

Abstract
Hsp70 proteins are key to maintaining intracellular protein homeostasis. To carry out this task, they employ a large number of cochaperones and adapter proteins. Here, we review what is known about the interaction between the chaperones and partners, with a strong slant toward structural biology. Hsp70s in general, and Hsc70 (HSPA8) in particular, display an amazing array of interfaces with their protein cofactors. We also review the known interactions between Hsp70s with lipids and with active compounds that may become leads toward Hsp70 modulation for treatment of a variety of diseases.
Keywords: Hsc70, Protein-protein interactions, Protein-drug interactions, Structural biology, Crystallography, NMR spectroscopy
Hsp70 chaperones are highly conserved in all kingdoms; in animals, they are an important member of the collection of protein chaperones including Hsp60, Hsp70, Hsp90, Hsp100, and small Hsps (Bar-Lavan et al. 2016). The archetypical Hsp70 is called DnaK in bacteria, and it functions in protein trafficking and protein refolding cycles, acting together with a nucleotide exchange factor (NEF), termed GrpE, and a J-protein, called DnaJ. In the presence of ATP, this trio is capable of refolding denatured luciferase in vitro and has thus been classified as a bona fide protein refolding machinery (Schroder et al. 1993). While the prokaryotic system has a single Hsp70, NEF, and J protein, the number of genes has expanded in eukaryotes. For example, the yeast genome codes for 14 Hsp70s, 20 J-proteins (Craig and Huang 2005), and 3 NEFs (Mayer and Bukau 2005). In humans, there are 13 Hsp70s, 50 J-proteins (Hsp40), and seven NEFs (Kampinga et al. 2009). The original protein-refolding function of the Hsp70/J/NEF trio seems to be maintained throughout the kingdoms, but eukaryotes require more isoforms to enable the system to have representatives in each organelle (e.g., ER, mitochondria, cytoplasm/nucleus). Moreover, an expansion of cochaperones seems to have allowed the system to adapt to a wider range of specialized functions, such as trafficking and signaling. Finally, higher eukaryotes have also added new partners for the Hsp70s, including HOP, HIP, CHIP, and other proteins, that couple it to additional cellular functionality, such as the proteosomal and autophagosomal degradation systems and to the Hsp90 protein folding machinery. There are likely more cochaperones to be found and some may be cell-specific. For example, a 16-kDa protein in the Nm23/nucleoside diphosphate kinase family binds to HSPA8 in both human and fish hepatocytes (Leung and Hightower 1997). This stress-inducible protein may affect HSPA8 functions by maintaining HSPA8 in a monomeric state and by aiding in the release of unfolded proteins from HSPA8 through direct protein-protein interactions or by supplying ATP to HSPA8 indirectly through phosphate transfer to bound ADP. The p16 cochaperone potentially links to energy metabolism in stressed and cytoprotected cells and tissues. A key to understanding Hsp70 biology, especially in eukaryotes, is to understand how it coordinates with an expanded number of cochaperones to enable its broad functions.
HSPA8 (Hsc70; constitutive) and HSPA1 (Hsp70-1; inducible) are the two major human Hsp70s of the cytoplasm and nucleus. Broadly speaking, these proteins are involved in four general activities in the cell: (i) binding and release of native and misfolded proteins to favor protein (re)folding cycles (Young et al. 2004), (ii) transporting unfolded proteins through membranes to enable delivery of cargo to organelles (Hohfeld and Hartl 1994), (iii) recruiting proteins to the proteasome for turnover (Demand et al. 1998), and (iv) bringing proteins to the endosome/lysosome for chaperone-mediated autophagy (Majeski and Dice 2004). These diverse functions are achieved by Hsc70 interactions with different molecular partners: J proteins, NEFs, and Hsp90 for function (i); membrane transporting systems for role (ii); interaction with ubiquitin ligases and NEFs for role (iii); and interactions with phosphoserine lipids and LAMP-2A for role (iv). Even from this simplified overview of Hsp70, it is clear that cochaperones and other partners are the keys to understanding function. Another lesson is that each of the categories of chaperone function involves multiple components.
Additional critical roles for HSPA8 have been suggested based on biophysical and biochemical data and these remain to be explored in depth. For example, HSPA8 may function as a thermal sensor capable of matching the level of chaperoning capacity in cells with the needs of proteostasis over the physio-logical temperature range of mammals. Leung et al. (1996) showed with isothermal calorimetry that HSPA8 undergoes an exothermic reversible transition over the 30–37°C temperature range, a range that includes the extremities of mammals. HSPA8 is protease resistant-resistant at 20°C, sensitive to chymotrypsin at 40°C, and resistant again when returned to 20°C, indicating that the thermal transition represents a reversible conformational change. For a variety of substrates tested, the amount of HSPA8-substrate complex formation increased with increasing temperature over the same range. Therefore, a conformational change in HSPA8 linked to modulating protein chaperone activity over physiolog-ically relevant temperatures suggests roles in balancing protein synthesis and turnover to maintain cellular proteostasis as well as a potential role in acquired thermotolerance.
The Hsp70s are increasingly being seen as drug targets across a range of diseases (Patury et al. 2009). For example, HSPA1 expression is induced in cancer cells, where it inhibits apoptosis (Nylandsted et al. 2000). The levels of HSPA1 are even further elevated in response to chemotherapy, which may partially limit the effectiveness of Hsp90 inhibitor treatments (Hubbard et al. 2011). In another example, HSPA8 enhances the lifetime of aberrant tau protein in neurons (Jinwal et al. 2013), likely contributing to neurodegenerative disease. Hence, in addition to the basic scientific interest in delineating the Hsp70 functional cycles, there are translational implications in better understanding these mechanisms. How might we selectively disrupt some disease-associated functions of Hsp70 and not other, housekeeping activities? Several synthetic modulators of Hsp70 have recently emerged that have begun to answer this question.
In this review, we will focus on the structural and biophysical aspects of how Hsp70s bind to their different partners, as revealed by X-ray crystallography and multidimensional NMR spectroscopy. For a complementary recent review that focuses on the biochemical aspects, see Radons (2016). Ideally, we would like to focus on the Hsp70s of one species, and preferentially Homo sapiens. However, much of the structural biology is focused on the E. coli system of DnaK/GrpE/DnaJ because these proteins are more easily handled in vitro. Herein, we want to cast a wide net, so we will assume that the level of homology between the different Hsp70 proteins and cofactors is sufficiently high so that structural knowledge can be safely extrapolated between orthologs and paralogs. For example, human HSPA8 is 80% homologous and 55% identical to E. coli DnaK. In this review, we will use the name Hsp70 if we do not want to discriminate between homologs and will use the systematic names as proposed by Kampinga et al. (2009) if we do.
The ATPase cycle of Hsp70s
As shown in Fig. 1, the prototypical Hsp70 chaperone E. coli DnaK consists of an N-terminal nucleotide-binding domain (NBD; residues 1–383) that binds and hydrolyzes ATP, a substrate-binding domain (SBD; residues 397–506) that binds hydrophobic substrates, a LID domain (LID; residues 507–602), and an unstructured C-terminal tail (residues 603–638). Human Hsp70 proteins closely resemble this domain distribution.
Fig. 1.

Structures of the E. coli Hsp70 protein DnaK in the ADP/NRLLLTG state (bottom, 2KHO) and in the ATP/Apo state (top, 4B9Q). The nucleotide-binding domains (NBDs in green) have the same orientations. The substrate-binding domain (SBD) is in red. Lid domain (blue). DnaK contains another 30 unstructured residues at the C-terminus of the lid (not shown)
In the ATP state, both SBD and LID are docked to the NBD, allowing the open hydrophobic cleft in the SBD to bind to the exposed hydrophobic regions of misfolded/unfolded proteins with a (weak) affinity of approximately 10 μM (Kityk et al. 2012; Qi et al. 2013). In the ADP state, the LID is docked to the SBD and covers the hydrophobic cleft in the SBD, which binds to captured misfolded/unfolded proteins with an affinity of approximately 100 nM. The SBD-LID combination is dynamically tethered to the NBD through a linker (Bertelsen et al. 2009).
A combination of DnaK and ATP with cochaperones DnaJ and GrpE can refold thermally denatured luciferase in vitro (Schroder et al. 1993), establishing this trio as a protein refolding machinery. A cartoon illustrating this process is shown in Fig. 2.
Fig. 2.

The Hsp70 substrate unfolding cycle based on data for E. coli DnaK. DnaK-ATP state (labeled K.ATP) (red and blue), DnaK-ADP state (labeled K.ADP) (brown and blue), DnaJ (pink), the nucleotide exchange factor GrpE (labeled E) (cyan), and substrate (green). ATP (red dot) and ADP (brown dot). The transparent Hsp70 shapes represent additional binding. The different steps in the cycle are indicated by labeled arrows which are explained in the text
The cycle starts with DnaK in the ATP state. The chaperone forms a triple complex with a misfolded substrate protein and the cochaperone DnaJ (steps 1a, 1b, 1c). DnaJ has a J-domain which is flexibly tethered to a substrate-binding domain (not homologous to DnaK). Interaction of the J-domain with DnaK accelerates ATP hydrolysis, while DnaJ’s substrate binding domain serves to stabilize the triple complex. DnaJ may also help recruit substrate to DnaK (Schroder et al. 1993). Whether or not DnaJ dissociates from the triple complex once it has reached the ADP state is not well established, but we will assume so in Fig. 2 for the sake of clarity (steps 2a and 2b). Evidence has been presented that multiple DnaK molecules can bind to s single substrate, ultimately all ending in the ADP state (steps 3a and 3b) (Noguchi et al. 2014). A process of entropic pulling, potentially involving several Hsp70s and DnaJs, progressively unfolds the substrate which remains bound (Goloubinoff and De Los Rios 2007; Kellner et al. 2014) (step 4.) Subsequently, the nucleotide exchange factor GrpE enables ADP ➔ ATP exchange (steps 5a and 5b), with a concomitant decrease in the affinity of Hsp70 for its substrate. Possibly aided by direct competition of the NEF with the bound substrate (Melero et al. 2015; Rauch et al. 2016), the substrate is now released in a folding-competent form, either to the cytosol (step 6a) or to GroEL for refolding (not shown). If the substrate unfolding failed the cycle may be repeated. DnaK releases ADP and GrpE (step 6b) and returns to the ATP state for a following cycle (step 6c).
The human Hsp70 system conforms to this basic ATPase cycle, although there are differences in complexity. As mentioned above, the human genome codes for 13 Hsp70 isoforms (Kampinga et al. 2009). The Hsp70s are highly homologous and consist of three constitutively expressed proteins, Hsc70 (HSPA8) in the cytosol and nucleus, Bip (HSPA5) in the endoplasmic reticulum, and Mortalin (HSPA9) in the mitochondria. The expression of the other Hsp70s, especially Hsp70–1 (HSPA1a/b), is induced upon cellular stress. These stress-induced factors appear to be dedicated to maintaining cellular proteostasis. For protein-refolding cycles the human Hsp70s utilize 50 different J-proteins and at least 8 different NEFs. The J-proteins are characterized by the inclusion of a conserved J-domain. Some of the human J-proteins (HDJA1 and HDJA2) are homologous with the E. coli DnaJ even outside the J-domain, while others have completely unrelated motifs outside the J-domain. It is believed that these different J-proteins can bestow tissue and functional specificity to the otherwise “generalist” Hsp70 family (Kampinga and Craig 2010). The human NEF family contains 6 BAG proteins (Bcl-2-associated AnthanoGene), a Hsp110 protein, HSPBP1, and HME. HME is restricted to the mitochondria and shows homology to E. coli GrpE. The other NEFs are quite different from the bacterial prototype and share no obvious sequence homology. Hsp110 is structurally related to Hsp70 itself, using its NBD to act as a NEF (Polier et al. 2008) (Schuermann et al. 2008). The BAG proteins use a helical BAG domain to interact with the top lobes of the NBD to favor nucleotide cycling (Sondermann et al. 2001). HSPBP1 is structurally unrelated to either Hsp110 or the BAG NEFs (Shomura et al. 2005). Regardless of the exact identity of the J-protein or NEF, each partner is thought to regulate the human Hsp70 protein refolding machinery in analogous fashion as the DnaK/DnaJ/GrpE cycle (Frydman and Hohfeld 1997). At the end of the cycle, HSPA1 or HSPA8 can transfer the substrate to Hsp90, either directly or in a complex with Hsp organizing protein (HOP) (Brinker et al. 2002). HOP contains two tetratricopeptide repeats (termed TPR1 and TPR2A), which bind to EEVD-COOH motifs at the C-terminal tails of Hsp70 and Hsp90, respectively. Unfolded Hsp70 substrate is also being transferred to TRIC, the mammalian GroEL homolog, for refolding (Frydman 2000).
Why does the human Hsp70 system have so many cochaperones? We use the BAG family of NEFs to illustrate the current ideas. The BAG proteins contain an alpha-helical BAG domain of approximately 100 amino acids which conveys the NEF function. However, BAG proteins have other functionalities as well (Kabbage and Dickman 2008). All of the BAG-family proteins function as adapter proteins forming complexes with different signaling molecules and the Hsp70’s. For instance, BAG-1 has besides its BAG domain a TXEEX domain involved in DNA binding and transcription activation, and a ubiquitin-like domain, which can target BAG1-Hsp70 complexes to the proteasome for degradation of specific client proteins. BAG3 is one of the largest BAG proteins and contains multiple protein-protein interaction motifs: a WW domain, multiple PXXP motifs, and two IPV motifs, allowing for PPxY protein binding, SH3 protein binding, and small heat shock protein binding, respectively (Kabbage and Dickman 2008). The non-BAG domains of BAG3 have recently been shown to also interact with HSPA8 SBD (Rauch et al. 2016). The BAG3-HSPA8 complex stabilizes several oncogenes and is a particularly interesting drug target (Li et al. 2015). BAG1 and BAG3 represent key players of cellular by stimulating the turnover of polyubiquitinated proteins by proteasomal and autophagic degradation pathways, respectively (Gamerdinger et al. 2009). BAG-4, containing a single BAG domain and another 350 residues for which no domain annotation is known as of yet, is a regulator preventing constitutive signaling by death domain receptors (Jiang et al. 1999). BAG-5 consists of four BAG domains (Takayama et al. 1999). Currently, not much is known about the role of BAG-5 in cells other that it binds to Hsp70. Thus, all of the BAG proteins enhance nucleotide exchange in Hsp70 through their BAG domains, yet they engender dramatically different functionality by the remainder of their sequences. In other words, depending on which of the BAGs 1–6 is engaged, the fate of the Hsp70-bound client protein might be quite different.
On top of the diversity of the BAG proteins, these proteins are all antagonized by the Hsp interacting protein (HIP), a homodimer containing several TPR domains (Hohfeld et al. 1995). HIP strongly stabilizes the Hsp70 ADP state. Surprisingly, it was shown that HSPA1, HDJ1, and HIP, without a NEF, can promote protein refolding (Hohfeld et al. 1995). The presence of TPR domains suggests that HIP has additional protein-protein interaction functions. Thus, the story of Hsp70 function seems to be one in which cochaperones and other partners combine with Hsp70 in dynamic ways, creating multivalent complexes that dictate outcomes.
HSPA8 shuttles proteins to the cellular degradation pathways
Another important aspect of Hsp70 function is that it bridges misfolded proteins (or “clients”) to distinct cellular fates. To illustrate this point, we briefly review how Hsp70s can direct clients to either the ubiquitin-proteasome system (UPS) or the chaperone-mediated autophagy (CMA) pathway. Indeed, it is because of Hsp70’s interactions with these divergent paths that it is sometimes called the “triage” chaperone (Wickner et al. 1999). In short, it interacts with unfolded proteins and helps determine whether it should be folded (and saved) or whether it is hopelessly misfolded (and should be removed). The chaperone also seems to determine which of the two major degradation pathways should be employed. How does Hsp70 do this for thousands of different polypeptide sequences and countless different protein folds?
One of the best-understood roles of HSPA8 are in proteosomal degradation. The C-terminal of Hsp70 interacting protein (CHIP) contains a TPR domain, which binds to the C-terminal IEEVD motif of HSPA1 and HSPA8 (Jiang et al. 2001). CHIP also contains a Ubox domain, which binds to ubiquitin ligases such as UBCH5. The latter ubiquitinates the HSPA8-bound substrate (and HSPA8 itself), which targets the complex to the proteasome for degradation (Atkin and Paulson 2014). Thus, CHIP links HSPA8 to the UPS and helps regulate protein turnover. Surprisingly, HSPA8 also plays a crucial role in CMA (Arias and Cuervo 2011) and endosomal microautophagy (eMI) (Sahu et al. 2011). In both CMA and eMI, HSPA8 recognizes a subset of cytosolic proteins containing a KFERQ motif (Terlecky et al. 1992). In CMA, the HSPA8-substrate complex docks to the lysosomal receptor LAMP-2A, after which substrate is translocated into the lysosome. In eMI, HSPA8 directly interacts with the endosomal membrane and delivers KFERQ-type proteins for ESCRT I- and III-mediated import into an endosome. Despite progress in understanding the players in these degradation pathways, the molecular mechanisms are mysterious. One of the most important, open questions is whether HSPA8 uses other, dedicated cochaperones to help make these triage decisions. In theory, assembly of a discrete collection of cochaperones, perhaps directed by structural features of the misfolded protein client, could be sufficient to explain how “decisions” are made. In other words, protein-protein interactions and molecular recognition by multiple chaperone surfaces might be the key.
Hsp70 internal interfaces: the differences between the ATP and ADP states
Hsp70s have an NBD, SBD, LID, and tail, connected by linkers in that order. The Hsp70 chaperone structural biology field is replete of crystal and solution structures of the individual NBDs (first: HSPA8 (Flaherty et al. 1990)) and SBDs (first: DnaK (Zhu et al. 1996)) of Hsp70 orthologs and paralogs but has been plagued by a paucity of valid structures for the full-length proteins, which became only available in 2009 for DnaK. No structures for full-length mammalian Hsp70s are available as of yet. And, even today, no structural comparison between ATP and ADP states for any Hsp70 can be made solely on basis of experimental data.
The first structure of a full-length Hsp70, DnaK(1–605), containing NBD, SBD, and LID (Bertelsen et al. 2009) was based on the crystal structures of the isolated SBD bound to the peptide NRLLLTG (1DKX) and the isolated NBD of DnaK without nucleotide and bound to GrpE (1DKG). NMR data provided the relative orientation and mobility range of these domains. A key finding was that the NBD and SBD domains were relatively free to move with respect to each other in this state, like “beads on a string” (Fig. 1). Structures of the ATP state of mutated NBD-SBD constructs of E. coli DnaK, without peptide, were determined by two groups (Kityk et al. 2012; Qi et al. 2013). These crystal structures show the SBD firmly docked to the NBD, and the alpha-helical LID domain docked to the NBD instead of the SBD (Fig. 1). NMR data indicated that the NBD-SBD docking in the ATP state occurs in solution as well (Swain et al. 2007) (Zuiderweg et al. 2013). The enormous differences in interdomain docking between the ATP- and ADP-bound states have been discussed and evaluated in detail in the original papers (Kityk et al. 2012; Qi et al. 2013) and will not be repeated here, except to point out that Hsp70 exposes and covers large and different interdomain surfaces in the two states.
The DnaK SBD in the peptide-bound state is very well ordered (Zhu et al. 1996), while the apo state is disordered around the substrate binding cleft (Pellecchia et al. 2000; Kityk et al. 2012; Qi et al. 2013). This is illustrated in Fig. 3a, where the SBD is colored according to the crystallographic B-factor. High B-factors indicate disorder, which can indicate either (frozen-out) dynamics or static disorder. The figure thus shows that the loops surrounding the substrate are highly disordered in the apo state. The NMR spectra of DnaK SBD showed much conformational exchange broadening for the resonances of the same loops, indicating molecular dynamics at the milli/micro-second time scale (Pellecchia et al. 2000). The structural and dynamical differences between DnaK SBD in the apo and peptide-bound state have been further studied by solution NMR by Zhuravleva and Gierasch (2015).
Fig. 3.

a Overlay of the SBD of DnaK E. coli in the NRLLLTG (1DKX) and apo (4B9Q) state. The figure ribbon is colored according to the B-factor ranging from blue (high order) to yellow (low order). In this representation, the NRLLLTG state is dark blue throughout. The asterisks indicate the loops forming the substrate binding cleft that are flexible without substrate and rigid with substrate. b Overlay of the NBD of HSPA8 in the ADP state (green, 3HSC), with a model of HSPA8 NBD based on DnaK in the ATP state (red, 4B9Q). The subdomain nomenclature is shown. c Model of the ADP state of the NBD of DnaK from Thermus thermophilus (green, 4B9Q) based on DnaK E. coli, and the experimental rotational changes occurring upon binding of AMPPNP state (red). In both b, c, the ∼10° clockwise rotation of lobe II with respect to lobe I between the NBD ATP and ADP state is indicated with curved arrows
There is no direct experimental comparison possible for the DnaK NBD in the ATP and ADP state, by account that no structure has been determined for the latter (the NBD of the 1KHO hybrid structure is based on the GrpE-bound NBD (1DKX)). So, what happens to the NBD between ATP and ADP states? Many crystal structures are available for the mammalian Hsp70 NBDs in the ADP state and ATP (analogue) states. Regretfully, these structures are all identical within experimental precision, providing no insight into ATP/ADP-induced conformational differences that must occur. The best we can do for now is to construct a homology model of HSPA8 NBD in the ATP state, based on the crystal structure of DnaK in the ATP state, and compare it with the crystal structure of HSPA8 NBD in the ADP state (Fig. 3b). The model and structure are superposed on subdomain IA (left bottom in figure). According to this model, the left and right hand lobes of the human HSPA8 NBD rotate by 10° with respect to each other between the states. Furthermore, the cleft between lobes I and II is compressed in the ATP state and major changes occur in domain IB (left top). This model likely has some validity: early NMR work indicated that ATP/ADP binding causes major conformational changes in the HSPA8 NBD (Zhang and Zuiderweg 2004). Furthermore, Bhattacharya et al. (2009) showed, also by NMR, that the left and right hand lobes of the NBD of DnaK Thermus thermophilus rotate by 10° with respect to each other between ADP and ATP states in solution.
No experimental structural comparison of HSPA8 SBD in the apo and peptide-bound state is available. However, there is strong NMR evidence that the Hsc70 SBD in the apo state is even more dynamically disordered than the DnaK SBD in the apo state (unpublished).
In summary, these less-than-perfect comparisons between different experimental structures reinforce each other, and it seems safe to conclude that the human Hsp70 domains are differently docked in ATP and ADP states, that the NBD subdomains rotate with respect to each other upon hydrolysis of the ATP gamma phosphate, and that the SBD is partially disordered without substrate bound. Nature seems to use these varying surfaces and dynamics to modulate the energetics of Hsp70 conformational equilibrium by cochaperones, e.g., by DnaJ. Further, it seems likely that more interactions, which are not yet fully investigated or described, will be found to exploit these dramatic differences as well.
Complexes with NEFs
The next step in building the structural picture of how chaperone complexes function is to examine the interaction between Hsp70s and the NEFs. This interaction is emblematic of the protein-protein contacts that form the next “layer” of dynamics and allostery in the system, and it has been extensively characterized by X-ray crystallography. The BAG proteins all bind to the “rear” of lobes IB and IIB and stabilize an open structure of the nucleotide-binding cleft (Sondermann et al. 2001), providing an explanation for the nucleotide exchange mechanism (Fig. 4). The Hsp110 proteins engage a much larger surface of HSPA8, still including lobes IB and IIB (Polier et al. 2008; Schuermann et al. 2008). Figure 4 also shows that the perturbation of the NBD cleft-opening varies significantly between the different NEFs. Whether such differences in extent of cleft opening also occur in solution is currently unknown. Similarly, the different BAG domains seem to have distinct orientations of their BAG helical bundles with respect to the HSPA8 NBD, but it is not yet clear if this reflects a meaningful divergence or if crystal packing interactions are skewing the structures. The crystal structure for the HSPBP1 complex contains only NBD lobe II (Shomura et al. 2005). It can therefore not really explain the mechanism of nucleotide exchange, but it does indicate that, at least in lobe II, the interaction surface is more extensive than that in the BAG series. No solution studies were published for the Hsp70 /NEF interactions. However, good quality NMR data could be collected on HSPA8 NBD in complex with BAG1, whereas addition of HSPBP1 to a solution of HSPA8 NBD caused massive precipitation (unpublished results). This appears to indicate that HSPBP1 perturbs the NEF structure much more then than BAG1 does. Put together, the interaction surfaces of the different NEFs (i.e., HSPBP1, BAGs, Hsp110) on the Hsp70 proteins have some similarities, but are also different (Fig. 5). In Fig. 5, the orientation is 180 around the vertical axis as compared to Fig. 4, to best illustrate the differences between the surfaces. Another way to look at the difference is by summarizing the residues that make contact in the structures (Table 1). All surfaces are extensive and will be quite difficult to perturb with small chemicals. In addition, the surfaces overlap considerably so that it will be difficult to affect NEF – Hsp70 interaction with any specificity bet-ween NEFs. The possible exceptions are HSPA8 residues 45–47, that are involved in the BAG1 interaction only.
Fig. 4.
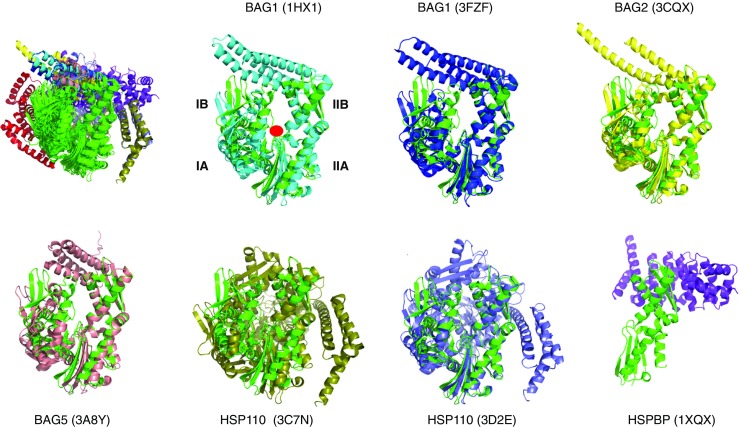
Comparison of the perturbation of Hsp70 NBD by nucleotide exchange factors. The HSPA8 NBD in ADP state (3HSC) are shown in green. The location of the nucleotide binding site is indicated with a red circle in the second panel. The structures are aligned on subdomain IIB. All structures compared (top left). The other panels show the individual structures. Bag1 (1HX1), Cyan; Bag1 (3FZF), blue; Bag2 (3J8F), yellow; Bag 5 (3A8Y), salmon; Hsp110 (3C7N), slate; Hsp110 3D2E), olive; HSBP1 (1XQS), purple
Fig. 5.

Interactions surfaces between Hsp70s and the different NEFs. The color coding is: yellow: subdomain IA; cyan, IIA; green, IB; purple, IIB. In red are those NBD atoms (including H) that are within 3 Å of NEF atoms (including H)
Table 1.
Summary of HSPA8 NBD contacts with NEFs
| HSP110 (3C7N) contacts | HSP110 (3D2E) contacts | HSPB (1XQS) contacts | BAG1 (1HX1) contacts | BAG1 (3FZF) contacts | BAG2 (3CQX) contacts | BAG5 (3A8Y) contacts | HIP (4J8F) contactsa |
|---|---|---|---|---|---|---|---|
| H23 | Q22 | R247 | T45 | D46 | Q33 | A60 | K25 |
| K25 | H23 | K248 | D46 | N47 | N35 | L61 | D32 |
| E27 | K25 | H249 | N57 | A60 | N57 | K257 | Q33 |
| A30 | D32 | K250 | A60 | M61 | Q58 | R258 | N35 |
| D32 | Q33 | R258 | M61 | R258 | A60 | R261 | N57 |
| Q33 | G34 | R262 | N62 | R261 | M61 | R262 | Q58 |
| G34 | R36 | T265 | R258 | R262 | R258 | T265 | L61 |
| R36 | L50 | E268 | R261 | T265 | R261 | R269 | Y134 |
| L50 | A54 | R269 | R262 | R269 | R262 | I284 | R258 |
| A54 | N57 | A270 | T265 | S281 | T265 | D285 | R262 |
| N57 | Q58 | R272 | R269 | E283 | A266 | S286 | R269 |
| M61 | L61 | T273 | S281 | I284 | R269 | D292 | T273 |
| A133 | A133 | S277 | I282 | D285 | E283 | Y294 | S281 |
| Y134 | Y134 | Q279 | E283 | S286 | I284 | E283 | |
| R258 | R262 | A280 | I284 | G290 | D285 | D285 | |
| R269 | T273 | S281 | D285 | D292 | S286 | D292 | |
| R272 | S276 | L282 | S286 | Y294 | D292 | Y294 | |
| T273 | T278 | E283 | G290 | Y294 | |||
| S276 | Q279 | D285 | D292 | ||||
| T278 | S281 | D292 | Y294 | ||||
| S281 | D285 | Y294 | |||||
| E283 | S286 | ||||||
| D285 | T298 | ||||||
| S286 | R299 | ||||||
| G290 | A300 | ||||||
| T298 | R301 | ||||||
| R299 | E304 | ||||||
| A300 | K348 | ||||||
| R301 | D352 | ||||||
| E303 | |||||||
| E304 | |||||||
| R342 |
Hsc70 residues within 3 Å of any residue of the cochaperone, any atoms, including hydrogens
aCrystal contacts between HSPA1 NBD and HIP as described in Li et al. (2013)
As mentioned above, HIP is an “anti-exchange” factor, which stabilizes the Hsp70 ADP-state, via interactions between Hsp70 and the middle domain of HIP (residues 77–247) (Hohfeld et al. 1995). Although a full-length structure is not available, X- ray structures of the domains have provided some clues as to how it might work. For example, a fusion between HSPA1 NBD (1–383) and the HIP middle domain (77–247) without a linker was crystallized (Li et al. 2013). In that structure, the “cis” interface between HSPA1 and HIP is dictated by the short connection between the domains and may not be relevant. However, a serendipitous “trans” crystal contact between HSPA1 of one fusion with HIP of another, partially overlaps with the interface of BAG1 with HSPA8 (Li et al. 2013) (see Fig. 6). This competition may explain the HIP-BAG1 antagonism. In addition, the NBD in this crystal structure is more closed than Hsc70 in the ADP state; hence, it seems that HIP closes the NBD cleft to stabilize the nucleotide-bound state (both ADP and ATP).
Fig. 6.

The figure shows two fusion constructs as found in 4J8F: HSPA1 NBD (blue) fused to HIP middle domain (red) and another HSPA1 NBD (cyan) fused to another HIP middle domain (orange). The crystal contact between the blue NBD and orange HIP may constitute a natural complex. Nucleotide binding site and subdomains are labeled for the blue NBD
While the focus of this section has been on NEF interactions at the Hsp70 NBD, some of the NEFs seem to operate by multivalent contacts. For example, a crystal structure between DnaK NBD and the bacterial NEF, GrpE, suggested that NEFs could interact with the SBD as well (Harrison et al. 1997). In that structure, the long helical bundle of GrpE lies along the DnaK NBD lobe I and points in the direction of the SBD. Indeed, the Valpuesta and Mura groups have shown using biophysical methods that there is an interaction between a disordered region at the terminus of the helical region and the DnaK SBD (Melero et al. 2015). In this model, GrpE directly competes with clients, helping to accelerate their removal. Very recently, it was shown by NMR that the N-terminal domain of BAG-3, outside the BAG domain, interacts with the HSPA8 SBD (Rauch et al. 2016). The interaction is likely with the hydrophobic cleft, in a manner somewhat analogous to what was reported for DnaK (Melero et al. 2015). Hence, some of the mammalian NEFs may also help substrate release in a bifunctional manner: stimulation of ATP binding and thereby increasing the substrate off-rate as well as direct competition with the substrate. Significantly, it was shown that the isolated BAG domain from BAG-3 was unable to release a peptide client from HSPA1 on its own (Rauch et al. 2016), suggesting that the multivalent contact is necessary. These observations contribute to a growing model in which chaperone interactions within the cochaperone complexes are more nuanced than previously imagined, especially in the mammalian systems.
Complexes with J proteins
In contrast to the Hsp70–NEF interaction, the interaction between Hsp70 and J-proteins has been much more elusive. The J-proteins are defined by the presence of a J-domain, which is a triple-helical bundle of ∼70 residues containing an invariant HPD loop between helices 2 and 3 (Bork et al. 1992; Szyperski et al. 1994). Human HdJ1 contains a J-domain, a long linker, and one or more substrate-binding domains, and a dimerization domain (Suzuki et al. 2010). Some J-proteins contain a J-domain connected to quite different protein-protein interaction domains, likely accounting for functional specificity (Kampinga and Craig 2010). The isolated J-domain has been shown to stimulate DnaK ATP hydrolysis but does bind less tightly to the DnaK proteins than full-length J-proteins (Suh et al. 1998). Besides the conserved HPD loop, the J-domains display conserved lysines and arginines on helix 2. Mutagenesis of the HPD loop and/or the Lys/Arg results in loss of DnaJ stimulation of Hsp70 ATP hydrolysis, suggesting a possible involvement in the interaction or positioning of the proper residues. Mutagenesis of DnaK suggest that corresponding residues between NBD lobes IA and IIA may be the opposing surface that makes contact with the HPD (Suh et al. 1999); however, this area on DnaK also mediates the NBD-SBD contacts in the ATP state, making the results difficult to interpret.
No crystal structure between an isolated J-domain and an Hsp70 has been reported, but a structure of a J-domain of auxillin and the HSPA8 NBD, covalently linked by a designed cross-link, was published. That structure showed a proximity of residues in and around the cross link (Jiang et al. 2007). Mutation of some of these residues does disrupt J-protein-driven allostery, but again, these results are convoluted by the roles of the lobe IA and lobe IIA region in many other aspects of the cycle. More recent solution NMR experiments may have revealed why it has been so difficult to characterize the Hsp70/J interactions: The affinity of the isolated J-domain (1–72) for wt-DnaK (ADP) is relatively weak (16 μM), while the binding surface on DnaK is found to be an extended area of negative electrostatic potential to which the positively charged helix II of DnaJ binds in a diffusive manner (Ahmad et al. 2011). The center of this rather ill-defined area is at DnaK residue Asp 211/214 (DnaK/HSPA8), located at the periphery of subdomain IIA (see Fig. 7 top-right). The DnaK and DnaJ contact areas correlate with those indicated by mutagenesis, except for the HPD loop. No evidence was found for the proximity of this loop to DnaK in the study by Ahmad et al. (2011), although transient contacts could not be excluded. DnaJ’s helix II also delocalizes to a negative area on the “face” of the SBD (Ahmad et al. 2011). DnaJ stimulates Hsp70 ATP hydrolysis and binds more strongly to DnaK in the ATP state than to the ADP state (Suh et al. 1998). Currently, there is no structural information elucidating how the J-domain binds to the ATP state.
Fig. 7.

Function-modifying interactions of small molecules with the HSPA8 NBD (3HSC). Subdomain IA: yellow; IB: green; IIA: cyan; IIB: blue. The interaction surfaces, obtained from NMR chemical shift perturbation studies, are colored red. MKT077 interaction with HSPA8 from (Rousaki et al. 2011). The following interaction surfaces were obtained for the E. coli DnaK NBD but displayed on the Hsc70 NBD. DnaJ-J domain interaction with DnaK from (Ahmad et al. 2011); Myricetin interaction with DnaK from (Chang et al. 2011); 115-7c interaction with DnaK from (Wisen et al. 2010); Telmisartan interaction with DnaK (Cesa et al. 2013). See also Table 2
TPR-protein interactions
Hsp70 proteins utilize the C-terminal IEEVD sequence to interact with the TPR proteins HOP, CHIP, PP5, DnaJC7, FKBP51, and FKBP52, with the exception of HIP (Assimon et al. 2015). Mutagenesis in the IEEVD sequence shows loss of binding, and alanine scanning has shown the residues important in the interaction. Further, details of the interaction of the CHIP TPR domain with GTIEEVD (Wang et al. 2011) and PTIEEVD (Zhang et al. 2015) peptides were elucidated by X-ray crystallography (See Fig. 8). The interaction is clearly stabilized by electrostatics involving HSPA8’s E643, D646, and the COO− terminus. E644 is not in direct contact with a positive charge, but resides in a positive potential. But there must be more to the interaction than electrostatics: HSPA8’s I641 is in a hydrophobic pocket formed by CHIP residues F98 and F131, and HSPA8 V645 sits in a similar pocket formed by CHIP’s Phe 37 and Leu 68.
Fig. 8.

The interaction of the CHIP TPR domain with the HSPA8 PTIEEVD C-terminal peptide (3Q49). CHIP is as a cartoon, while PTIEEVD is as sticks. Residues on CHIP contacting the HSPA8 peptide are rendered in spacefill. The color coding represents chemical properties: gray, polar; green; hydrophobic; red, negative; blue, positive; olive, mixed (Thr and Tyr)
NMR studies by Zhang et al. (2015) indicate that CHIP interacts besides the C-terminal tail also with the HSPA8 SBD itself. NMR studies of the interaction of full-length CHIP with full-length HSPA8 (Smith et al. 2013), on the other hand, show no evidence of the latter interaction, although transient touches cannot be excluded. The hallmark of the interaction as determined by the latter NMR studies shows CHIP to be dynamically tethered to Hsc70 through the IEEVD clamp, with HSPA8 residues 610–638 remaining flexible and unaffected by the presence of CHIP. This type of scenario may be biologically advantageous (Smith et al. 2013), because the E2 ubiquitin ligase bound to CHIP can reach more residues of the HSPA8 substrate for ubiquitination than if the complex was rigid. A tethered binding mode was also observed for the protein phosphatase 5, which uses its TPR domain to interact with the HSPA8 IEEVD sequence as well (Connarn et al. 2014). This seems equally desirable: A tethered phosphatase can dephosphorylate many more HSPA8 substrate residues than a statically bound one.
Complexes for chaperone-mediated autophagy
Lysosomes take up and degrade intracellular proteins in response to serum deprivation or other nutrient signals in a process called autophagy. There are many types of autophagy, including micro-autophagy and macro-autophagy. By injecting a variety of proteins into fibroblasts, Dice (1990) discovered that certain proteins containing a peptide sequences biochemically related to KFERQ are preferentially targeted for hydrolysis by micro autophagy. In subse-quent work, his group determined that these KFERQ proteins were recognized by HSPA8 (Terlecky et al. 1992). Further, it was discovered that the HSPA8-bound KFERQ proteins are (1) targeted to the LAMP2A receptor on lysozomes for internalization and hydrolysis, in a process termed Chaperone-Mediated Autophagy (CMA) (Cuervo and Dice 1996) or (2) to late endosomes for complete or partial degradation, called endosomal microautophagy (eMI) (Sahu et al. 2011). Protein cargo selection is mediated by the chaperone HSPA8 and requires the SBD of HSPA8 for electrostatic interactions with the endosomal membrane (Sahu et al. 2011; Morozova et al. 2016). The intermolecular interactions required for both processes are just recently being investigated.
The KFERQ sequence does not resemble the prototypical hydrophobic substrates such as NRLLLTG. In on-going NMR work, we observe spectacular changes in the NMR spectrum of the isolated HSPA8 SBD upon binding of known substrates such as NRLLLTG (Rauch et al. 2016), but we do not observe any change upon adding an Ac-KFERQ-NH2 peptide (to be published). Additionally, RnaseA, which is the prototypical CMA client (Dice 1990), also fails to induce perturbations in the HSPA8 SBD NMR spectrum. Accordingly, as of now, the mechanism of recognition between the KFERQ sequence and HSPA8 remains elusive. However, it is absolutely clear that this interaction is not the same as a canonical hydrophobic peptide-SBD binding.
Another example of the binding of two different peptides having dramatically different effects on HSPA8 has been reported by (Takenaka et al. 1995). They screened a 15-mer phage display random peptide library and found two classes of peptides, those obeying the motif Hy(W/X)HyXHyXHy, rich in large hydrophobic residues such as FYQLALT used in the Takenaka study, now known to bind in the SBD, and peptides rich in both hydrophobic and basic residues like NIVRKKK. It was noted that this latter class resembled organellar targeting sequences particularly mitochondrial ones and that DNAJA1 (formerly human DnaJ2) contained the sequence KIVREKK, a possible interaction site with HSPA8. NIVRKKK is a very weak stimulator of the HSPA8 ATPase activity compared to FYQLALT. Interestingly, this peptide blocked the binding of NIVRKKK to HSPA8, likely by inducing a conformational change that blocked the NIVRKKK binding site. This was indicated by the much more rapid migration of two forms of HSPA8-FYQLALT on native polyacrylamide gels compared to HSPA8-NIVRKKK. Again, it is clear that the interactions of these two peptides with HSPA8 are very different.
The endosomal membrane contains a large fraction of phosphoserine lipids, which carry a net negative charge. (Sahu et al. 2011) had previously determined that the HSPA8 SBD is involved in the interaction between membrane and protein necessary for the ESCORT-mediated translocation of Hsc70-bound client into endosomes. Very recently, Morozova et al. (2016), using NMR and mutagenesis, narrowed the interaction site down to a cluster of positive charges on the HSPA8 LID domain. This interaction site, quite different from any other known site, is shown in Fig. 9, which also summarizes all interactions.
Fig. 9.

Summary of all interaction sites discussed in this review on a homology model for HSPA8 based on DnaK SBD in the ADP state (1DKX), extended with an unstructured tail comprising residues 605–646. NBD, gray; SBD: Blue; LID domain: cyan; tail: yellow. In magenta, substrate peptide NRLLLTG and nucleotide ADP. Interaction sites: all NEFs: beige; all Hsc70 and E. coli DnaK NBD-binding compounds with activity: black. CHIP: orange from (Smith et al. 2013); Phosphoserine lipids: red (Morozova et al. 2016); J-domain – E. coli DnaK interaction from (Ahmad et al. 2011) pink; E. coli DnaK interaction with PET-16 (4R5G): green
Finally, CMA involves interaction of a HSPA8-KFERQ substrate complex with the LAMP-2A receptor located in the lysosomal membrane surface (Cuervo and Dice 1996). LAMP-2A is integral membrane protein that serves as pore for cargo transport. The HSPA8-KFERQ substrate complex interacts with the LAMP-2A C-terminal cytosolic peptide GLKRHHTGYEQ (Cuervo and Dice 1996). Recent NMR data demonstrates that the cytosolic tail of LAMP-2A may bind HSPA8 and its cargo simultaneously (Rout et al. 2014). However, at present it remains unknown how HSPA8 recognizes the LAMP-2A receptor and it remains unclear how these interactions lead to CMA.
Lipid interactions
Interest in the association of HSPA family proteins with individual lipids and with membranes has been growing. The initial report that HSPA1A and HSPA8 could interact with lipids came from studies that found the proteins associated with fatty acids during purification (Guidon and Hightower 1986). Subsequently, the De Maio group reported that both HSPA1A and HSPA8 form stable ion-conductance pathways in artificial lipid membranes (Arispe and De Maio 2000; Vega et al. 2008), which has been corroborated by (Macazo and White 2014). It is believed that the interactions of HSPA8 with membranes involve changes in the conformations and self-association of the protein (Armijo et al. 2014). Recently, Guzhova and coworkers (Komarova et al. 2015) provided evidence of a cationic amino acid sequence of 20 amino acids (amino acids 493–512, between SBD and LID) on HSPA1A that has cell-penetrating activity similar to the full-size protein. This peptide termed KST can carry into cells cargo having a molecular mass 30 times greater than its own. KST peptide was also shown to effectively compete with HSPA1A transport and that both used an active intracellular transport mechanism including negatively charged lipid membrane domains and vesicles as well. As mentioned above, a positively charged region of HSPA8 was also found responsible for the interaction with internal endosomal vesicles (Morozova et al. 2016).
Despite the fact that DnaK and HSPA1A share many structural features, they can behave differently structurally and biologically. De Maio and colleagues compared their interactions with unilamellar liposomes with different lipid compositions. Whereas HSPA1A was highly specific for binding to negatively charged phospholipids, particularly phosphatidylserine, DnaK bound to all the lipid types tested regardless of their charge (Lopez et al. 2016). In addition, these two proteins associated with the liposome membrane differently. HSPA1A had several regions imbedded in the membrane, and the N-terminus was exposed on the exterior of the liposome, whereas DnaK had its N-terminal segment inserted into the membrane and a C-terminal segment was exposed on the outside of the liposome. Using protease protection of protein fragments by the membrane as a test for insertion, four DnaK 11 sequences were protected and four of these sequences had little or no overlap with HSPA1A sequences. For HSPA1A, 13 sequences were protease-protected and six of these had little or no overlap with protected sequences of DnaK. The KST sequence in HSPA1A described above was partially protected, and a different part of the comparably aligned sequence of DnaK was protected. The homo-multimeric complexes that formed were different as well. HSPA1A formed large oligomeric complexes when inserted into the membrane and DnaK formed dimeric complexes within the membrane. It is clear that much experimentation has to be carried out to characterize the structural aspects of these interactions.
Compound interactions
One of the reasons for better understanding the structure and function of the Hsp70 complexes is to enable the discovery of chemical modulators of Hsp70 function. However, this class of chaperones is a significant challenge. Inhibiting the nucleotide binding with a competitive inhibitor is difficult because of the high conservation between the ATP-binding fold in Hsp70 NBD and the fold in other abundant proteins, such as actin. Further, the tight affinity of Hsp70 for ATP and the high cellular concentration of ATP/ADP make it difficult to effectively compete for this interaction. On the other end of the chaperone, any molecule capable of directly competing with the client-binding cleft of the SBD would, likewise, need to contend with the high cellular concentration of possible clients. Moreover, such a molecule would need to be quite hydrophobic and difficult to handle. On top of these problems, it is not clear how one might specifically target a single paralog of Hsp70, owing to the high homology.
Despite these obstacles, several groups have made progress in developing inhibitors of Hsp70 ATP hydrolysis. Here, we review these compound-protein interactions, in much the same way as we reviewed the cochaperone interactions. Again, the goal is to elucidate the broader concepts across different observations and groups.
A group at Vernalis, Inc., was the first to report nucleotide analogs that inhibit HSPA1 (Williamson et al. 2009; Cheeseman et al. 2016). These molecules were developed through a fragment-based and structure-guided program, resulting in molecules that make contacts with key residues in the NBD (Fig. 10). As designed, the nucleosides of the three analogues occupy the same location as ADP in HSPA8. Only VER-155008 contacts other parts of the protein and offers some hope for being Hsp70 chaperone selective. Its properties were discussed in detail by Schlecht et al. (2013), who showed it to be a competitive ATP-binding inhibitor.
Fig. 10.

Nucleotide-analogue inhibitors of HSPA8. HSPA8 NBD (3HSC) is shown as a cartoon in gray. ADP + Mg + Na + PO4 bound to the HSPA8 NBD are in blue; compound SGV in cyan (5AQZ); compound GB8 in red (5AR0); compound 3FD (VER-155008) in magenta (4I08). Nucleotides and analogs interact with all four NBD subdomains as indicated
Hsp70 has evolved to use a great many of its surfaces and crevices to engage in both intramolecular and intermolecular interactions. This “economical” approach gives hope that one can affect Hsp70 function using molecules that bind to less obvious, allosteric regions. One of the first compounds identified to inhibit DnaK’s ATP hydrolysis was 115-7C and was found to locate to the interface of DnaK that is involved in J protein interactions (Wisen et al. 2010) (see Fig. 7). Clearly, this molecule modulates Hsp70 function in an indirect way by interfering with J-domain binding (see Fig. 7). This binding site may allow the compound class to eventually have better selectivity, because of the relatively lower conservation in that region. While 115-7c acts on the J-protein interface, several other Hsp70 modulating compounds are likely to be involved in modulating Hsp70’s NEF interactions (Chang et al. 2011; Cesa et al. 2013) (see Fig. 7). A third class of allosteric inhibitors is based on the rhodocyanine MKT077. MKT077 had been in Phase I clinical trial in the 1990s as a very promising, high-therapeutic window tumor suppressor, but the trial failed because of the compound’s renal toxicity (Britten et al. 2000). By NMR, the binding site of MKT077 was found to be a deep pocket on the HSPA8 NBD between domains IA and IIA (Fig. 7), a location that is adjacent to, but not at, the nucleotide-binding site (Rousaki et al. 2011). Significantly, the binding occurs only in the ADP state, which could be rationalized because this pocket is occluded in the ATP state of the protein (Bhattacharya et al. 2009). Hence, serendipitously, an allosteric modulator was found that maintains/drives HSPA8 to the ADP state/conformation where it has high affinity for its substrates. Thus, MKT077 can stall the HSPA8 refolding cycle in the substrate-bound state, increasing the chance that the complex will be recognized by CHIP and guided to the proteasome. This mechanism may be underlying the now well-documented ability of MKT077 derivatives to help clear tangles of aberrant tau protein in Alzheimer’s model cells (Fontaine et al. 2015).
Recently, fragment-based screening monitored by crystallography was used to find additional binding sites in proteins (Ludlow et al. 2015). Amidst 20 other proteins, HSPA2 NBD was also included in the test, and four compounds were obtained that bind to HSPA2 in three different binding locations (see Fig. 11). Compound IWT (red) binds to domain IA and may compete with the ATP-dependent docking of the SBD on the NBD. This interesting compound could thus modulate the NBD-SBD allostery and functionality. The other two compounds, PZA and KYD, bind to HSPA2 locations that do not change conformation between ATP and ADP state. Their binding locations are also not involved in known chaperone-cochaperone interactions. Hence, we do not expect these compounds to affect HSPA2 function.
Fig. 11.

Non-canonical fragment binding to HSPA2 NBD, not tested for activity (Ludlow et al. 2015). Cyan, NBD with compound PZA (5FPD.pdb); red, NBD with compound IWT (5FPM.pdb); magenta: NBD with compound KYD (5FPN.pdb). The compounds are rendered in spacefill
At present, two non-peptide compounds were identified that interact with the Hsp70 SBD. The inhibitor 2-phenylethynesulfonamide (PES) interacts with the SBD of HSPA1 (Leu et al. 2009). Schlecht et al. (2013) tested the compound in folding and binding assays and found that its activity was detergent-like. Another compound, PET-16, affects Hsp70 function by allostery (Leu et al. 2014). The hydrophobic compound nestles itself within the hydrophobic core of the SBD, at a location that likely disturbs the allosteric communication between the substrate in the SBD and NBD interface on the SBD (see Fig. 9). This binding site was discovered before by (Wang et al. 1998) as the only free peptide binding site in a SBD construct that was self-associated in its primary binding site.
One of the fascinating aspects of the small molecule interactions with Hsp70 is the diversity of binding sites. The Hsp70 residues that interact with the different chemicals discussed are listed in Table 2, illustrating this point. Much like the cochaperone interactions that are spread across the entire surface of the protein, the compound interaction sites seem to be similarly distributed. The “best” ones and their effects on chaperone functions in disease are only beginning to be made clear.
Table 2.
Summary of HSPA8 contacts with CHIP, J, and various chemicals
| CHIPa | MKT-077b | DOPSc | J-domaind | PET-16e | Myricytinf | 115-7cg | Telmisartanh | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T641 | R76 | K569 | D208 | E213 | L392 | L394 | K55 | K56 | T173 | T177 | R56 | N57 |
| I642 | A148 | K573 | D211 | D214 | D393 | D395 | R56 | N57 | A174 | A178 | M89 | W90 |
| E643 | Y149 | K583 | G212 | G215 | V394 | V396 | Q57 | Q58 | A176 | A180 | F91 | F92 |
| E644 | F150 | K589 | T215 | I216 | T395 | T397 | A58 | V59 | L177 | I181 | R261 | R264 |
| V645 | T222 | K601 | F216 | F217 | P396 | P398 | V59 | A60 | G180 | G184 | ||
| D646 | A223 | E217 | E218 | L397 | L399 | L66 | V67 | L181 | L185 | |||
| G224 | D326 | D323 | L399 | L401 | F67 | F68 | D182 | D186 | ||||
| D225 | I418 | I420 | G74 | G75 | K183 | K187 | ||||||
| T226 | Q442 | E444 | I88 | H89 | N187 | E192 | ||||||
| H227 | I478 | I480 | M89 | W90 | R188 | R193 | ||||||
| L228 | A480 | A482 | F91 | F92 | T189 | N194 | ||||||
| D481 | N483 | K92 | M93 | A191 | L196 | |||||||
| G482 | G484 | V103 | V105 | V192 | I197 | |||||||
| A503 | N505 | K106 | K108 | S203 | S208 | |||||||
| S504 | D506 | A111 | S113 | I204 | I209 | |||||||
| N222 | A223 | I205 | L210 | |||||||||
| G223 | G224 | D211 | D214 | |||||||||
| E230 | E231 | G212 | G215 | |||||||||
| I237 | V238 | T215 | I216 | |||||||||
| M259 | V260 | F216 | F217 | |||||||||
| Q260 | R261 | E217 | E218 | |||||||||
| L262 | L263 | |||||||||||
| E267 | E268 | |||||||||||
aFrom NMR intensity perturbations of human wt-HSPA8
bFrom NMR Chemical Shift Perturbation (CSP) in human HSPA8 (1–386)
cFrom NMR CSP in human HSPA8 (387–604) and mutagenesis of full-length HSPA8
dFrom Paramagnetic Relaxation Enhancement NMR for E. coli DnaK(1–608). Left column: DnaK count, right column: human HSPA8 count
e3A proximities between compound and E. coli DnaK in the crystal structure (4R5G). Left DnaK count, right HSPA8 count
fFrom NMR CSP in E. coli DnaK (1–388) Left DnaK count, Right: HSPA8 count
gFrom NMR CSP in E. coli DnaK (1–388) Left DnaK count, Right: HSPA8 count
hFrom NMR CSP in E. coli DnaK (1–388) Left DnaK count, Right: HSPA8 count
Conclusions
In conclusion, the Hsp70s have evolved to utilize many different interfaces to accommodate many different binding partners. Figure 9 serves as an overview of the different binding sites discussed here. Even the chaperone itself uses completely different interfaces between its domains (NBD, SBD, LID) in the different allosteric states. While it is likely that most of the intermolecular interfaces have now been identified, there are still several known unknowns that need to be hunted down: (1) the interaction of NEFs with SBD, (2) the true interaction of HIP, (3) possible Hsp70-Hsp70 interactions when bound to substrate, (4) the interaction with DnaJ in the ATP state, (5) the interaction of Hsp70 proteins with autophagy (KFERQ proteins) clients, (6) the interaction with the LAMP-2A receptor and (7) lipid interactions. Likely, there are several more for now unknown interfaces with for now unknown Hsp70 partners to be chased in the future.
Acknowledgements
This work was supported by NIH grant 5-R01-NS-059690-01-08.
References
- Ahmad A, Bhattacharya A, McDonald RA, Cordes M, Ellington B, Bertelsen EB, Zuiderweg ER. Heat shock protein 70 kDa chaperone/DnaJ cochaperone complex employs an unusual dynamic interface. Proc Natl Acad Sci U S A. 2011;108:18966–18971. doi: 10.1073/pnas.1111220108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011;23:184–189. doi: 10.1016/j.ceb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N, De Maio A. ATP and ADP modulate a cation channel formed by Hsc70 in acidic phospholipid membranes. J Biol Chem. 2000;275:30839–30843. doi: 10.1074/jbc.M005226200. [DOI] [PubMed] [Google Scholar]
- Armijo G, et al. Interaction of heat shock protein 70 with membranes depends on the lipid environment. Cell Stress Chaperones. 2014;19:877–886. doi: 10.1007/s12192-014-0511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assimon VA, Southworth DR, Gestwicki JE. Specific binding of tetratricopeptide repeat proteins to heat shock protein 70 (Hsp70) and heat shock protein 90 (Hsp90) is regulated by affinity and phosphorylation. Biochemistry. 2015;54:7120–7131. doi: 10.1021/acs.biochem.5b00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkin G, Paulson H. Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci. 2014;7:63. doi: 10.3389/fnmol.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Lavan Y, Shemesh N, Ben-Zvi A. Chaperone families and interactions in metazoa. Essays Biochem. 2016;60:237–253. doi: 10.1042/EBC20160004. [DOI] [PubMed] [Google Scholar]
- Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci U S A. 2009;106:8471–8476. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Kurochkin AV, Yip GN, Zhang Y, Bertelsen EB, Zuiderweg ER. Allostery in Hsp70 chaperones is transduced by subdomain rotations. J Mol Biol. 2009;388:475–490. doi: 10.1016/j.jmb.2009.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P, Sander C, Valencia A, Bukau D (1992) A module of the DnaJ heat shock proteins found in malaria parasites. Trends Biochem Sci 17:129 [DOI] [PubMed]
- Brinker A, et al. Ligand discrimination by TPR domains - relevance and selectivity of EEVD-recognition in Hsp70 center dot hop center dot Hsp90 complexes. J Biol Chem. 2002;277:19265–19275. doi: 10.1074/jbc.M109002200. [DOI] [PubMed] [Google Scholar]
- Britten CD, et al. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000;6:42–49. [PubMed] [Google Scholar]
- Cesa LC, Patury S, Komiyama T, Ahmad A, Zuiderweg ER, Gestwicki JE. Inhibitors of difficult protein-protein interactions identified by high-throughput screening of multiprotein complexes. ACS Chem Biol. 2013;8:1988–1997. doi: 10.1021/cb400356m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, et al. Chemical screens against a reconstituted multiprotein complex: myricetin blocks DnaJ regulation of DnaK through an allosteric mechanism. Chemistry & biology. 2011;18:210–221. doi: 10.1016/j.chembiol.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman MD, et al. Exploiting protein conformational change to optimize adenosine-derived inhibitors of HSP70. J Med Chem. 2016;59:4625–4636. doi: 10.1021/acs.jmedchem.5b02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connarn JN, et al. The molecular chaperone Hsp70 activates protein phosphatase 5 (PP5) by binding the tetratricopeptide repeat (TPR) domain. J Biol Chem. 2014;289:2908–2917. doi: 10.1074/jbc.M113.519421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig EA, Huang P. Cellular functions of Hsp70 chaperones. Protein folding handbook. Weinheim: Wiley-V C H Verlag Gmbh; 2005. [Google Scholar]
- Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science (New York, NY) 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- Demand J, Luders J, Hohfeld J. The carboxy-terminal domain of Hsc70 provides binding sites for a distinct set of chaperone cofactors. Mol Cell Biol. 1998;18:2023–2028. doi: 10.1128/MCB.18.4.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305–309. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- Flaherty KM, Deluca-Flaherty C, McKay DB. 3-dimensional structure of the ATPase fragment of a 70 k heat-shock cognate protein. Nature. 1990;346:623–628. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- Fontaine SN, et al. Isoform-selective genetic inhibition of constitutive cytosolic Hsp70 activity promotes client tau degradation using an altered Co-chaperone complement. J Biol Chem. 2015;290:13115–13127. doi: 10.1074/jbc.M115.637595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2000;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- Frydman J, Höhfeld J (1997) Chaperones get in touch: the Hip-Hop connection. Trends Biochem Sci. 22(3):87-92 [DOI] [PubMed]
- Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009;28:889–901. doi: 10.1038/emboj.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloubinoff P, De Los Rios P. The mechanism of Hsp70 chaperones: (entropic) pulling the models together. Trends Biochem Sci. 2007;32:372–380. doi: 10.1016/j.tibs.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Guidon PT, Jr, Hightower LE. Purification and initial characterization of the 71-kilodalton rat heat-shock protein and its cognate as fatty acid binding proteins. Biochemistry. 1986;25:3231–3239. doi: 10.1021/bi00359a023. [DOI] [PubMed] [Google Scholar]
- Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science (New York, NY) 1997;276:431–435. doi: 10.1126/science.276.5311.431. [DOI] [PubMed] [Google Scholar]
- Hohfeld J, Hartl FU. Post-translational protein import and folding. Curr Opin Cell Biol. 1994;6:499–509. doi: 10.1016/0955-0674(94)90068-X. [DOI] [PubMed] [Google Scholar]
- Hohfeld J, Minami Y, Hartl FU. Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell. 1995;83(4):589–598. doi: 10.1016/0092-8674(95)90099-3. [DOI] [PubMed] [Google Scholar]
- Hubbard J1, Erlichman C, Toft DO, Qin R, Stensgard BA, Felten S, Ten Eyck C, Batzel G, Ivy SP, Haluska P (2011) Phase I study of 17-allylamino-17 demethoxygeldanamycin, gemcitabine and/or cisplatin in patients with refractory solid tumors. Invest New Drugs 29(3):473-80. doi:10.1007/s10637-009-9381-y [DOI] [PMC free article] [PubMed]
- Jiang Y, Woronicz JD, Liu W, Goeddel DV. Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science (New York, NY) 1999;283:543–546. doi: 10.1126/science.283.5401.543. [DOI] [PubMed] [Google Scholar]
- Jiang JH, Ballinger CA, Wu YX, Dai Q, Cyr DM, Hohfeld J, Patterson C. CHIP is a U-box-dependent E3 ubiquitin ligase - identification of Hsc70 as a target for ubiquitylation. J Biol Chem. 2001;276:42938–42944. doi: 10.1074/jbc.M101968200. [DOI] [PubMed] [Google Scholar]
- Jiang J, Maes EG, Taylor AB, Wang L, Hinck AP, Lafer EM, Sousa R. Structural basis of J cochaperone binding and regulation of Hsp70. Mol Cell. 2007;28:422–433. doi: 10.1016/j.molcel.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinwal UK, Akoury E, Abisambra JF, O'Leary JC 3rd, Thompson AD, Blair LJ, Jin Y, Bacon J, Nordhues BA, Cockman M, Zhang J, Li P, Zhang B, Borysov S, Uversky VN, Biernat J, Mandelkow E, Gestwicki JE, Zweckstetter M, Dickey CA (2013) Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J 27(4):1450-9. doi:10.1096/fj.12-220889 [DOI] [PMC free article] [PubMed]
- Kabbage M, Dickman MB. The BAG proteins: a ubiquitous family of chaperone regulators. Cellular and molecular life sciences : CMLS. 2008;65:1390–1402. doi: 10.1007/s00018-008-7535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga H, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chap. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner R, Hofmann H, Barducci A, Wunderlich B, Nettels D, Schuler B. Single-molecule spectroscopy reveals chaperone-mediated expansion of substrate protein. Proc Natl Acad Sci U S A. 2014;111:13355–13360. doi: 10.1073/pnas.1407086111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kityk R, Kopp J, Sinning I, Mayer MP. Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol Cell. 2012;48:863–874. doi: 10.1016/j.molcel.2012.09.023. [DOI] [PubMed] [Google Scholar]
- Komarova EY, Meshalkina DA, Aksenov ND, Pchelin IM, Martynova E, Margulis BA, Guzhova IV. The discovery of Hsp70 domain with cell-penetrating activity. Cell Stress Chaperones. 2015;20:343–354. doi: 10.1007/s12192-014-0554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Zhang P, Murphy ME, Marmorstein R, George DL. Structural basis for the inhibition of HSP70 and DnaK chaperones by small-molecule targeting of a C-terminal allosteric pocket. ACS Chem Biol. 2014;9:2508–2516. doi: 10.1021/cb500236y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung SM, Senisterra G, Ritchie KP, Sadis SE, Lepock JR, Hightower LE. Thermal activation of the bovine Hsc70 molecular chaperone at physiological temperatures: physical evidence of a molecular thermometer. Cell Stress Chaperones. 1996;1:78–89. doi: 10.1379/1466-1268(1996)001<0078:TAOTBH>2.3.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung SM, Hightower LE (1997) A 16-kDa protein functions as a new regulatory protein for Hsc70 molecular chaperone and is identified as a member of the Nm23/nucleoside diphosphate kinase family. J Biol Chem. 272(5):2607-14 [DOI] [PubMed]
- Li Z, Hartl FU, Bracher A. Structure and function of hip, an attenuator of the Hsp70 chaperone cycle. Nat Struct Mol Biol. 2013;20:929–935. doi: 10.1038/nsmb.2608. [DOI] [PubMed] [Google Scholar]
- Li X, et al. Validation of the Hsp70-Bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol Cancer Ther. 2015 doi: 10.1158/1535-7163.MCT-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez V, Cauvi DM, Arispe N, De Maio A. Bacterial Hsp70 (DnaK) and mammalian Hsp70 interact differently with lipid membranes. Cell Stress Chaperones. 2016;21:609–616. doi: 10.1007/s12192-016-0685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow RF, Verdonk ML, Saini HK, Tickle IJ, Jhoti H. Detection of secondary binding sites in proteins using fragment screening. Proc Natl Acad Sci U S A. 2015;112:15910–15915. doi: 10.1073/pnas.1518946112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macazo FC, White RJ. Monitoring charge flux to quantify unusual ligand-induced ion channel activity for use in biological nanopore-based sensors. Anal Chem. 2014;86:5519–5525. doi: 10.1021/ac500832a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2435–2444. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B (2005) Regulation of Hsp70 chaperones by co-chaperones. Protein folding handbook
- Melero R, et al. Modulation of the chaperone DnaK allosterism by the nucleotide exchange factor GrpE. J Biol Chem. 2015;290:10083–10092. doi: 10.1074/jbc.M114.623371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova K, et al. Structural and biological interaction of hsc-70 protein with phosphatidylserine in endosomal microautophagy. J Biol Chem. 2016;291:18096–18106. doi: 10.1074/jbc.M116.736744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi A, Ikeda A, Mezaki M, Fukumori Y, Kanemori M. DnaJ-promoted binding of DnaK to multiple sites on sigma32 in the presence of ATP. J Bacteriol. 2014;196:1694–1703. doi: 10.1128/JB.01197-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylandsted J, Rohde M, Brand K, Bastholm L, Elling F, Jäättelä M (2000) Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc Natl Acad Sci U S A. 97(14):7871-6 [DOI] [PMC free article] [PubMed]
- Patury S, Miyata Y, Gestwicki JE. Pharmacological targeting of the Hsp70 chaperone. Curr Top Med Chem. 2009;9:1337–1351. doi: 10.2174/156802609789895674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellecchia M, Montgomery DL, Stevens SY, Vander Kooi CW, Feng HP, Gierasch LM, Zuiderweg ER. Structural insights into substrate binding by the molecular chaperone DnaK. Nat Struct Biol. 2000;7:298–303. doi: 10.1038/74062. [DOI] [PubMed] [Google Scholar]
- Polier S, Dragovic Z, Hartl FU, Bracher A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell. 2008;133:1068–1079. doi: 10.1016/j.cell.2008.05.022. [DOI] [PubMed] [Google Scholar]
- Qi R, et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat Struct Mol Biol. 2013;20:900–907. doi: 10.1038/nsmb.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radons J. The human HSP70 family of chaperones: where do we stand? Cell Stress Chaperones. 2016;21:379–404. doi: 10.1007/s12192-016-0676-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch JN, Zuiderweg ER, Gestwicki JE. Non-canonical interactions between heat shock cognate protein 70 (Hsc70) and Bcl2-associated Anthanogene (BAG) Co-chaperones are important for client release. J Biol Chem. 2016;291:19848–19857. doi: 10.1074/jbc.M116.742502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ER. Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol. 2011;411:614–632. doi: 10.1016/j.jmb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout AK, Strub MP, Piszczek G, Tjandra N. Structure of transmembrane domain of lysosome-associated membrane protein type 2a (LAMP-2A) reveals key features for substrate specificity in chaperone-mediated autophagy. J Biol Chem. 2014;289:35111–35123. doi: 10.1074/jbc.M114.609446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu R, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–139. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlecht R, et al. Functional analysis of Hsp70 inhibitors. PLoS One. 2013;8:e78443. doi: 10.1371/journal.pone.0078443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder H, Langer T, Hartl FU, Bukau B. Dnak, Dnaj and Grpe form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 1993;12:4137–4144. doi: 10.1002/j.1460-2075.1993.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuermann JP, et al. Structure of the Hsp110:Hsc70 nucleotide exchange machine. Mol Cell. 2008;31:232–243. doi: 10.1016/j.molcel.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shomura Y, et al. Regulation of Hsp70 function by HspBP1: structural analysis reveals an alternate mechanism for Hsp70 nucleotide exchange. Mol Cell. 2005;17:367–379. doi: 10.1016/j.molcel.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Smith MC, et al. The E3 ubiquitin ligase CHIP and the molecular chaperone Hsc70 form a dynamic, tethered complex. Biochemistry. 2013;52:5354–5364. doi: 10.1021/bi4009209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondermann H, Scheufler C, Schneider C, Hohfeld J, Hartl FU, Moarefi I. Structure of a bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science (New York, NY) 2001;291:1553–1557. doi: 10.1126/science.1057268. [DOI] [PubMed] [Google Scholar]
- Suh WC, Burkholder WF, Lu CZ, Zhao X, Gottesman ME, Gross CA. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc Natl Acad Sci U S A. 1998;95:15223–15228. doi: 10.1073/pnas.95.26.15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh WC, Lu CZ, Gross CA. Structural features required for the interaction of the Hsp70 molecular chaperone DnaK with its cochaperone DnaJ. J Biol Chem. 1999;274:30534–30539. doi: 10.1074/jbc.274.43.30534. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Noguchi S, Arakawa H, Tokida T, Hashimoto M, Satow Y. Peptide-binding sites as revealed by the crystal structures of the human Hsp40 Hdj1 C-terminal domain in complex with the octapeptide from human Hsp70. Biochemistry. 2010;49:8577–8584. doi: 10.1021/bi100876n. [DOI] [PubMed] [Google Scholar]
- Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyperski T, Pellecchia M, Wall D, Georgopoulos C, Wüthrich K (1994) NMR structure determination of the Escherichia coli DnaJ molecular chaperone: secondary structure and backbone fold of the N-terminal region (residues 2-108) containing the highly conserved J domain. Proc Natl Acad Sci USA 91:11343–11347 [DOI] [PMC free article] [PubMed]
- Takayama S, Xie Z, Reed JC. An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J Biol Chem. 1999;274:781–786. doi: 10.1074/jbc.274.2.781. [DOI] [PubMed] [Google Scholar]
- Takenaka IM, Leung SM, McAndrew SJ, Brown JP, Hightower LE. Hsc70-binding peptides selected from a phage display peptide library that resemble organellar targeting sequences. J Biol Chem. 1995;270:19839–19844. doi: 10.1074/jbc.270.34.19839. [DOI] [PubMed] [Google Scholar]
- Terlecky SR, Chiang HL, Olson TS, Dice JF. Protein and peptide binding and stimulation of in vitro lysosomal proteolysis by the 73-kDa heat shock cognate protein. J Biol Chem. 1992;267:9202–9209. [PubMed] [Google Scholar]
- Vega VL, et al. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. Journal of immunology (Baltimore, Md : 1950) 2008;180:4299–4307. doi: 10.4049/jimmunol.180.6.4299. [DOI] [PubMed] [Google Scholar]
- Wang H, Pang Y, Kurochkin AV, Hu W, Flynn GC, Zuiderweg ERP. The solution structure of the 21 kDa chaperone protein DnaK substrate binding domain: a preview of chaperone - protein interaction. Biochemistry. 1998;37:7929–7940. doi: 10.1021/bi9800855. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. Molecular mechanism of the negative regulation of Smad1/5 protein by carboxyl terminus of Hsc70-interacting protein (CHIP) J Biol Chem. 2011;286:15883–15894. doi: 10.1074/jbc.M110.201814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: folding, refolding, and degrading proteins. Science (New York, NY) 1999;286:1888–1893. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- Williamson DS, et al. Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. J Med Chem. 2009;52:1510–1513. doi: 10.1021/jm801627a. [DOI] [PubMed] [Google Scholar]
- Wisen S, et al. Binding of a small molecule at a protein-protein interface regulates the chaperone activity of hsp70-hsp40. ACS Chem Biol. 2010;5:611–622. doi: 10.1021/cb1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zuiderweg ER. The 70-kDa heat shock protein chaperone nucleotide-binding domain in solution unveiled as a molecular machine that can reorient its functional subdomains. Proc Natl Acad Sci U S A. 2004;101:10272–10277. doi: 10.1073/pnas.0401313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, et al. A bipartite interaction between Hsp70 and CHIP regulates ubiquitination of chaperoned client proteins. Structure (London, England: 1993) 2015;23:472–482. doi: 10.1016/j.str.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XT, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Structural analysis of substrate binding by the molecular chaperone DnaK. Science (New York, NY) 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuravleva A, Gierasch LM. Substrate-binding domain conformational dynamics mediate Hsp70 allostery. Proc Natl Acad Sci U S A. 2015;112:E2865–E2873. doi: 10.1073/pnas.1506692112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuiderweg ER, Bertelsen EB, Rousaki A, Mayer MP, Gestwicki JE, Ahmad A. Allostery in the Hsp70 chaperone proteins. Top Curr Chem. 2013;328:99–153. doi: 10.1007/128_2012_323. [DOI] [PMC free article] [PubMed] [Google Scholar]