

Abstract
Background
Endophenotypes are laboratory-based measures hypothesized to lie in the causal chain between genes and clinical disorder, and to serve as a more powerful way to identify genes associated with the disorder. One promise of endophenotypes is that they may assist in elucidating the neurobehavioral mechanisms by which an associated genetic polymorphism affects disorder risk in complex traits. We evaluated this promise by testing the extent to which variants discovered to be associated with schizophrenia through large-scale meta-analysis show associations with psychophysiological endophenotypes.
Method
We genome-wide genotyped and imputed 4905 individuals. Of these, 1837 were whole-genome-sequenced at 11× depth. In a community-based sample, we conducted targeted tests of variants within schizophrenia-associated loci, as well as genome-wide polygenic tests of association, with 17 psychophysiological endophenotypes including acoustic startle response and affective startle modulation, antisaccade, multiple frequencies of resting electroencephalogram, electrodermal activity and P300 event-related potential.
Results
Using single variant tests and gene-based tests we found suggestive evidence for an association between contactin 4 (CNTN4) and antisaccade and P300. We were unable to find any other variant or gene within the 108 schizophrenia loci significantly associated with any of our 17 endophenotypes. Polygenic risk scores indexing genetic vulnerability to schizophrenia were not related to any of the psychophysiological endophenotypes after correction for multiple testing.
Conclusions
The results indicate significant difficulty in using psychophysiological endophenotypes to characterize the genetically influenced neurobehavioral mechanisms by which genome-wide association study-identified risk loci affect disorder risk.
Keywords: Endophenotypes, gene-based tests, polygenic risk scores, schizophrenia, single variant association
Introduction
A major goal in mental health research is to identify endophenotypes that are neurobehavioral measures of psychiatric disorder liability. While complex in their application, endophenotypes are conceptually simple: in the causal chain between genes and clinical outcome, endophenotypes are a laboratory-based measure of a component of that chain that are causally closer to gene action than the eventual clinical outcome. Endophenotypes are usually defined as laboratory-based measures that are associated with a clinical outcome, heritable, manifest in an affected individual even when the disorder is not active, and cosegregate with the associated clinical outcome in families (Gottesman & Gould, 2003). We have also argued previously that to qualify as an endophenotype, v. a putative endophenotype, the laboratory-based measure should also show robust and reliable associations with genetic variants through genetic association studies (Iacono et al. 2016). Endophenotypes are also thought to be more genetically homogeneous than their associated clinical outcomes (Cannon & Keller, 2006), and so effects of individual genetic variants on endophenotypes are suspected to be larger and possibly easier to detect than the effect of those variants on the clinical outcome. Recent work suggests, however, that the effects of individual genetic variants on endophenotypes may not be appreciably larger than for complex and distal psychiatric or medical outcomes (Iacono et al. 2016). Using a community sample of over 4900 individuals from the Minnesota Twin Family Study (MTFS) (Iacono et al. 2014a), we have previously evaluated the genetic basis of 17 putative psychophysiological endophenotypes, including antisaccade eye movements, P300, electrodermal activity (EDA), resting electroencephalogram (EEG), and acoustic eyeblink startle, all of which were found to have moderate to high heritability and which, in past research, have demonstrated associations with various clinical outcomes, including schizophrenia. We conducted genome-wide association analyses (Malone et al. 2014a, b; Vaidyanathan et al. 2014a, b, c), analyses of array-based non-synonymous exonic variants (Vrieze et al. 2014b) and 11 whole genome sequencing (Vrieze et al. 2014c). Ultimately, we were neither able to convincingly corroborate any prior finding for any gene or variant previously associated with any of the 17 psychophysiological endophenotypes nor were we able to conclusively identify novel genetic associations. Our findings suggested that endophenotypes will not easily deliver novel genetic discoveries of relevance to clinical psychopathological outcomes.
The present study is a crucial direct extension of our prior work with these 17 endophenotypes. Here, the question is not whether psychophysiological endophenotypes can aid in initial discovery of disorderrelevant genes, but whether such endophenotypes can inform us about the role and mechanisms of action of genes already known to be associated with a major neurodevelopmental mental disorder. To answer this question, we took the results from a recent schizophrenia genetic association meta-analysis by the Psychiatric Genomics Consortium (PGC), which discovered 128 independent genetic variants in 108 genomic loci associated with schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014), and tested them for association with the same 17 endophenotypes described above. Many of these endophenotypes have demonstrated substantial construct validity as endophenotypes for schizophrenia (Iacono et al. 2016); others less so, but nevertheless have been associated with schizophrenia, found to be heritable in twin studies, and show deviation in schizophrenia patient first-degree relatives (Fukushima et al. 1988; Braff et al. 1992; Iacono et al. 1999; Hong et al. 2012; Kam et al. 2013; Chen et al. 2014; Narayanan et al. 2014; Qiu et al. 2014). The only two endophenotypes that have not been linked to schizophrenia are our measures of affectively modulated startle (Iacono et al. 2016). All 17 of our endophenotypes are measures of basic neurological and physiological function (e.g. resting EEG, EDA, startle) that, broadly speaking, may be expected to be disrupted in the presence of high genetic liability for schizophrenia.
The MTFS is a powerful sample in which to test endophenotype utility, especially for schizophrenia and other relatively rare disorders. Endophenotypes are hypothesized to be present in unaffected family members of probands, and before the advent of cheap genotyping and known risk loci, familial relatedness was an excellent proxy for genetic risk for disorder. With the help of the PGC results we can rank-order individuals in a community sample by their predicted genetic risk according to measured genetic variants. Thus, even though schizophrenia rates in the present sample are nearly zero, we still have an estimate of each individual’s genetic liability to schizophrenia and, therefore, to endophenotypes truly related to schizophrenia. In the present article we use this sample combined with array-based genotypes, whole genome sequencing, and a variety of statistical approaches to test for relationships between the 108 PGC schizophrenia-associated loci and 17 putative endophenotypes to address these questions:
Are any of the 128 PGC schizophrenia-associated single nucleotide polymorphisms (SNPs) also associated with any of our 17 endophenotypes, especially those endophenotypes that show robust phenotypic associations with schizophrenia?
Is an aggregate index of schizophrenia genetic liability (obtained by summing risk alleles, weighted by strength of association from the PGC findings) associated with any of these endophenotypes?
Are there rare variants within the 108 PGC-identified loci that have reliable associations with any of these endophenotypes?
Method
Participants
All participants were taken from the MTFS, a community-representative study of twins and their parents. Participants completed psychiatric assessments, self-report questionnaires and laboratory-based measures (Braff et al. 1992; Iacono et al. 1999, 2014a, 2016). The total number of individuals available with genotypes and at least one psychophysiological measure was 4905. All individuals were genotyped on the Illumina 660W-Quad, with genotyping protocols and quality control described extensively previously (Miller et al. 2012; Vrieze et al. 2014a). After quality control, the Illumina 660W-Quad contained a total of 527 829 variants.
Of those individuals with array genotypes, 1431 were selected for 11× whole genome sequencing. Among the 1431 sequenced participants, 406 were members of a monozygotic twin pair and, for each monozygotic twin pair, we sequenced only one member of the pair and copied the sequence to the other twin resulting in a total of 1837 individuals for whom whole-genome sequence variant calls were available. Monozygotic twin genomes were copied under the assumption that their genomes are identical or, at the very least, so similar that any genetic differences between the two twins would be indistinguishable from error at 11× depth sequencing. Table 1 contains descriptive information on the sample. Online Supplementary Table S1 shows the 17 × 17 endophenotype correlation matrix. All of the endophenotypes shows moderate to strong twin-based heritability except the two startle difference scores.
Table 1.
Descriptive statistics for the 17 endophenotypesa
| Endophenotype | n (Array) | n (Whole-genome) | % Female | Mean age, years | Twin-based heritability (95% CI) | GCTA SNP heritability (S.E.) |
|---|---|---|---|---|---|---|
| Antisaccade | 4469 | 1285 | 44.2 | 23.6 | 0.51 (0.42–0.56) | 0.43 (0.17) |
| P300 | 4166 | 1244 | 43.4 | 27.0 | 0.50 (0.32–0.66) | 0.19 (0.19) |
| P300 genetic factor | 3088 | 859 | 43.2 | 30.3 | 0.54 (0.36–0.71) | 0.27 (0.20) |
| Alpha at Cz power | 3948 | 1133 | 44.3 | 23.0 | 0.80 (0.65–0.86) | 0.31 (0.19) |
| Alpha at O1O2 power | 3966 | 1147 | 44.1 | 23.0 | 0.77 (0.62–0.82) | 0.58 (0.19) |
| Alpha at O1O2 frequency | 3966 | 1147 | 44.1 | 23.0 | 0.84 (0.69–0.86) | 0.58 (0.19) |
| Beta at Cz zpower | 3948 | 1133 | 44.3 | 23.0 | 0.85 (0.76–0.87) | 0.22 (0.19) |
| Theta at Cz power | 3948 | 1133 | 44.3 | 23.0 | 0.73 (0.57–0.76) | 0.05 (0.19) |
| Delta at Cz power | 3948 | 1133 | 44.3 | 23.0 | 0.56 (0.42–0.61) | 0.12 (0.19) |
| Total at Cz power | 3948 | 1133 | 44.3 | 23.0 | 0.78 (0.64–0.81) | 0.09 (0.19) |
| Skin conductance level | 3791 | 1281 | 43.0 | 29.9 | 0.66 (0.46–0.71) | 0.34 (0.17) |
| Skin conductance amplitude | 4102 | 1200 | 43.6 | 23.1 | 0.47 (0.27–0.58) | 0.32 (0.18) |
| Skin conductance frequency | 4299 | 1235 | 43.9 | 23.5 | 0.53 (0.43–0.57) | 0.25 (0.18) |
| Electrodermal activity factor | 4424 | 1285 | 43.8 | 23.8 | 0.58 (0.47–0.62) | 0.35 (0.18) |
| Overall startle | 3323 | 939 | 50.3 | 19.7 | 0.37 (0.16–0.57) | 0.49 (0.22) |
| Aversive difference | 3321 | 939 | 50.3 | 19.7 | 0 (0.00–0.16) | 0 (0.21) |
| Pleasant difference | 3322 | 939 | 50.3 | 19.7 | 0.01 (0.00–0.18) | 0.01 (0.21) |
CI, Confidence interval; GCTA, genetic complex trait analysis; SNP, single nucleotide polymorphism; S.E., standard error.
The heritability estimates were from a series of previous papers (2, 3, 4 and 5). The relatedness threshold used in that prior work to estimate the SNP heritability was 0.025.
Measures
We evaluated 17 psychophysiological measures, all of which, except affectively modulated startle, have received support as endophenotypes for schizophrenia. All measures have been described in detail previously (Iacono et al. 2014a; Vrieze et al. 2014b). All measures were corrected for sex, age, parent/child and technical artifacts. Measures were inverse-normalized prior to genetic association to mitigate outlier effects.
Antisaccade
Participants viewed a spot of light in the center of a screen. A light then appeared in the left or right visual field and participants were instructed to look away from the stimulus. Antisaccade here is measured as the proportion of trials in which the participant initially looked toward the spot of light, rather than away from it. Antisaccade eye movement has long been a strong candidate endophenotype for schizophrenia, replicated in dozens of studies over recent decades (Radant et al. 2010; Leonard et al. 2013). Antisaccade eye tracking error is highly heritable (Vaidyanathan et al. 2014b), present in first-degree relatives of individuals with schizophrenia, and co-segregates within families (Calkins et al. 2004; Radant et al. 2010).
P300 event-related potential (ERP)
Participants completed a rotated heads visual oddball task (Begleiter et al. 1984). Amplitude of the P300 wave was obtained at a midline parietal electrode (Pz) from the average ERP waveform derived from all target trials. P3 genetic factor score was generated from a factor analysis of P300 amplitude from multiple scalp electrode sites in four-member families (Malone et al. 2014b). P300 has been studied extensively in schizophrenia with visual and auditory tasks in case–control studies producing over 100 journal articles in the last 10 years (Bramon et al. 2004). Individuals with schizophrenia tend to have reduced P300 amplitude during the decision-making process as compared with controls (Jeon & Polich, 2003).
Resting EEG
Participants were asked to relax with eyes closed for 5 min while listening to soft white noise. EEG was recorded from the vertex of the head (Cz), referenced to the two ears, and from two bipolar derivations: O1-P7 and O2-P8. Power in the alpha, beta, theta and delta frequency bands were obtained from a fast Fourier transform of EEG recorded at the Cz electrode. Alpha power and peak frequency were also obtained for the two bipolar leads and averaged together. Schizophrenia is robustly associated with increased low-frequency power in particular (Kam et al. 2013; Narayanan et al. 2014), a finding that is also observed in their relatives (Narayanan et al. 2014).
EDA
EDA was measured from the fingertips as participants viewed scenes from a closed-captioned movie while loud tones were intermittently delivered. Four measures of EDA were derived. Skin conductance level was a measure of the participants’ tonic resting level measured at the end of the session. Skin conductance response frequency is a count of the number of tones to which the participant responded. Skin conductance response amplitude is the mean magnitude for trials that participants had an observable response. EDA factor score is a global measure using a factor score derived from a model fit to the three skin conductance measures. Electrodermal hyporesponsiveness is characteristic of schizophrenia (Bernstein et al. 1982), and deviations in skin conductance activity are present in the first-degree relatives of schizophrenia patients (Iacono et al. 1999).
Acoustic startle
Participants viewed a series of standard images from the International Affective Picture System (Lang et al. 2008). Images are categorized as affectively pleasant or aversive or as affectively neutral. Eye blink reactions to loud noise probes during viewing of the images were recorded from the orbicularis oculi muscle, and three measures of reactivity derived. Overall startle is the overall magnitude of the integrated electromyographic response averaged over all the trials; overall startle is moderately heritable in this MTFS sample (Vaidyanathan et al. 2014a) and the acoustic startle response has been reported as deviant in schizophrenia patients (Braff et al. 1992). Aversive difference startle is the z-score difference in mean startle magnitude between aversive and neutral images whereas pleasant difference startle is the difference between the pleasant and neutral images.
Selected PGC variants
All variants were selected based on their inclusion in the PGC’s 2014 meta-analysis of schizophrenia. The variant list was downloaded in July 2014 (https://www.med.unc.edu/pgc/downloads).
In their meta-analysis, the PGC conducted imputation using 1000 Genomes phase 1, the standard reference panel available at that time. In the present study, we used 1000 Genomes phase 3 as our imputation reference panel, as phase 3 is an improvement over phase 1. Imputation was conducted using Minimac3 (Das et al. 2016) on an imputation webserver (https://imputationserver.sph.umich.edu). In total, 45 994 018 variants were imputed, including 124 of the 128 PGC genome-wide significant variants. Imputation quality is shown in online Supplementary Table S2. Of these 124 variants, 90% had a Minimac R2 (RSQ) higher than 0.9. The four remaining variants failed because they did not exist in 1000 Genomes phase 3. According to the 1000 Genomes Project Consortium, a total of 2.3 million variants were present in phase 1 but absent from phase 3 (http://www.1000genomes.org/faq/why-are-phase-1-sites-missing-phase-3-dataset/). Differences between phase 1 and 3 are due to platform differences, a few samples from phase 1 not included in phase 3, and improved variant calling pipelines.
Of the 128 variants, 124 were successfully imputed. However, one of the 124 variants (an indel on chromosome 6) was monomorphic in the MTFS. To replace the monomorphic and remaining four variants, we used surrogate variants from 1000 Genomes phase 3 that were in high linkage disequilibrium with the missing variants but were still significantly associated with schizophrenia in the PGC results. We used rs281768 (association with schizophrenia in PGC: p = 3.47 × 10−14) as a surrogate for an indel on chromosome 2, position 200 825 237 (p = 1.78 × 10−14); rs3798869 (p = 1.15×10−9) for an indel on chromosome 6, position 84 280 274 (p = 8.57 × −10); and an indel on chromosome 22, position 39 987 024 (p = 8.91 × 10−11) for an indel on chromosome 22, position 39 987 017 (p = 2.20 × 10−11). Since two of the unimputed variants (indel at chromosome 10, position 104 957 618 and rs7907645; p = 1.04 × 10−13 and p = 5.82 × 10−11, respectively) were within the same locus and we do not have access to conditional genetic association results from the PGC, we replaced those two variants with a single variant (rs12414028; PGC p = 1.96 × 10−12). In sum, we analysed 123 variants directly and four surrogate variants out of the 128 total target PGC schizophrenia-associated variants.
Whole genome sequencing
Sequencing details are included in the online Supplementary material. We used the integrated GotCloud program for alignment and variant calling (Jun et al. 2015). After alignment, read clipping and duplicate removal, average sequencing depth genome-wide was 11×. Quality-control information is shown in online Supplementary Table S3. Variant calling was done with GotCloud defaults, using Beagle for linkage disequilibrium refinement of called genotypes (online Supplementary Table S4). To estimate power of 11× sequencing for rare variant discovery we compared the sequence variant calls to Illumina HumanExome array variant calls. The sequenced calls identified 11484 of the 15791 singletons on an Illumina HumanExome array genotyped on the same sample (73%), 10 308 of the 10 990 doubletons (94%) and 6488 of 6573 tripletons (99%). Of 151 860 monomorphic sites on the exome array, sequencing called 2951 of these sites polymorphic (about 2%). We next estimated genotype concordance between the sequence variant calls and the Illumina HumanExome and 660W-Quad genotypes. Autosomal genotypes showed 99.91% concordance (online Supplementary Fig. S1). In sum, the sequence data provided accurate genotype calls, even for rare variants including singletons and doubletons.
Single variant association tests
For each of the 127 successfully imputed schizophrenia-associated variants we tested association with each of the 17 endophenotypes. We also conducted single variant tests of all whole-genome-sequenced variants within the 108 PGC-identified loci. In both cases, we used a linear mixed model, EMMAX, that accounts for both family structure and population stratification using an empirical kinship matrix (Kang et al. 2010).
Gene-based aggregate tests
We conducted gene-based burden tests of rare nonsynonymous variants identified in the whole genome sequences. Non-synonymous variants were annotated using EPACTS (Kang et al. 2010) against GENCODE v11. Two types of gene-based tests were conducted, a variable threshold burden test (VT), and the sequence kernel association test (SKAT; Price et al. 2010; Wu et al. 2011). In the VT approach, each participant is assigned a burden score for each gene. The burden score is simply the sum of the number of non-synonymous minor alleles under a certain minor allele frequency (MAF). The MAF is allowed to vary until the sum score shows the strongest association with the trait, and the resulting p value is corrected for the number of MAF thresholds considered. The SKAT approach is more complex, but the basic intuition is that it conducts a test of the variance of variant effects within a gene, and whether that variance is larger than expected by chance. That is, do the variant effects differ from zero more than expected by chance? In SKAT we tested both MAF = 0.01 and MAF = 0.05. The purpose of using both VT and SKAT is that they each achieve greater statistical power in complementary scenarios. VT is most powerful when all minor alleles within a gene have the same direction of effect and has low power when minor alleles have opposite directions of effect. SKAT has the same power regardless of whether the minor alleles within a gene have opposite or the same directions of effect.
Polygenic risk scores
Finally, we computed polygenic risk scores and tested for association between the score and each endophenotype. A polygenic risk score is a weighted sum of risk alleles across many variants, with the weight of each variant equal to the effect size for that variant in the PGC schizophrenia results. More formally, for individual i, their polygenic risk score for variants j through n is:
The odds ratio and effect allele coding were taken from the PGC results described earlier. First, we considered a polygenic risk score from the imputed PGC genome-wide-significant variants, corresponding to a p value threshold of 5 × 10−8. At this threshold, we included all the variants (and their surrogates) identified by the PGC. We then computed polygenic risk scores with increasing numbers of variants by relaxing the inclusion p value threshold at each order of magnitude below 5 × 10−8. For these scores, we only considered variants where the reference and alternate alleles matched between our imputed genotypes and the PGC-reported genotypes. Of the variants, 93.12% matched, and the majority of variants that did not match were indels or multi-allelic sites. We chose to exclude indels due to their allelic complexity and the multiple ways that indel alleles have been coded in sequence VCF files (Tan et al. 2015). It would be difficult if not impossible to ensure that we accurately coded effect alleles for indels and multi-allelic sites for all the variants. We also removed low-frequency SNPs (MAF < 10%) and low-quality imputed variants (Minimac RSQ<0.9). For each p value threshold, we then rank-ordered all candidate SNPs by reported p value for association with schizophrenia in the PGC. We considered the most significant SNP and removed all variants in linkage disequilibrium r2 > 0.1 within 1 megabase of this variant from our rank-ordered list. We then moved on to the next most significant variant and again removed variants in linkage disequilibrium, repeating this process until no more variants remained in the rank-ordered list. This approach is similar to that used in the PGC’s own analysis (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). To derive statistically valid p values for the correlations between the polygenic risk scores and each endophenotype in the context of our twin design, we estimated in OpenMx (Boker et al. 2011) correlations and their 95% confidence intervals (Neale & Miller, 1997) using a standard twin family model (Martin & Eaves, 1977) with a shared environmental component estimated in the twins and no shared environment in the parent generation.
Results
Single variant associations of 127 PGC variants
Accounting for multiple testing only within a single endophenotype (0.05/127 = 3.9 × 10−4), we found two SNPs associated with an endophenotype. SNP rs17194490 (reference = G, alternate = T) was associated both with antisaccade and P300. The alternate allele decreased antisaccade errors (β = −0.12, p = 2.3 × 10−4, alternate allele frequency = 0.163) and increased amplitude on the P300 genetic factor (β = 0.12, p = 2.7 × 10−4). The alternate allele in SNP rs7267348 (reference allele = T, alternate = C) decreased beta EEG power (β = −0.11, p = 3.8 × 10−4, alternate allele frequency = 0.25). Neither variant survived multiple testing for more than a single endophenotype. Summary statistics for all 127 variant associations with all 17 endophenotypes are shown in online Supplementary Table S5.
Whole genome sequence fine-mapping of the 108 PGC loci
In the whole genome sequence variant call set we discovered 203 905 variants within the 108 loci identified by and defined by the PGC (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). We again employed a liberal multiple-testing correction (0.05/203 905 = 2.5 × 10−7) that corrected only for the number of SNPs within these loci and ignored multiple phenotypes. Even at this liberal threshold, we found no significant association between any variant in these loci and any of the 17 endophenotypes.
Gene-based associations
We identified 403 genes that are either within or overlap the 108 loci identified by the PGC. Once more at a liberal threshold of 1.2 × 10−4, correcting only for the number of genes and ignoring multiple endophenotypes, the VT identified no significant association between any gene and any of the endophenotypes. SKAT with a MAF threshold of 0.01 also failed to identify associations, although changing the MAF threshold to 0.05, we report one gene (strawberry notch homolog 1; SBNO1) on chromosome 12 that was significantly associated with pleasant difference startle (p = 4.19 × 10−5), although this did not survive correction for multiple testing of 403 genes.
Polygenic risk score
Fig. 1 illustrates results from the polygenic risk score analysis. The figure shows, for each p value threshold the number of variants included in the risk score, the p value threshold, and the correlation estimate between the endophenotype and the polygenic risk score. The only polygenic risk score association approaching significance was that between the score based on the top 127 variants and overall startle (p ~ 0.005), which does not survive a correction for multiple testing of 17 endophenotypes (0.05/17 = 0.003), let alone for multiple testing of 17 endophenotypes and nine p value thresholds.
Fig. 1.
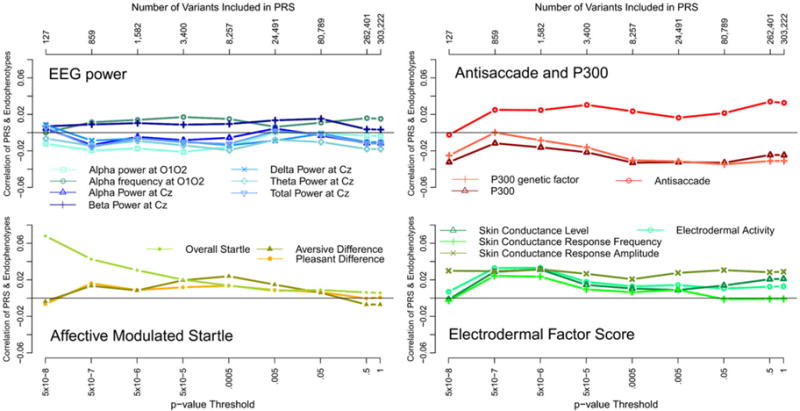
Correlations between schizophrenia polygenic risk scores (PRS) and each of the endophenotypes. To avoid clutter and improve readability, the figure is organized into four panels, where each panel shares the x and y axes. No correlation between polygenic risk and any endophenotype was significant after accounting for multiple testing. EEG, Electroencephalogram.
Discussion
One argument for the utility of endophenotypes has been that they may aid in gene discovery for clinical outcomes. Previous genome-wide association work by our group suggests that this is not true for common psychophysiological endophenotypes in large community samples (Iacono et al. 2014b; Vrieze et al. 2014c). Another compelling argument for the utility of endophenotypes is that, for a given gene known to be associated with a clinical disorder, an endophenotype may clarify, pinpoint, or suggest causal neurobehavioral mechanisms by which the gene exerts influence on the clinical outcome. Even if endophenotypes can elucidate neurobehavioral etiology in this way, it will require much larger samples than are routinely used for this purpose. The present study of about 4500 individuals, the largest of its kind to date, produced null results across a wide variety of analyses and genotyping technologies. None of the known schizophrenia variants showed a robust association with any of the endophenotypes. One suggestive single-variant result was for an intronic variant in the gene contactin 4 (CNTN4) associated with both antisaccade and P300, although this association did not survive multiple testing correction for all phenotypes. CNTN4 is a member of the contactin family of immunoglobulins that functions in neuronal network formation and plasticity. Deletion or mutation in CNTN4 has been found to be associated with autism spectrum disorders (Fernandez et al. 2004), and, of course, schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). The other variant was in an intron of the gene for prostaglandin-I synthase (PTGIS) which encodes for proteins in the cytochrome P450 enzyme superfamily.
The present study is also the largest of its kind to date to use whole genome sequencing to fine-map the 108 loci identified by the PGC genome-wide association study (GWAS) meta-analysis of schizophrenia. Our one significant gene-based association was with SBNO1, a gene that is highly conserved across species and has a function in regulating development (Egan et al. 1998), but no obvious biological link to neurobehavioral abnormalities relevant to affective modulated startle. In addition, this particular startle measure is perhaps the least strong endophenotype studied here (heritability = 0.01), casting further doubt on the utility of this finding. The lack of additional findings suggests that there are no rare or low-frequency variants of large effect on any of these 17 endophenotypes within the 108 PGC loci. This finding is perhaps not surprising given difficulties in finding rare variant associations with schizophrenia from other, larger exome sequencing studies (Purcell et al. 2014; Singh et al. 2016).
The traditional definition of an endophenotype requires that cases show differences from controls but also that the genetic relatives of cases show differences from controls, in essence a requirement that the endophenotype have a genetic basis. One strength of the present study is that we were able to use a large number of known schizophrenia risk variants to rank individuals according to their genetic risk for schizophrenia in multiple ways. (1) For each of the PGC variants, we tested whether the schizophrenia risk allele was also associated with any of our endophenotypes. (2) We used a polygenic risk score to obtain a weighted sum of the number of risk alleles that each participant harbored. Ultimately, this procedure rank-ordered each individual in our sample according to their genetic risk for schizophrenia, and we correlated this rank ordering with each of the endophenotypes. While these results do not rule out the possibility that these endophenotypes share a genetic basis with schizophrenia, they do indicate that much larger sample sizes than those employed here will be required to detect such an association.
The utility of our 17 endophenotypes for characterizing genetic effects on neurobehavioral mechanisms in clinical samples remains to be seen, and is currently being pursued through work such as that being done by the Consortium on the Genetics of Schizophrenia (COGS). COGS is also evaluating a greater number of putative endophenotypes for study. COGS uses clinical probands to recruit families, thus increasing the variance of endophenotype expression in their sample and ensuring high levels of genetic risk in the unaffected individuals. They have reported on multiple heritable endophenotypes that differentiate cases and controls but it is currently unclear the extent to which they have discovered individual genetic loci robustly associated with any of their schizophrenia-related endophenotypes after one employs standard multiple testing and ancestry corrections (Greenwood et al. 2011, 2016). Case–control samples like COGS and the PGC, because they employ an extreme sampling design, are more powerful than community samples for investigations of genetic association with disorders like schizophrenia. However, when the goal is to test known schizophrenia genetic loci for association with known schizophrenia-relevant endophenotypes, a case–control study of schizophrenia necessarily confounds diagnostic status and endophenotype level, resulting in the possibility of spurious gene–endophenotype correlations (Iacono et al. 2016). That is, if the gene is associated with schizophrenia, and cases are known to have higher levels of the endophenotype than controls, then the gene will also be associated with the endophenotype regardless of its causal role in schizophrenia. The same will be true for any phenotype (e.g. education level, marital status, etc.) that is systematically different in individuals with schizophrenia.
By current standards in complex trait genetics, our study was small. Our hypothesis was that the size of genetic effects for endophenotypes would overcome the relatively small sample size. Clearly, genetic effects on these endophenotypes are not large enough to be detected in this sample. Given the cost of collecting laboratory-based psychophysiological endophenotypes like those used in the present research it appears that endophenotypes are less efficient for gene discovery than simply assessing and genotyping more cases and controls. However, it remains to be seen the extent to which endophenotypes can illuminate the mechanistic pathways between genes and clinical outcomes. While antisaccade and P300 are convincing putative endophenotypes in schizophrenia and EEG and electrodermal measures have also received significant support, multiple promising putative endophenotypes for schizophrenia were not available in the present research (Allen et al. 2009). Some of these endophenotypes may be associated with genetic loci of substantially larger effect sizes than any of our 17, although there is little evidence that psychophysiological or imaging-based endophenotypes are associated with larger effects than other complex traits and diseases (Iacono et al. 2016). Despite the fact that our study could not definitively test all candidate endophenotypes in schizophrenia, we suggest that our findings indicate that mechanism studies of the present kind will not be effective in community samples of the size investigated here, because none of the variants known to be associated with clinical outcome has any appreciable association with any of the endophenotypes. Samples of larger sizes with the endophenotypes measured here may yet uncover that these endophenotypes are indeed associated with schizophrenia risk loci, but the question remains whether such findings warrant the expensive in-person laboratory-based procedures required to measure these particular endophenotypes.
Supplementary Material
Acknowledgments
This research was supported through National Institutes of Health grants AA023974, HG008983, DA037904, DA040177, DA041120, DA036216, DA005147, AA009367 and DA024417.
Footnotes
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0033291716003184
Declaration of Interest
None.
References
- Allen AJ, Griss ME, Folley BS, Hawkins KA, Pearlson GD. Endophenotypes in schizophrenia: a selective review. Schizophrenia Research. 2009;109:24–37. doi: 10.1016/j.schres.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begleiter H, Porjesz B, Bihari B, Kissin B. Event-related brain potentials in boys at risk for alcoholism. Science (New York, NY) 1984;225:1493–1496. doi: 10.1126/science.6474187. [DOI] [PubMed] [Google Scholar]
- Bernstein AS, Frith CD, Gruzelier JH, Patterson T, Straube E, Venables PH, Zahn TP. An analysis of the skin conductance orienting response in samples of American, British, and German schizophrenics. Biological Psychology. 1982;14:155–211. doi: 10.1016/0301-0511(82)90001-1. [DOI] [PubMed] [Google Scholar]
- Boker S, Neale M, Maes H, Wilde M, Spiegel M, Brick T, Spies J, Estabrook R, Kenny S, Bates T, Mehta P, Fox J. OpenMx: an open source extended structural equation modeling framework. Psychometrika. 2011;76:306–317. doi: 10.1007/s11336-010-9200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Archives of General Psychiatry. 1992;49:206–215. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- Bramon E, Rabe-Hesketh S, Sham P, Murray RM, Frangou S. Meta-analysis of the P300 and P50 waveforms in schizophrenia. Schizophrenia Research. 2004;70:315–329. doi: 10.1016/j.schres.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Calkins ME, Curtis CE, Iacono WG, Grove WM. Antisaccade performance is impaired in medically and psychiatrically healthy biological relatives of schizophrenia patients. Schizophrenia Research. 2004;71:167–178. doi: 10.1016/j.schres.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Keller MC. Endophenotypes in the genetic analyses of mental disorders. Annual Review of Clinical Psychology. 2006;2:267–290. doi: 10.1146/annurev.clinpsy.2.022305.095232. [DOI] [PubMed] [Google Scholar]
- Chen KC, Lee IH, Yang YK, Landau S, Chang WH, Chen PS, Lu RB, David AS, Bramon E. P300 waveform and dopamine transporter availability: a controlled EEG and SPECT study in medication-naive patients with schizophrenia and a meta-analysis. Psychological Medicine. 2014;44:2151–2162. doi: 10.1017/S0033291713002808. [DOI] [PubMed] [Google Scholar]
- Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, Schlessinger D, Stambolian D, Loh P-R, Iacono WG, Swaroop A, Scott LJ, Cucca F, Kronenberg F, Boehnke M, Abecasis GR, Fuchsberger C. Next-generation genotype imputation service and methods. Nature Genetics. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan SE, St-Pierre B, Leow CC. Notch receptors, partners and regulators: from conserved domains to powerful functions. Current Topics in Microbiology and Immunology. 1998;228:273–324. doi: 10.1007/978-3-642-80481-6_11. [DOI] [PubMed] [Google Scholar]
- Fernandez T, Morgan T, Davis N, Klin A, Morris A, Farhi A, Lifton RP, State MW. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. American Journal of Human Genetics. 2004;74:1286–1293. doi: 10.1086/421474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima J, Fukushima K, Chiba T, Tanaka S, Yamashita I, Kato M. Disturbances of voluntary control of saccadic eye movements in schizophrenic patients. Biological Psychiatry. 1988;23:670–677. doi: 10.1016/0006-3223(88)90050-9. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. American Journal of Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Greenwood TA, Lazzeroni LC, Calkins ME, Freedman R, Green MF, Gur RE, Gur RC, Light GA, Nuechterlein KH, Olincy A, Radant AD, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Sugar CA, Swerdlow NR, Tsuang DW, Tsuang MT, Turetsky BI, Braff DL. Genetic assessment of additional endophenotypes from the Consortium on the Genetics of Schizophrenia Family Study. Schizophrenia Research. 2016;170:30–40. doi: 10.1016/j.schres.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood TA, Lazzeroni LC, Murray SS, Cadenhead KS, Calkins ME, Dobie DJ, Green MF, Gur RE, Gur RC, Hardiman G, Kelsoe JR, Leonard S, Light GA, Nuechterlein KH, Olincy A, Radant AD, Schork NJ, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Swerdlow NR, Tsuang DW, Tsuang MT, Turetsky BI, Freedman R, Braff DL. Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the Consortium on the Genetics of Schizophrenia. American Journal of Psychiatry. 2011;168:930–946. doi: 10.1176/appi.ajp.2011.10050723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong LE, Summerfelt A, Mitchell BD, O’Donnell P, Thaker GK. A shared low-frequency oscillatory rhythm abnormality in resting and sensory gating in schizophrenia. Clinical Neurophysiology. 2012;123:285–292. doi: 10.1016/j.clinph.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nature Genetics. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono WG, Ficken JW, Beiser M. Electrodermal activation in first-episode psychotic patients and their first-degree relatives. Psychiatry Research. 1999;88:25–39. doi: 10.1016/s0165-1781(99)00071-2. [DOI] [PubMed] [Google Scholar]
- Iacono WG, Malone SM, Vaidyanathan U, Vrieze SI. Genome-wide scans of genetic variants for psychophysiological endophenotypes: a methodological overview. Psychophysiology. 2014a;51:1207–1224. doi: 10.1111/psyp.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono WG, Malone SM, Vrieze SI. Endophenotype best practices. International Journal of Psychophysiology: Official Journal of the International Organization of Psychophysiology. 2016 doi: 10.1016/j.ijpsycho.2016.07.516. Published online 27 July 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono WG, Vaidyanathan U, Vrieze SI, Malone SM. Knowns and unknowns for psychophysiological endophenotypes: integration and response to commentaries. Psychophysiology. 2014b;51:1339–1347. doi: 10.1111/psyp.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon Y-W, Polich J. Meta-analysis of P300 and schizophrenia: patients, paradigms, and practical implications. Psychophysiology. 2003;40:684–701. doi: 10.1111/1469-8986.00070. [DOI] [PubMed] [Google Scholar]
- Jun G, Wing MK, Abecasis GR, Kang HM. An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data. Genome Research. 2015;25:918–925. doi: 10.1101/gr.176552.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam JW, Bolbecker AR, O’Donnell BF, Hetrick WP, Brenner CA. Resting state EEG power and coherence abnormalities in bipolar disorder and schizophrenia. Journal of Psychiatric Research. 2013;47:1893–1901. doi: 10.1016/j.jpsychires.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Sul JH, Service SK, Zaitlen NA, Kong S, Freimer NB, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nature Genetics. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang PJ, Bradley MM, Cuthbert BN. International Affective Picture System (IAPS): Affective Ratings of Pictures and Instruction Manual Technical Report A-8. University of Florida; Gainesville, FL: 2008. [Google Scholar]
- Leonard CJ, Robinson BM, Kaiser ST, Hahn B, McClenon C, Harvey AN, Luck SJ, Gold JM. Testing sensory and cognitive explanations of the antisaccade deficit in schizophrenia. Journal of Abnormal Psychology. 2013;122:1111–1120. doi: 10.1037/a0034956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone SM, Burwell SJ, Vaidyanathan U, Miller MB, McGue M, Iacono WG. Heritability and molecular–genetic basis of resting EEG activity: a genome-wide association study. Psychophysiology. 2014a;51:1225–1245. doi: 10.1111/psyp.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone SM, Vaidyanathan U, Basu S, Miller MB, McGue M, Iacono WG. Heritability and molecular–genetic basis of the P3 event-related brain potential: a genome-wide association study. Psychophysiology. 2014b;51:1246–1258. doi: 10.1111/psyp.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin NG, Eaves LJ. The genetical analysis of covariance structure. Heredity. 1977;38:79–95. doi: 10.1038/hdy.1977.9. [DOI] [PubMed] [Google Scholar]
- Miller MB, Basu S, Cunningham J, Eskin E, Malone SM, Oetting WS, Schork N, Sul JH, Iacono WG, McGue M. The Minnesota Center for Twin and Family Research Genome-Wide Association Study. Twin Research and Human Genetics: the Official Journal of the International Society for Twin Studies. 2012;15:767–774. doi: 10.1017/thg.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan B, O’Neil K, Berwise C, Stevens MC, Calhoun VD, Clementz BA, Tamminga CA, Sweeney JA, Keshavan MS, Pearlson GD. Resting state electroencephalogram oscillatory abnormalities in schizophrenia and psychotic bipolar patients and their relatives from the bipolar and schizophrenia network on intermediate phenotypes study. Biological Psychiatry. 2014;76:456–465. doi: 10.1016/j.biopsych.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale MC, Miller MB. The use of likelihood-based confidence intervals in genetic models. Behavior Genetics. 1997;27:113–120. doi: 10.1023/a:1025681223921. [DOI] [PubMed] [Google Scholar]
- Price AL, Kryukov GV, de Bakker PIW, Purcell SM, Staples J, Wei L-J, Sunyaev SR. Pooled association tests for rare variants in exon-resequencing studies. American Journal of Human Genetics. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kähler A, Duncan L, Stahl E, Genovese G, Fernández E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PKE, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SGN, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll SA, Sklar P. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Tang Y, Chan RC, Sun X, He J. P300 aberration in first-episode schizophrenia patients: a meta-analysis. PLOS ONE. 2014;9:e97794. doi: 10.1371/journal.pone.0097794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radant AD, Dobie DJ, Calkins ME, Olincy A, Braff DL, Cadenhead KS, Freedman R, Green MF, Greenwood TA, Gur RE, Gur RC, Light GA, Meichle SP, Millard SP, Mintz J, Nuechterlein KH, Schork NJ, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Swerdlow NR, Tsuang MT, Turetsky BI, Tsuang DW. Antisaccade performance in schizophrenia patients, their first-degree biological relatives, and community comparison subjects: data from the COGS study. Psychophysiology. 2010;47:846–856. doi: 10.1111/j.1469-8986.2010.01004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, Suvisaari J, Chheda H, Blackwood D, Breen G, Pietiläinen O, Gerety SS, Ayub M, Blyth M, Cole T, Collier D, Coomber EL, Craddock N, Daly MJ, Danesh J, DiForti M, Foster A, Freimer NB, Geschwind D, Johnstone M, Joss S, Kirov G, Körkkö J, Kuismin O, Holmans P, Hultman CM, Iyegbe C, Lönnqvist J, Männikkö M, McCarroll SA, McGuffin P, McIntosh AM, McQuillin A, Moilanen JS, Moore C, Murray RM, Newbury-Ecob R, Ouwehand W, Paunio T, Prigmore E, Rees E, Roberts D, Sambrook J, Sklar P, Clair DS, Veijola J, Walters JTR, Williams H, Swedish Schizophrenia Study; INTERVAL Study; DDD Study; UK10 Consortium. Sullivan PF, Hurles ME, O’Donovan MC, Palotie A, Owen MJ, Barrett JC. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nature Neuroscience. 2016;19:571–577. doi: 10.1038/nn.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A, Abecasis GR, Kang HM. Unified representation of genetic variants. Bioinformatics. 2015;31:2202–2204. doi: 10.1093/bioinformatics/btv112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan U, Isen JD, Malone SM, Miller MB, McGue M, Iacono WG. Heritability and molecular genetic basis of electrodermal activity: a genome-wide association study. Psychophysiology. 2014a;51:1259–1271. doi: 10.1111/psyp.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan U, Malone SM, Donnelly JM, Hammer MA, Miller MB, McGue M, Iacono WG. Heritability and molecular genetic basis of antisaccade eye tracking error rate: a genome-wide association study. Psychophysiology. 2014b;51:1272–1284. doi: 10.1111/psyp.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan U, Malone SM, Miller MB, McGue M, Iacono WG. Heritability and molecular genetic basis of acoustic startle eye blink and affectively modulated startle response: a genome-wide association study. Psychophysiology. 2014c;51:1285–1299. doi: 10.1111/psyp.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze SI, Feng S, Miller MB, Hicks BM, Pankratz N, Abecasis GR, Iacono WG, McGue M. Rare nonsynonymous exonic variants in addiction and behavioral disinhibition. Biological Psychiatry. 2014a;75:783–789. doi: 10.1016/j.biopsych.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze SI, Malone SM, Pankratz N, Vaidyanathan U, Miller MB, Kang HM, McGue M, Abecasis G, Iacono WG. Genetic associations of nonsynonymous exonic variants with psychophysiological endophenotypes. Psychophysiology. 2014b;51:1300–1308. doi: 10.1111/psyp.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze SI, Malone SM, Vaidyanathan U, Kwong A, Kang HM, Zhan X, Flickinger M, Irons D, Jun G, Locke AE, Pistis G, Porcu E, Levy S, Myers RM, Oetting W, McGue M, Abecasis G, Iacono WG. In search of rare variants: preliminary results from whole genome sequencing of 1325 individuals with psychophysiological endophenotypes. Psychophysiology. 2014c;51:1309–1320. doi: 10.1111/psyp.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. American Journal of Human Genetics. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.