DMSO-d6 mixture (1:4, v/v)) and B) ATR-FTIR spectrum.
3.2 Chemical and enzymatic hydrolytic release of DX from HA-DXM conjugates
To evaluate the release of DX by chemical and enzymatic hydrolysis, HA-DXM conjugate was incubated in PBS buffer or PBS buffer with Ease or Ease/Hase for 7 days. Because DX was conjugated to HA through a double ester linkage, DX could be released as free DX or DXM, depending upon which ester bond was hydrolyzed. HPLC analysis of day 7 release samples showed that DXM was the primary product released by chemical hydrolysis during incubation in buffer alone (Figure 3). In the presence of Ease/Hase, peaks corresponding to both DXM and free DX were observed, along with a peak at approximately 1 minute retention time that likely corresponds to HA-DXM degraded by Hase to low molecular weight oligomers that were not precipitated by methanol treatment. These results demonstrate that enzymatic degradation of HA-DXM can release free DX.
Figure 3.
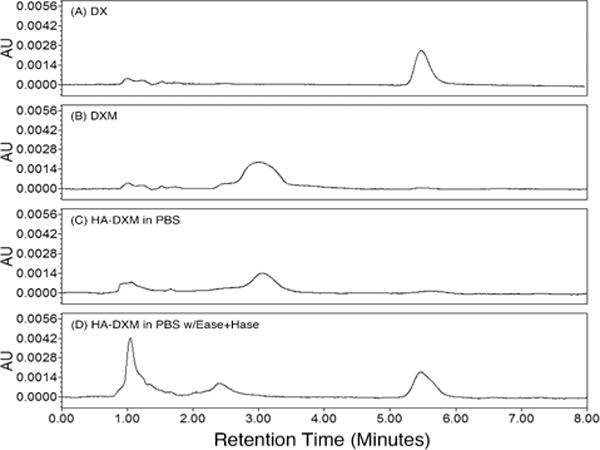
HPLC chromatogram of dexamethasone (DX, A), dexamethasone monosuccinate (DXM, B), and released DX and DXM from HA-DXM incubated in PBS alone (C) or with esterase (Ease) and hyaluronidase (Hase) (D). HA-DXM conjugates were incubated in PBS (pH 7.4) buffer or PBS (pH 7.4) buffer with Ease and Hase (5 units/ml each) for 7 days, then treated with methanol, and analyzed by HPLC along with DX and DXM.
DX release kinetics were evaluated by ELISA assay. DXM itself was tested by ELISA and exhibited less than 1% cross-reactivity, indicating that the ELISA was highly specific for free DX (data not shown). HA-DXM incubated in PBS buffer alone showed no significant increase in the concentration of released DX over 7 days and only 4.6% total release, suggesting that HA-DXM exhibits high stability with respect to chemical hydrolysis (Figure 4A). In the presence of Ease alone, approximately 75% of DX was released within three days. Inclusion of Hase further increased release with significantly higher levels of DX measured at days one through three from HA-DXM exposed to Ease/Hase relative to esterase alone. Covalent conjugation to HA has been a widely utilized strategy to increase solubility and alter biodistribution of hydrophobic drugs.46–49 Susceptibility to hydrolytic degradation is dependent upon the types of chemical bonds and their position relative to the polymer backbone. For example, Ito et al. synthesized dexamethasone-conjugated HA using an adipic dihydrazide-succinic spacer with ester linkage. Although high initial DX loading was obtained (~13.1%), the vast majority was lost due to chemical hydrolysis during dialysis with the remaining 0.1–0.2% released over 5–8 days.50 In contrast, Wang et al. synthesized heparin-paclitaxel conjugates and found that a direct ester bond between carrier and drug was resistant to both chemical and enzymatic hydrolysis, while release could be facilitated by addition of various amino acid spacers.51,52 In our study, DX was conjugated to HA through double ester bond using succinic anhydride spacer. This relatively short spacer length may account for resistance to chemical hydrolysis, while sufficiently overcoming steric hindrance to allow enzymatic cleavage.
Figure 4. In vitro.
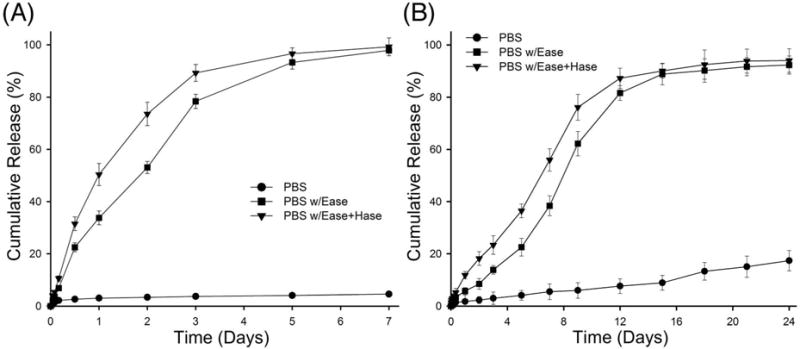
chemical and enzymatic hydrolytic release of dexamethasone from HA-DXM conjugates (A) and PEG-bis-AP/HA-DXM semi-IPNs (B). HA-DXM conjugates and PEG-bis-AP/HA-DXM semi-IPNs were incubated in PBS (pH 7.4) buffer, PBS (pH 7.4) buffer with esterase (Ease), and PBS (pH 7.4) buffer with Ease and hyaluronidase (Hase) (5 units/ml each). The concentration of released dexamethasone was measured by ELISA.
The release of DX from PEG-bis-AP/HA-DXM semi-IPNs was also evaluated. The % cumulative DX release was much higher in the presence of enzymes than physiological buffer alone (Figure 4B), but substantially slower compared to release from soluble HA-DXM. For example, DX release from soluble HA-DXM in the presence of Ease/Hase was greater than 80% within 3 days, while only approximately 25% of DX was released from PEG-bis-AP/HA-DXM semi-IPNs at 3 days and 12–16 days were required for complete release. Previously, we have shown that PEG-diacrylate/HA semi-IPNs exhibit polymerization-induced phase separation.38 This may be responsible for the slower release of DX from HA-DXM within semi-IPNs than in solution, as the concentration of HA-DXM resulting from phase separation may reduce its accessibility to degradative enzymes. DX release from semi-IPNs incubated in buffer alone remained low, with less than 20% release occurring after 24 days. Slow release of DX from the PEG-bis-AP/HA-DXM semi-IPNs in PBS alone and increased release in the presence of enzymes demonstrates that HA-DXM incorporated within semi-IPNs is relatively resistant to chemical hydrolysis and that DX release in the presence of cells may be controlled by cell-secreted enzymes.
In order to investigate the role of cell-mediated enzymatic degradation, DX release was measured from PEG-bis-AP/HA-DXM semi-IPNs with and without encapsulated hMSCs (Figure 5A). The concentration of released DX from semi-IPNs containing hMSCs was significantly higher than acellular controls from day 1 to day 12, demonstrating the contribution of cell-mediated activity to DX release. In the presence of cells, the DX concentration was maintained over 100 nM throughout the first 20 days, while in the absence of cells, it did not exceed this concentration until day 13. Although DX can stimulate osteogenic differentiation of marrow stromal cells at concentrations as low as 1 nM, 100 nM has been shown to increase mineralization.20 To investigate the potential mechanism of cell-mediated release, DX release was also measured from acellular semi-IPNs containing HA-DXM incubated in cell culture medium in the presence and absence of exogenous Hase (Figure 5B). DX release was substantially increased in the presence of Hase and highly consistent with the cell-mediated release results, with the maximum rate of release observed between days 4–8 that corresponds to the rapid rise in DX concentration in the presence of cells. These results suggest that the underlying mechanism of cell-mediated DX release involves initial Hase-mediated degradation of the HA backbone that then likely facilities accelerated chemical or Ease-mediated hydrolysis to release free DX. While previous studies have demonstrated sustained release of DX from polymeric particles and networks through chemical hydrolysis and cell-mediated release of bioactive DX derivatives, this is to our knowledge, the first demonstration of a scaffold system providing cell-mediated release of free DX.
Figure 5.
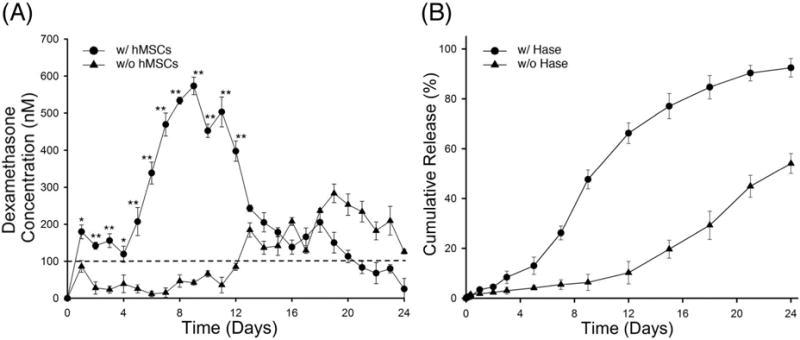
The concentration of released dexamethasone (DX) from PEG-bis-AP/HA-DXM semi-IPNs with and without encapsulated hMSCs (A) and cumulative DX release from acellular PEG-bis-AP/HA-DXM semi-IPNs in presence and absence of hyaluronidase (Hase, 5 U/ml) (B). Statistical comparisons between groups are indicated as * (p < 0.05) or ** (p < 0.01).
3.3 Osteogenic Differentiation of hMSCs Encapsulated in PEG-bis-AP/HA-DXM Semi-IPNs
In previous studies, we have shown that semi-IPNs composed of hydrolytically degradable PEG-diacrylates and native HA possess several properties that make them attractive candidates as scaffolds for orthopaedic applications. These include allowing cells to rapidly spread and attain a polarized morphology and elastic modulus of approximately 10 kPa at the polymer concentrations used in this study,35,38 both of which are scaffold properties that have been shown to increase osteogenic differentiation.53–55 To examine whether DX released from HA-DXM conjugate was bioactive and capable of supporting osteogenic differentiation, hMSCs were photo-encapsulated within semi-IPN hydrogels composed of PEG-bis-AP/HA or PEG-bis-AP/HA-DXM hydrogels and cultured in growth media with (+DX) or without (−DX) soluble DX supplementation for 21 days.
Osteogenic differentiation was first assessed by measuring changes in mRNA expression levels of bone-specific genes (Figure 6). Expression levels of all genes at all time points were significantly higher in the PEG-bis-AP/HA (+DX) and PEG-bis-AP/HA-DXM (−DX) than the PEG-bis-AP/HA (-DX) negative control. Runt-related transcription factor 2 (RUNX2) expression levels in the PEG-bis-AP/HA (+DX) and PEG-bis-AP/HA-DXM (−DX) groups were increased throughout the culture period with a peak at day 14, while Osterix/SP7 expression levels peaked at day 7 and significantly decreased at the later time points. Both genes encode transcription factors that have been identified as early markers of the osteoblast phenotype and shown to exhibit maximum expression levels during the early stages of osteogenic differentiation.56,57 Expression levels of type I collagen, alpha 1 (COL1A1), ALP, bone sialoprotein (IBSP), and osteocalcin (OCN) that serve as intermediate/late markers of the osteoblast phenotype increased with increasing time in culture in the PEG-bis-AP/HA (+DX) and PEG-bis-AP/HA-DXM (−DX) groups. Importantly, there were no significant differences throughout the culture period between expression levels in the PEG-bis-AP/HA (+DX) and PEG-bis-AP/HA-DXM (−DX) groups except for Osterix/SP7 at day 7. These results demonstrate that DX released from HA-DXM and soluble DX supplementation exhibited similar bioactivity for the induction of osteogenic gene expression. to supplemental soluble DX for induction of osteogenic gene expression.
Figure 6.
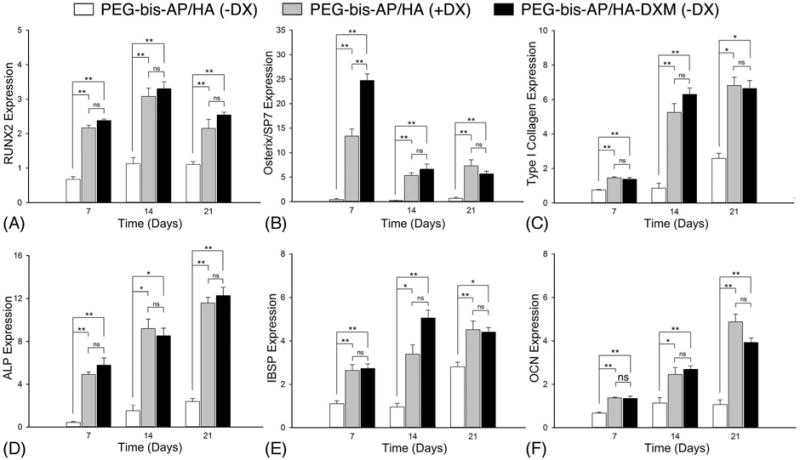
Relative mRNA expression levels of osteogenic markers: RUNX2 (A), Osterix/SP7 (B), Type I collagen (C), ALP (D), IBSP (E), and OCN (F). hMSCs were encapsulated in PEG-bis-AP semi-IPNs containing native hyaluronic acid (HA) or dexamethasone-conjugated HA (HA-DXM) and cultured in growth media containing ascorbic acid and β-glycerophosphate with (+DX) and without (−DX) soluble dexamethasone supplementation (100 nM) for 7, 14, and 21 days. Statistical comparisons between groups are indicated as * (p < 0.05), ** (p < 0.01), or ns (p > 0.05).
We also measured ALP activity by enzymatic assay (Figure 7). As observed in the mRNA results, ALP activity of hMSCs in both the PEG-bis-AP/HA (+DX) and PEG-bis-AP/HA-DXM (−DX) groups increased with time in culture and was significantly greater than the PEG-bis-AP/HA (−DX) control at all time points. There were no significant differences in ALP activity at any time point between the groups receiving DX through media supplementation or release from HA-DXM.
Figure 7.
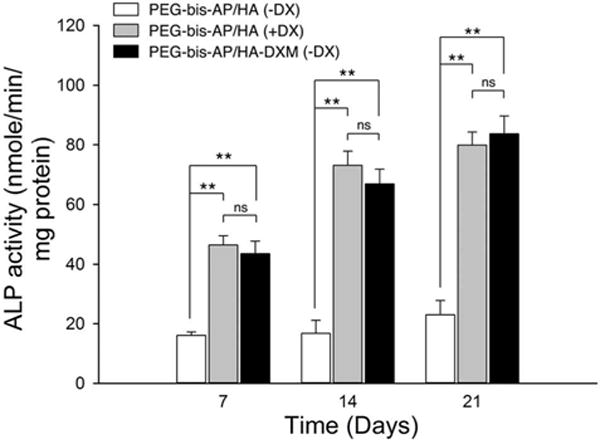
Alkaline phosphatase (ALP) activity of hMSCs encapsulated in PEG-bis-AP semi-IPNs containing native hyaluronic acid (HA) or dexamethasone-conjugated HA (HA-DXM) and cultured in growth media containing ascorbic acid and β-glycerophosphate with (+DX) and without (−DX) soluble dexamethasone supplementation (100 nM) for 7, 14, and 21 days. Statistical comparisons between groups are indicated as * (p < 0.05), ** (p < 0.01), or ns (p > 0.05).
Matrix mineralization by encapsulated hMSCs in semi-IPNs was assessed by histological analysis and calcium quantification. Alizarin red S and von Kossa staining of histological sections is shown in Figure 8. At 7 days, there was little detectable mineralization in any of the three groups. However, by days 14 and 21, calcium deposition within the semi-IPNs was clearly visible in PEG-bis-AP/HA (+DX) and PEG-bis-AP/HA-DXM (−DX) groups, but remained limited in the PEG-bis-AP/HA (−DX) negative control.
Figure 8.
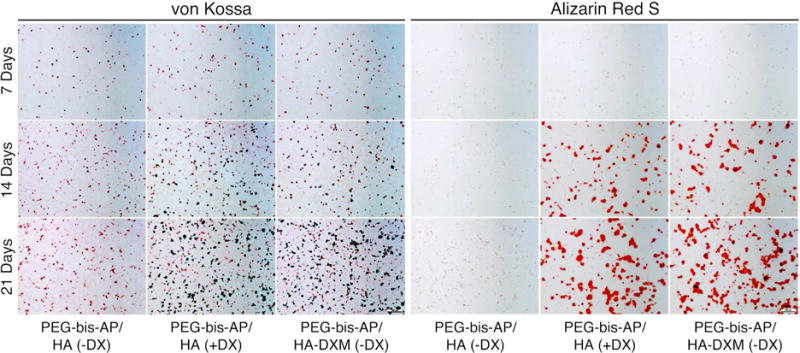
Histological analysis of mineralization in semi-IPNs by von Kossa and Alizarin Red S staining. hMSCs were encapsulated in PEG-bis-AP semi-IPNs containing native hyaluronic acid (HA) or dexamethasone-conjugated HA (HA-DXM) and cultured in growth media containing ascorbic acid and β-glycerophosphate with (+DX) and without (−DX) soluble dexamethasone supplementation (100 nM) for 7, 14, and 21 days. Original magnification: 100×, scale bars: 100 μm.
Quantitative analysis also demonstrated that calcium content increased with increasing time in culture (Figure 9). At all time points, the calcium content in PEG-bis-AP/HA-DXM (−DX) and PEG-bis-AP/HA (+DX) semi-IPNs was significantly greater than the PEG-bis-AP/HA (−DX) negative control, while no significant differences were detected between these two groups. A limited amount of mineralization was detected in the PEG-bis-AP/HA (−DX) group by day 21, which may be attributable to spontaneous hMSC differentiation in the absence of DX or non-specific mineralization. However, calcium levels in the groups receiving DX via soluble supplementation or release from HA-DXM were approximately 7-fold higher at day 21, demonstrating that mineralization was predominantly derived from DX-induced osteogenic differentiation.
Figure 9.
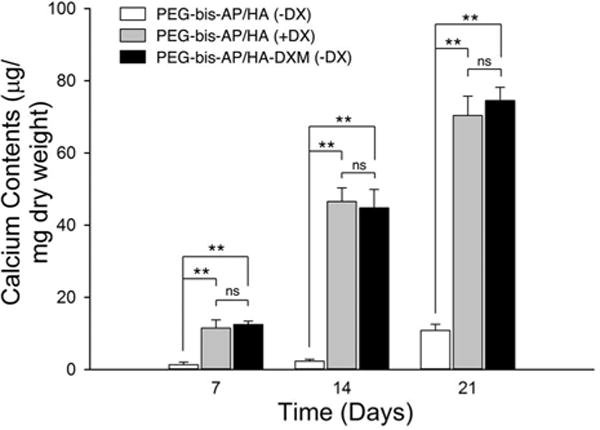
Quantification of calcium deposition by hMSCs encapsulated in PEG-bis-AP semi- IPNs containing native hyaluronic acid (HA) or dexamethasone-conjugated HA (HA-DXM) and cultured in growth media containing ascorbic acid and β-glycerophosphate with (+DX) and without (−DX) soluble dexamethasone supplementation (100 nM) for 7, 14, and 21 days. Statistical comparisons among groups are indicated as * (p < 0.05), ** (p < 0.01), or ns (p > 0.05).
Despite the positive outcomes of these studies, there are some limitations to use of PEG-bis-AP/HA-DXM as a scaffold for bone tissue engineering. One is that semi-IPNs formed from the PEG-bis-AP macromer undergo chemical hydrolytic degradation over approximately three weeks. As a potential improvement, we have recently shown that PEG-bis-AP can be combined with two other PEG-diacrylate derivatives possessing faster and slower degradation rates to produce semi-IPNs with more finely controlled degradation over 5 weeks.38 A second potential limitation is that cell spreading within the networks via HA degradation accelerates between about 4–12 days post-encapsulation, leading to a rapid rise in DX release. In this study, we conjugated DX to HA at a relatively low degree of substitution. Previous studies have shown that HA degradation can be prolonged via increasing the degree of chemical modification.58 By preparing HA-DXM at higher DS and mixing with native HA, we believe it should be possible to further adjust the DX release rate to obtain a more stable concentration of released drug. Ultimately, the proper dosing and delivery rate will need to be tested in an animal bone defect model.
4. Conclusions
Dexamethasone-conjugated hyaluronic acid (HA-DXM) was successfully synthesized and incorporated into PEG diacrylate hydrogels. Free DX was released from PEG-bis-AP/HA-DXM semi-IPNs via cell-mediated degradation and induced osteogenic differentiation of encapsulated hMSCs characterized by increased osteogenic gene expression, elevated alkaline phosphatase activity, and calcium deposition. By all quantitative measures, HA-DXM was equally effective (p>0.05) as conventional osteogenic culture protocols in which soluble DX is added to the culture medium in stimulating hMSC osteogenic differentiation. These results show that PEG-diacrylate/HA-DXM semi-IPNs can simultaneously serve as a scaffold for osteogenic cells and delivery system for osteoinductive molecules and therefore may be applicable in point-of-care cell transplantation strategies for orthopaedic repair. In addition, the general approach may be useful for delivery of inductive small molecules for other applications such as angiogenesis and neural regeneration.
Supplementary Material
Acknowledgments
This study was supported by the National Institute of Health (Grant 8P20GM103444-04) through the SCBioCRAFT COBRE Center of Regeneration and Formation of Tissues.
Footnotes
ASSOCIATED CONTENT
Primer sequences of osteoblast-specific genes. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bose S, Roy M, Bandyopadhyay A. Trends Biotechnol. 2012;30:546–554. doi: 10.1016/j.tibtech.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amini AR, Laurencin CT, Nukavarapu SP. Crit Rev Biomed Eng. 2012;40:363–408. doi: 10.1615/critrevbiomedeng.v40.i5.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma J, Both SK, Yang F, Cui F-Z, Pan J, Meijer GJ, Jansen JA, Van Den Beucken JJJP. Stem Cells Transl Med. 2014;3:98–107. doi: 10.5966/sctm.2013-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sensebé L, Bourin P, Tarte K. Hum Gene Ther. 2011;22:19–26. doi: 10.1089/hum.2010.197. [DOI] [PubMed] [Google Scholar]
- 5.Sipp D, Turner L. Science. 2012;338:1296–1297. doi: 10.1126/science.1229918. [DOI] [PubMed] [Google Scholar]
- 6.Coelho MB, Cabral JMS, Karp JM. Annu Rev Biomed Eng. 2012;14:325–349. doi: 10.1146/annurev-bioeng-071811-150041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon E, Ph D, Dhar S, Chun DE. Tissue Eng. 2007;13:619–627. doi: 10.1089/ten.2006.0102. [DOI] [PubMed] [Google Scholar]
- 8.Choi YS, Park S-N, Suh H. Biomaterials. 2005;26:5855–5863. doi: 10.1016/j.biomaterials.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 9.Song I-H, Caplan AI, Dennis JE. J Orthop Res. 2009;27:916–921. doi: 10.1002/jor.20838. [DOI] [PubMed] [Google Scholar]
- 10.Gothard D, Smith EL, Kanczler JM, Rashidi H, Qutachi O, Henstock J, Rotherham M, El Haj A, Shakesheff KM, Oreffo ROC. Eur Cells Mater. 2014;28:166–208. doi: 10.22203/ecm.v028a13. [DOI] [PubMed] [Google Scholar]
- 11.White AP, Vaccaro AR, Hall JA, Whang PG, Friel BC, McKee MD. Int Orthop. 2007;31:735–741. doi: 10.1007/s00264-007-0422-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKay WF, Peckham SM, Badura JM. Int Orthop. 2007;31:729–734. doi: 10.1007/s00264-007-0418-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaidya R, Sethi A, Bartol S, Jacobson M, Coe C, Craig JG. J spinal Disord Tech. 2008;21:557–562. doi: 10.1097/BSD.0b013e31815ea897. [DOI] [PubMed] [Google Scholar]
- 14.Vaidya R, Carp J, Sethi A, Bartol S, Craig J, Les CM. Eur spine J. 2007;16:1257–1265. doi: 10.1007/s00586-007-0351-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong DA, Kumar A, Jatana S, Ghiselli G, Wong K. Spine J. 2008;8:1011–1018. doi: 10.1016/j.spinee.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 16.Comer GC, Smith MW, Hurwitz EL, Mitsunaga Ka, Kessler R, Carragee EJ. Spine J. 2012;12:881–890. doi: 10.1016/j.spinee.2012.09.040. [DOI] [PubMed] [Google Scholar]
- 17.Shimer AL, Oner FC, Vaccaro AR. Injury. 2009;40:S32–S38. doi: 10.1016/S0020-1383(09)70009-9. [DOI] [PubMed] [Google Scholar]
- 18.Lo KW-H, Ashe KM, Kan HM, Laurencin CT. Regen Med. 2012;7:535–549. doi: 10.2217/rme.12.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Segar CE, Ogle ME, Botchwey E. Curr Pharm Des. 2013;19:3403–3419. doi: 10.2174/1381612811319190007. [DOI] [PubMed] [Google Scholar]
- 20.Jaiswal N, Haynesworth SE, Caplan A, Bruder SP. J Cell Biochem. 1997;64:295–312. [PubMed] [Google Scholar]
- 21.Maniatopoulos C, Sodek J, Melcher AH. Cell Tissue Res. 1988;254:317–330. doi: 10.1007/BF00225804. [DOI] [PubMed] [Google Scholar]
- 22.Dawes GJS, Fratila-Apachitei LE, Necula BS, Apachitei I, Witkamp GJ, Duszczyk J. J Mater Sci Mater Med. 2010;21:215–221. doi: 10.1007/s10856-009-3846-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dawes G, Fratila-Apachitei L, Necula B, Apachitei I, van Leeuwen J, Duszczyk J, Eijken M. J Biomater Appl. 2012;27:477–483. doi: 10.1177/0885328211412634. [DOI] [PubMed] [Google Scholar]
- 24.Shi X, Ren L, Tian M, Yu J, Huang W, Du C, Wang D-A, Wang Y. J Mater Chem. 2010;20:9140–9148. [Google Scholar]
- 25.Shi X, Wang Y, Varshney RR, Ren L, Gong Y, Wang DA. Eur J Pharm Sci. 2010;39:59–67. doi: 10.1016/j.ejps.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 26.Nuttelman CR, Tripodi MC, Anseth KS. J Biomed Mater Res Part A. 2006;76:183–195. doi: 10.1002/jbm.a.30537. [DOI] [PubMed] [Google Scholar]
- 27.Koehler KC, Alge DL, Anseth KS, Bowman CN. Biomaterials. 2013;34:4150–4158. doi: 10.1016/j.biomaterials.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benoit DSW, Nuttelman CR, Collins SD, Anseth KS. Biomaterials. 2006;27:6102–6110. doi: 10.1016/j.biomaterials.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 29.Siepmann J, Göpferich A. Adv DrugDeliv Rev. 2001;48:229–247. doi: 10.1016/s0169-409x(01)00116-8. [DOI] [PubMed] [Google Scholar]
- 30.Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmokel H, Bezuidenhout D, Djonov V, Zilla P, Hubbell JA. FASEB J. 2003;17:2260–2262. doi: 10.1096/fj.02-1041fje. [DOI] [PubMed] [Google Scholar]
- 31.Wilson AN, Guiseppi-Elie A. Int J Pharm. 2014;461:214–222. doi: 10.1016/j.ijpharm.2013.11.061. [DOI] [PubMed] [Google Scholar]
- 32.Phelps EA, Landázuri N, Thulé PM, Taylor WR, García AJ. Proc Natl Acad Sci U S A. 2010;107:3323–3328. doi: 10.1073/pnas.0905447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kyburz KA, Anseth KS. Ann Biomed Eng. 2015;43:489–500. doi: 10.1007/s10439-015-1297-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang C, Mariner PD, Nahreini JN, Anseth KS. J Control Release. 2012;162:612–618. doi: 10.1016/j.jconrel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kutty JK, Cho E, Soo Lee J, Vyavahare NR, Webb K. Biomaterials. 2007;28:4928–4238. doi: 10.1016/j.biomaterials.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 36.Kutty JK, Ph D, Webb K. J Biomater Sci. 2009;20:737–756. doi: 10.1163/156856209X426763. [DOI] [PubMed] [Google Scholar]
- 37.Cho E, Kutty JK, Datar K, Lee JS, Vyavahare NR, Webb K. J Biomed Mater Res Part A. 2009;90:1073–1082. doi: 10.1002/jbm.a.32172. [DOI] [PubMed] [Google Scholar]
- 38.Lee HJ, Sen A, Bae S, Lee JS, Webb K. Acta Biomater. 2015;14:43–52. doi: 10.1016/j.actbio.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oh EJ, Park K, Choi J-S, Joo C-K, Hahn SK. Biomaterials. 2009;30:6026–6034. doi: 10.1016/j.biomaterials.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 40.Sahoo S, Chung C, Khetan S, Burdick J. Biomacromolecules. 2008;9:1088–1092. doi: 10.1021/bm800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Everts M, Kok RJ, Asgeirsdóttir SA, Melgert BN, Moolenaar TJM, Koning GA, van Luyn MJA, Meijer DKF, Molema G. J Immunol. 2002;168:883–889. doi: 10.4049/jimmunol.168.2.883. [DOI] [PubMed] [Google Scholar]
- 42.Hern DL, Hubbell JA. J Biomed Mater Res. 1998;39:266–276. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 43.Livak KJ, Schmittgen TD. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 44.Ramp WK, Demaree DN. Am J Physiol. 1984;246:C30–C36. doi: 10.1152/ajpcell.1984.246.1.C30. [DOI] [PubMed] [Google Scholar]
- 45.Vyavahare N, Hirsch D, Lerner E, Baskin JZ, Schoen FJ, Bianco R, Kruth HS, Zand R, Levy RJ. Circulation. 1997;95:479–488. doi: 10.1161/01.cir.95.2.479. [DOI] [PubMed] [Google Scholar]
- 46.Pouyani T, Prestwich GD. Bioconjug Chem. 1994;5:339–347. doi: 10.1021/bc00028a010. [DOI] [PubMed] [Google Scholar]
- 47.Luo Y, Prestwich GD. Bioconjug Chem. 1999;10:755–763. doi: 10.1021/bc9900338. [DOI] [PubMed] [Google Scholar]
- 48.Rosato A, Banzato A, De Luca G, Renier D, Bettella F, Pagano C, Esposito G, Zanovello P, Bassi P. Urol Oncol Semin Orig Investig. 2006;24:207–215. doi: 10.1016/j.urolonc.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 49.Lee H, Lee K, Park TG. Bioconjug Chem. 2008;19:1319–1325. doi: 10.1021/bc8000485. [DOI] [PubMed] [Google Scholar]
- 50.Ito T, Fraser IP, Yeo Y, Highley CB, Bellas E, Kohane DS. Biomaterials. 2007;28:1778–1786. doi: 10.1016/j.biomaterials.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Xin D, Liu K, Xiang J. Pharm Res. 2009;26:785–793. doi: 10.1007/s11095-008-9762-5. [DOI] [PubMed] [Google Scholar]
- 52.Xin D, Wang Y, Xiang J. Pharm Res. 2010;27:380–389. doi: 10.1007/s11095-009-9997-9. [DOI] [PubMed] [Google Scholar]
- 53.Engler AJ, Sen S, Sweeney HL, Discher D. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 54.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 55.Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Rivera-Feliciano J, Mooney DJ. Nat Mater. 2010;9:518–526. doi: 10.1038/nmat2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Park BW, Kang EJ, Byun JH, Son MG, Kim HJ, Hah YS, Kim TH, Kumar MB, Ock SA, Rho GJ. Differentiation. 2012;83:249–259. doi: 10.1016/j.diff.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 57.Decaris ML, Leach JK. Ann Biomed Eng. 2011;39:1174–1185. doi: 10.1007/s10439-010-0217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Campoccia D, Doherty P, Radice M, Brun P, Abatangelo G, Williams DF. Biomaterials. 1998;19:2101–2127. doi: 10.1016/s0142-9612(98)00042-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.