

Abstract
Genome editing of human induced pluripotent stem cells (iPSCs) offers unprecedented opportunities for in vitro disease modeling and personalized cell replacement therapy. The introduction of Cas9-directed genome editing has expanded adoption of this approach. However, marker free genome editing using standard protocols remains inefficient, yielding desired targeted alleles at a rate of approximately 1–5%. We developed a protocol based on a doxycycline-inducible Cas9 transgene carried on a piggyBac transposon to enable robust and highly efficient Cas9-directed genome editing, so that a parental line can be expeditiously engineered to harbor many separate mutations. Treatment with doxycycline and transfection with gRNA, donor DNA, and piggyBac transposase resulted in efficient, targeted genome editing and concurrent scarless transgene excision. Using this approach, in seven weeks it is possible to efficiently obtain genome edited clones with minimal off-target mutagenesis and with indel mutation frequencies of 40–50% and homology-directed repair frequencies of 10–20%.
Keywords: Genome editing, Cas9, Stem Cells
Introduction
Human induced pluripotent stem cells (hiPSCs) offer an unprecedented opportunity for in vitro disease modeling and for personalized cell replacement therapy1. Applications of iPSCs have been greatly expanded by the advent of genome editing, in which the genomic sequence at a target site is altered by insertion or deletion (“indel”) mutations, or by introduction of precisely programmed (“knockin”) modifications2. Here we present a highly efficient and reproducible protocol to edit the genome of hiPSCs through the combined use of the CRISPR/Cas9 RNA-guided nuclease and piggyBac transposase3–5. This protocol is best suited to applications in which a common starting cell line is edited many different times to yield isogenic daughter cell lines that differ by the introduced mutations.
Genome editing relies on introduction of a double strand break at a target locus using “designer nucleases” that selectively target one site in the genome. The cell repairs the double strand break through either non-homologous end joining (NHEJ), creating indel mutations, or homology-directed repair (HDR), resulting in knockin modification near the nuclease cutting site. Potential nuclease platforms include zinc finger nucleases and transcription activator-like effector nucleases (TALENs)6. However, designing these nucleases is labor intensive and not readily multiplexed. More recently, the CRISPR/Cas9 nuclease has emerged as a powerful and malleable tool to introduce targeted double strand breaks6. Unlike zinc finger nucleases and TALENs, Cas9 specificity is determined by Watson-Crick base pairing between an engineered guide RNA (gRNA) and the target site7,8. As a result, Cas9 targeting is easily achieved by synthesizing the desired gRNA.
Although Cas9 efficiently directs target site cleavage, the efficiency of targeted genome modification was initially reported to be approximately 1–2% in human iPSCs7,9. At this efficiency, recovery of properly targeted clones without positive selection is labor intensive and inconsistent. We and others previously showed that low transfection efficiency of the relatively large Cas9 expression construct limits yield of targeted clones in pluripotent stem cells10. Procedures that select for Cas9 transfected cells, such as cell sorting for a fluorescent protein expressed from a co-transfected plasmid, increase the recovery of modified clones11. However, cell sorting is stressful for stem cells, exposes them to contamination risks, and can be cumbersome when performing modifications on multiple cell lines in parallel. Gonzalez et al. showed that knockin of inducible Cas9 into a safe harbour locus enhances genome editing efficiency12. However, this strategy consumed the AAVS safe harbour locus and the Cas9 transgene was not exciseable. We have refined this strategy and present here an optimized protocol to permit footprint-free, highly efficient and consistent genome modification in human iPSCs. This procedure can be used to develop isogenic cell lines that differ from each other by sequence variations introduced by genome editing, as we described for an iPSC-based model of Barth Syndrome3. The high efficiency of the procedure can also be used to simultaneously disrupt multiple genes or sequences that are present multiple times in the genome. For example, we used this strategy to simultaneously disrupt 62 copies of porcine endogenous retrovirus in a porcine cell line5. The inducibility of Cas9 in our system might also be exploited to permit temporally controlled gene inactivation in cells differentiated from iPSC, thereby potentially circumventing the need to establish stable mutant cell lines.
Development of the protocol: Dox-inducible Cas9 transgene encapsulated on a piggyBac transposon
We reasoned that Cas9 genome editing efficiency could be enhanced by generating a stable cell line that harbors an inducible Cas9 transgene encapsulated on a piggyBac transposon (Fig. 1a). Genome editing is performed by Cas9 induction accompanied by transfection of gRNA and homology-directed repair DNA donor template. The Cas9 transgene can be removed by transient transfection with piggyBac transposase.
Figure 1. Enhanced genome editing with Dox-inducible Cas9.

a. Overview. A piggy-Bac transposon encapsulating the reverse tet activator, a tet-activator responsive promoter driving humanized Cas9, and a puromycin resistance cassette were integrated into the genome of wild-type human iPSCs. Treatment with Dox and co-transfection with gRNA and donor DNA oligonucleotide efficiently yields mutant iPSC clones. The transposon is efficiently removed by transfection with an excision-only piggyBac transposase, either as a separate step or concurrently with the gRNA and donor oligo. b. PCR genotyping of PGP1 cells confirming hCas9-PB stable integration.
We generated a doxycycline-inducible, human codon optimized Cas9 (hCas9) construct contained within a piggyBac transposon7. The piggyBac transposon construct was stably introduced into the male PGP1-iPS cell line by co-transfecting it with a plasmid encoding the piggyBac transposase (Fig. 1b). The resulting PGP1-hCas9-PB stable cell line showed greater than 1000-fold induction of Cas9 by doxycycline (Dox) treatment (Fig. 1c).
Stable expression of hCas9 allowed us to efficiently target a human disease gene in iPSCs. We targeted Tafazzin (TAZ), a gene on the X chromosome that is mutated in Barth syndrome (BTHS), a mitochondrial cardiomyopathy13. We designed a gRNA and a homology directed repair (HDR) template to introduce a known BTHS mutation (c.517delG)14 into TAZ exon 6 and co-transfected them into PGP1-hCas9-PB with Dox treatment. The surveyor mutation detection assay suggested efficient TAZ gene modification with Dox treatment, and no detectable modification in the absence of Dox (Fig. 2a). High throughput sequencing of the targeted locus from pooled genomic DNA9 showed that 30% of cells had an indel near the engineered double strand break, while 50% had undergone HDR and harbored the sequence variant programmed by the HDR donor (Fig. 2b).
Figure 2. Efficiency of genome editing with Dox-inducible Cas9.

a. Surveyor mutation assay to detect genome modification. The indicated hiPSC lines were treated or not treated with Dox. Modification efficiency in genomic DNA at the TAZ locus was assessed using Surveyor nuclease followed by native gel separation of reaction products. Arrowheads indicate nuclease cleavage products. b. Deep sequencing analysis of the frequency of HDR or NHEJ genome modification at the TAZ locus. A PCR amplicon encompassing the TAZ gRNA target site was sequenced using a MiSeq Illumina sequencer at a minimum depth of 100,000 reads per amplicon. Amount of gRNA expression construct is shown in μg. c. After Dox-induced genome editing at the TAZ locus on the X chromosome of a male iPSC line, individual clones were picked and genotyped by Sanger sequencing. The pie chart displays the frequency of TAZ modification by HDR or NHEJ. d. Representative Sanger sequencing chromatograms, showing a clone that underwent HDR-mediated genomic modification (red arrow indicating one base HDR-programmed deletion) compared to a control.
We evaluated recovery of individual TAZ-modified clones. After transfection with gRNA and HDR donor, cells were plated at low density and treated with Dox. Colonies were then picked and genotyped by DNA sequencing. Out of 42 clones sequenced, 13 (31%) contained an indel and 16 (38%) contained the donor-programmed sequence variant (Fig. 2c–d). The efficiency of our strategy and protocol has been further tested in a different human embryonic stem cell line and at different loci, with HDR rates of ~20–35% and NHEJ rates of ~50% (Suppl. Fig. 1).
Development of the protocol: Excision of Dox-inducible Cas9 transgene by piggyBac transposase
Encapsulating the hCas9 transgene on a piggyBac transposon enabled its efficient excision. To illustrate this, we transiently transfected PGP1-hCas9-PB-TAZc.517delG with an excision competent, integration defective piggyBac expression plasmid15 and assessed hCas9 transgene excision by loss of puromycin resistance, encoded on the piggyBac transposon. PiggyBac transposase reduced the frequency of puromycin resistant clones, as assessed by crystal violet visualization of puromycin-resistant clones, demonstrating efficient transposon excision (Fig. 3a). Most individual clones recovered after transient piggyBac transposase expression were negative for the hCas9 transgene, as determined by PCR genotyping. For establishment of the PGP1-TAZc.517delG line lacking the hCas9 transgene, we genotyped 34 clones and 22 (64%) had undergone successful transgene removal (Fig. 3b). We have further streamlined the protocol by introducing piggyBac transposase into Dox-induced cells in the same transfection as gRNA and donor DNA. We found that co-transfection of the excision-only piggyBac mutant did not substantially reduce the yield of genome-edited clones, yet most of the recovered clones had still successfully undergone piggyBac transgene excision (Suppl. Fig. 2). Thus, including the excision-only piggyBac mutant into the transfection mix with gRNA and donor DNA permits efficient, single step genome editing and transgene excision.
Figure 3. Excision of Cas9-bearing transposon using piggyBac transposase.

a. PGP1-hCas9-PB-TAZc.821delG cells were transfected with piggyBac expression vector. Puromycin resistant clones, the clones that failed to undergo transposon excision, were visualized by crystal violet staining. b. PCR genotyping of individual clones with or without transfection of piggyBac expression vector. Representative examples of genotyping results of positive and negative clones are shown. Pie chart summarizes the genotyping results of 34 clones.
Development of the protocol: Quality control of recovered clones
We performed quality control on the genome-edited cell lines. PGP1e-TAZc.517delG cells had a normal karyotype (Suppl. Fig. 3a), expressed the pluripotency genes OCT4 and NANOG at levels comparable to the human ES cell line H7 (Suppl. Fig. 3b–c), and differentiated into all three germ layers in teratoma assays (Suppl. Fig. 3d–g). The cell lines differentiated efficiently into cardiomyocytes using a common directed differentiation protocol (Suppl. Fig. 3h).16 Indeed, we showed that the genome-edited PGP1e-TAZc.517delG iPSC line effectively recapitulates hallmarks of Barth syndrome (Suppl. Fig. 4).
A concern of Cas9-based genome-editing strategies has been off-target mutagenesis. Recently several studies used whole genome sequencing to demonstrate that Cas9 genome editing does not significantly impact the mutation burden of iPSCs4,17,18. We confirmed that our strategy is not substantially mutagenic by deep sequencing of 31 potential off-target sites (Fig. 4a). At each site we sequenced a minimum of 100,000 amplicons, which we previously showed yields a detection sensitivity of 0.07%4. At the 31 computationally predicted potential off-target sites, 30 sites had 3 nucleotide mismatches to the reference genome. Significant off-target activity was not detected at these sites. The final site, site 28, was designed to also have 3 nucleotide mismatches to the reference sequence, but as we reported previously4 a single nucleotide polymorphism in the PGP1 genome sequence eliminated one mismatch. This site with two mismatches was frequently mutated, highlighting the potential for SNPs to affect off-target mutagenesis at specific sites. Whole genome sequencing of six individual clones isolated using this protocol from separate genome editing experiments at three loci (TAZ, DNAJC19, JUP) showed that Cas9 does not induce frequent off-target mutagenesis. However, between 1–3 mutations that likely were linked to Cas9 were identified in each of the clones (Fig. 4b). With only one exception, we did not detect significant off-target activity at sites with 3 or more gRNA mismatches. The exception (site B, Fig. 4b) consisted of indel mutation at a genomic site on chromosome 14 that matched the gRNA at bases 8–19 (where position 1 is adjacent to the PAM). However, the “seed” nucleotides 1–7 did not match, and the PAM sequence, GGT, has no predicted activity19. An identical sequence also exists on chromosome 3, and this site also contained an indel mutation in one of the two clones (site C, Fig. 4b), suggesting that mutation at these sites was related to Cas9 activity. In addition, we detected clear off-target activity at a site (site A, Fig. 4b) with a 1 bp gRNA mismatch but a variant PAM sequence, CAG, with known partial activity19. Together these results suggest that Cas9 does not induce widespread genomic instability or rearrangements, but does induce off-target mutagenesis at rare sites that cannot be fully anticipated by current prediction rules.
Fig. 4. Analysis of Cas9 off-target activity.

a. Off-target activity at 31 computationally predicted potential off-target sites for TAZ gRNA. Cas9-PB iPSCs were treated with DOX + TAZ gRNA + donor. Off-target activity at 31 sites was analyzed by PCR amplification of the candidate sites from pooled genomic DNA followed by deep sequencing (greater than 100,000 sequences per site). The candidate sites had 3 mismatches from the gRNA, with the exception of site 28, which had a single nucleotide polymorphism that created a 2 bp mismatch as we previously reported.4 “Cas9 no PB” and “Cas9 with PB” refer to omission or inclusion of piggyback transposase expression plasmid in the transfection of gRNA and DNA donor. “TAZ HDR” and “TAZ NHEJ” indicate the frequency of on target homology directed repair and non-homologous end joining at the TAZ locus. b. Whole genome sequencing on 6 independently isolated clones derived from PGP1-Cas9-PB (listed under WGS sample) after targeting at 3 loci (TAZ, DNAJC19, and JUP). HDR or NHEJ indicates the type of mutation found at the target site. In each whole genome sequence we identified 10–15 indels. These were analyzed for homology to the gRNA, presence of proto-spacer adjacent motf (PAM) sequences (blue) and recurrence in multiple clones or genomic locations. Based on this analysis, most of the indels were unlikely to be related to the gRNA sequences and may have arisen spontaneously during clonal expansion from a single cell. Those that were related to the gRNA sequence are listed as “off-target indels” and named with a number if it was among the 31 predicted potential off target sites (site 28, panel a) or a letter if it was not among these top predicted off target sites. Red letters indicate differences from the gRNA, the sequence of which is shown next to the locus name. The three separate TAZ clones sequenced all shared indels at the same two sites, Site 28 and Site A. Interestingly site A differs from the gRNA target by only one nucleotide, but was not computationally predicted because of its atypical PAM (CAG, pink underline). The two DNAJC19 clones had different indels at the same site, Site B. An identical genomic sequence on a different chromosome (Site C) also contained in indel in one of these clones. However, Sites B and C had neither a functional PAM (orange underline) nor close homology to the gRNA withing the Cas9 seed sequence. ^ indicates a site that is listed for completeness but may not have arisen from Cas9 activity (single occurrence; lack of PAM; multiple gRNA mismatches). CNV analysis of the whole genome sequencing data found no significant copy number variation in these clones.
We also investigated the potential for piggyBac excision to leave residual genomic scars. Our analysis (Suppl. Data) confirmed scarless piggyBac excision in all clones that we examined.
Limitations
Off-target mutagenesis is one potential consequence of genome editing. Whole genome sequencing of several mutant iPSC lines generated through application of this protocol did not reveal a substantial burden of off-target mutation, although each of the 6 cell lines that underwent whole genome sequencing did acquire 10–15 off-target mutations each, of which 1–3 were attributable to gRNA-related Cas9 activity. Interestingly, the mutation sites were in some cases poorly predicted by current algorithms.
Presently whole genome sequencing of each genome edited clone is not feasible. Rather, we suggest that at least two independent clones be recovered for each genome editing experiment. This will help to control for potential confounding effects of off-target mutations, although our whole genome sequencing data shows that some off-target sites are recurrently mutated in independent clones. Therefore it is optimal to use two independent gRNAs. Rescue of mutant cells by transient cDNA expression is an alternative strategy to control for off-target mutation3.
This protocol requires establishing a parental cell line with the stably integrated transgene, which represents an extra step compared to methods based on transient transfection coupled with cell sorting-based enrichment of transfected cells. However, the benefit of our strategy is increased consistency and higher efficiency for recovering genome edited clones once the parental line is established. Therefore we use this strategy in situations where it is desirable to make many different genome-edited cell lines from a common parent, such as when generating a series of cell lines that are isogenic except for introduced mutations. On the other hand, this strategy would be cumbersome for editing many different cell lines, such as for correction of mutations in patient-derived iPSC lines.
Our method involves stable, random integration of Cas9 followed by scarless excision. It may be desirable in future iterations of this protocol to target a piggyBac-exciseable Cas9 transgene to a safe harbour locus, such as AAVS. This would eliminate problems with transgene copy number and reduce the chance of deleterious integration sites. Although our data suggest that piggyBac excision is robust and that the frequency of leaving a scar at the excision sites was below our detection limits (0/75 excision sites evaluated; Suppl. Data), a targeted integration strategy would nevertheless reduce the risk and facilitation validation of scar-free excision.
Experimental Design
gRNA and donor template design
Critical parameters that should be optimized for genome editing are the design of the gRNA and the HDR donor (Box 1). As we noted previously, SNPs not represented in the reference genome may lead to off-target sites not predicted by such design algorithms4.
Box 1. Design of gRNAs, HDR donor DNA oligonucleotide, and genotyping primers.
Several excellent resources are available that provide detailed instructions on designing genome editing reagents. Some of these are:
Graham and Root, Resources for the design of CRISPR gene editing experiments.23
Addgene e-book, CRISPR 101: A Desktop Resource (www.addgene.org)
Yang et al., CRISPR/Cas9-Directed Genome Editing of Cultured Cells24
gRNA expression construct
This procedure is described in greater detail in ref. 24. Although computational prediction of effective gRNAs has improved, there is still substantial variation in gRNA efficiencies. Therefore it can be more efficient to generate several different gRNA constructs in parallel and then use them in Steps 28–47. Subsequent steps could then focus on the most efficient gRNA.
Find genomic sites of the form 5′-N19NGG-3′ (see Fig. 5a) within ± 50 bp of your intended target site (optimally within ±10 bp). The sequence can be on either strand. Publicly available gRNA design tools such as http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design are useful to select the most active gRNAs with the lowest chance of off-target activity. Additional design tools are described in ref. 23. For HDR, give priority to gRNAs located closest to the intended target site.
- Replace the bold, red X in the following sequence with the best N19 target sequence to yield the complete 455 bp sequence which contains a U6 promoter and the gRNA. The U6 promoter prefers a G as the transcribed base, and we have incorporated this into the sequence as indicated by the green G:
TGTACAAAAA AGCAGGCTTT AAAGGAACCA ATTCAGTCGA CTGGATCCGG TACCAAGGTC GGGCAGGAAG AGGGCCTATT TCCCATGATT CCTTCATATT TGCATATACG ATACAAGGCT GTTAGAGAGA TAATTAGAAT TAATTTGACT GTAAACACAA AGATATTAGT ACAAAATACG TGACGTAGAA AGTAATAATT TCTTGGGTAG TTTGCAGTTT TAAAATTATG TTTTAAAATG GACTATCATA TGCTTACCGT AACTTGAAAG TATTTCGATT TCTTGGCTTT ATATATCTTG TGGAAAGGAC 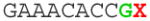
GTTTTAGAGC TAGAAATAGC AAGTTAAAAT AAGGCTAGTC CGTTATCAAC TTGAAAAAGT GGCACCGAGT CGGTGCTTTT TTTCTAGACC CAGCTTTCTT GTACAAAGTT GGCATTA Synthesize the gRNA as a double stranded DNA. For example, use gBlocks from Integrated DNA Technologies.
Clone the synthesized dsDNA into an empty backbone vector such as pCR-Blunt II-TOPO from Life Technologies. Validate the resulting clone by sequencing and prepare a midiprep. Resuspend the DNA in 10 mM Tris pH 8.0, 1 mM EDTA at a concentration of greater than 0.5 mg/ml.
HDR donor oligonucleotide (see Fig. 5b)
To generate donor DNA for oligo-mediated HDR, identify the positions targeted by the gRNA for cleavage and the desired modification site. Include the 45 nt on either side of this modification region as the homology arms of the donor DNA. 45 nt homology arms are sufficient for base changes or small insertions (< 40 nt). Larger insertions require larger homology arms10. If possible within the constraints of the desired experiment, introduce additional modifications in the HDR oligo that will alter the gRNA recognition sequence in the HDR donor oligo so that the oligo and the corrected genomic DNA will be refractory to gRNA-Cas9 cleavage. For instance, place silent mutations in the gRNA seed sequence, or disrupt the PAM sequence. It is also desirable to introduce or eliminate a restriction enzyme cleavage site to facilitate genotyping of candidate clones.
Synthesize the sequence as a single stranded oligonucleotide at 25 nmole scale. Desalted oligonucleotide is acceptable quality. Resuspend the oligonucleotide in ddH20 at a stock concentration of 10 μg/μl.
Genotyping primers (see Fig. 5b)
Surveyor assay primers: forward and reverse primers should be designed to produce an amplicon of approximately 700 bp, positioned so that predicted cleavage products will be easily resolved (e.g. 200 bp and 500 bp). Each primer should be at least 70 bp from the target modification site. Resuspend at 100 μM stock concentration, 10 μM working concentration. PCR using the primers should yield a robust single band.
Sanger sequencing/PCR primers: forward and reverse PCR primers should be designed to be approximately 150 bp from the target modification site. These will be used to both PCR amplify the target region and to perform Sanger sequencing of the PCR amplicon. Resuspend at 100 μM stock concentration, 10 μM working concentration.
Genotyping strategy
It is critical to design the genome editing strategy to facilitate genotyping of clones, since this step can otherwise be costly and rate-limiting. We have written this protocol using the most generic case, in which the genome editing strategy does not incorporate specific features that facilitate identification of the desired modification. As a result, Steps 37–47 use the Surveyor nuclease to determine if efficient modification occurred in pooled genomic DNA. Sanger sequencing is used in Steps 51–54 to genotype individual clones.
However, it is preferable to design features into the genome editing strategy that facilitate identification of clones with the desired genotype without relying on Sanger sequencing. In cases where a simple deletion is desired, using two gRNAs spaced ~100 bp apart may slightly increase mutation efficiency10 and will facilitate genotyping of individual clones. For HDR, the DNA donor could be designed to insert or remove a restriction endonuclease site (Box 1). If these features are incorporated into the genome editing strategy, then they can be substituted for Steps 37–47 and 51–54.
Controls
A negative control (omission of gRNA) should be included in the workflow. A useful positive control for newcomers to the protocol is to target the TAZ locus using the sequences outlined in Suppl. Table 1. TAZ, located on the X chromosome, is present in a single copy in XY iPSCs.
MATERIALS
REAGENTS
Pluripotent stem cell line. For example, PGP1 iPSC line (Coriell Institute) or CHB10 hESC line (Daley lab at Boston Children’s Hospital). CAUTION: The cell lines used should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
Human Stem Cell Nucleofector® Kit 1 (Lonza, cat. no. VPH-5012)
mTeSR1 complete kit for hES maintenance (StemCell Technologies, cat. no. 05850). Critical! avoid warming the mTeSR1 stock to 37 °C to prevent degradation of bFGF contained in the media.
mFreSR media (Stemcell Technologies, cat. no. 05854)
Knockout DMEM (Thermo Scientific, cat. no. 11330-032)
Dulbecco’s phosphate-buffered saline (DPBS; Life Technologies cat. no. 21600)
Corning Matrigel hESC-qualified Matrix (Corning cat. no. 354277)
Versene (Thermo Scientific, cat. no.15040-066)
Penicillin-streptomycin, 100× (Life Technologies, cat. no. 15140-163). CRITICAL: All cell culture media used in this protocol contains 1x Penicillin-streptomycin.
Puromycin dihydrochloride (Life Technologies, cat. no. A11138-03)
Rho-associated protein kinase (ROCK) inhibitor Y-27632 (Millipore, cat. no. SCM075)
Doxycycline (Dox; Sigma, cat. no. D3072-1ML). Critical! Dox is light sensitive. The sterile solution should be stored in a light-shielded container.
DNA loading dye, 6× (Fermentas/Thermo Scientific, cat. no. R0611)
Ethidium bromide (TianGen, cat. no. RT203). CAUTION: ethidium bromide is a mutagen. Handle with care and use personal protective equipment.
Proteinase K (Roche, cat no. 03115887001)
Tris pH 8.0 (Boston BioProducts cat. no. BBT-423)
EDTA (Boston BioProducts cat no. BM-150)
NaCl (Boston BioProducts cat. no. BM-244)
sarcosyl (Sigma cat. no. L7414)
Glycoblue (Life Technologies AM9515)
Plasmid Mini Kit (Life Technologies, cat. no. K2100-10)
Plasmid Midi Kit (Life Technologies, cat no. K2100-04)
TBE Gels, 4–20%, 1.0 mm, 15 well (Life Technologies, cat. no. EC62255BOX)
Agarose for gel electrophoresis (BioExpress GeneMate LE agarose, E-3120-125)
QIAquick PCR Purification Kit (50) (Qiagen, cat. no. 28104)
KAPA HIFI Hot Start ReadyMix PCR Kit (KAPA, cat. no. KK2601)
Quantitative PCR master mix (Affymetrix Veriquest probe and Sybr qPCR 2x master mix, 75650 and 75600)
Surveyor Mutation Detection kit (IDT, S25, 706025)
pPB-rtTA-hCas9-puro-PB plasmid: Dox-inducible hCas9 on a piggyBac transposon with a puromycin resistance marker. This plasmid is available from the authors upon request.
Excision only piggyBac Transposase expression vector (System Biosciences, cat. no. PB220PA-1)
Super piggyBac Transposase expression vector (System Biosciences, cat. no. PB210PA-1)
pCR-Blunt II-TOPO (Life Technologies, cat. no. K2800-02)
gRNA expression constructs and HDR donor oligonucleotide; see Box 1 and Fig. 5.
PCR primers for SURVEYOR analysis or sequencing; see Box 1 and Fig. 5.
-
hCas9 genotyping primers (amplicon size = 571bp; Integrated DNA Technologies)
hCas9-F: aggtggcgtaccatgaaaag
hCas9-R: gctttggtgatctccgtgtt
-
20x hCas9 qPCR probe assay (Bio-rad custom assay). The primers and probe are premixed in TE at a final concentration of 5 μM each.
hCas9-qPCR-F: aagaacggcctgtttggtaa
hCas9-qPCR-R: gttgaagcttggcatcttcg
hCas9-probe (FAM): cgccctgtcactcgggctgacc
20x EIF2C1 qPCR probe assay, Hex-labeled (Bio-rad, assay ID dHsaCP2500349).
Tissue culture plates (6-, and 24-well; Nunc, cat. no. 140675 and 142475)
Sterilized Pasteur pipettes (Fisher, cat. no. 13-678-20D)
Microcentrifuge tube (1.7 ml; Fisher Scientific, cat. no. 05-408-129)
Conical centrifuge tube, polypropylene, 15 ml (BD Falcon, cat. no. 352097)
Conical centrifuge tube, polypropylene, 50 ml (BD Falcon, cat. no. 352070)
Serological pipettes (5, 10 and 25 ml; Fisher Scientific, cat. no. 13-678-11D, 13-678-11E and 13-678-11)
Stericup filtration system (500 ml; Millipore, cat. no. SCGPU05RE)
Steriflip-HV filter unit (0.45 μm (50 ml); Millipore, cat. no. SE1M003M00)
Filter tip pipet tips (20 μl, 200 μl, 1 ml; ISC-BioExpress)
Fig. 5. Design of genome editing reagents.

A. gRNA recognition site. Red arrow indicates the gRNA cleavage site. Underline indicates the 12 nucleotide “seed” sequence that is generally considered most important for Cas9 targeting. The PAM (protospacer adjacent motif) sequence is required for Cas9 targeting. B. A gRNA is designed to introduce a double strand break (DSB, red lines) close to the desired modification site (blue). The HDR donor oligo is design to have homology arms that are at least 45 nt long on either side of the desired sequence modification (purple). Two additional sequence modifications are desirable if they can be achieved without impacting the ultimate experimental goal. First, introduce “silent” sequence variants that eliminate the gRNA recognition sequence after HDR (purple). Second, introduce or remove a restriction enzyme site so that HDR genomes can be identified by restriction digestion of PCR products (green). Genotyping PCR primers are designed flanking the targeted region. These genotyping primers should not overlap the homology arms.
Equipment
Humidified tissue culture incubator (37 °C, 5% CO2)
Tissue culture hood
Hemocytometer (Hausser Scientific, cat. no. 02-671-52)
Inverted phase-contrast microscope (Nikon, ECLIPSE TS100)
Nucleofector™ 2b Device (Lonza, cat. no. AAB-1001)
Bench top cell culture centrifuge (Thermo Scientific, cat. no. 004260F)
Microfuge (Eppendorf 5424)
Gel electrophoresis tanks, vertical and horizontal
Gel electrophoresis power supply
Gel documentation system
PCR thermocycler
Pipettors, 20 μl, 200 μl, 1 ml
37°C bacterial incubator and shaker
Coolcell LX cell freezing box (Biocision, cat. no. BCS-405)
Reagent Setup
Matrigel-coated dishes. Prepare as described in Box 2
Y-27632 stock solution (5 mM). Add 1 mg Y-27632 to 624 μl sterile dH20. Store at −20 °C. Aliquot to avoid repeated freeze/thaw. Stable for 6 months.
Y-27632 is freshly added to culture media with each use. Per 10 ml of mTeSR1, add 20 μl Y-27632 stock solution.
Culture media. Add Penicillin-streptomycin to all cell culture media to a final concentration of 1x. Media containing penicillin-streptomycin is stable for several months at 4 °C.
Lysis buffer: 10 mM Tris pH 8.0, 10 mM EDTA, 10 mM NaCl, 0.5% sarcosyl, 40 μg/mL proteinase K. Buffer without proteinase K can be stored at 4 °C for up to a year. Add Proteinase K freshly before use.
Box 2. Matrigel coating of dishes.
Preparing Matrigel Aliquots
Thaw 5 ml vial of Matrigel overnight at 4 °C.
Pre-chill the pipette tips and sterile 50 ml tubes to 4 °C.
Aliquot into each 50 ml tube the volume that will be diluted to 25 ml. This volume, typically between 300–500 μl, is lot specific and specified by the manufacturer.
Freeze at −20 °C. PAUSE POINT: frozen aliquots are stable for over one year.
Coating dishes with Matrigel
Thaw sufficient aliquots of Matrigel in 50 ml tubes at 4 °C overnight. Each will prepare 25 ml of diluted Matrigel.
Pre-chill pipet tips.
Add the 25 ml cold knockout DMEM to the matrigel in the 50 ml tubes. Gently mix. Do not vortex.
-
Add sufficient volume of diluted Matrigel to cover bottom of culture dishes, as follows:
TC Dish Format Volume of Diluted Matrigel 10 cm dish 8 ml/dish 6 cm dish 2 ml/dish 6 well dish 1 ml/well 24 well dish 0.25 ml/well To use the dish immediately, keep the dish at 37 °C for at least 20 min and not more than 2 hours. Alternatively, the plate can be stored at 4 °C for up to 1 week.
Prepare coated dish for use
To prepare a plate for use, put the plate into the tissue culture incubator for at least 20 min but no longer than 2 hours. Just before adding cells suspended in culture media, aspirate the diluted Matrigel without scratching the surface coating.
Procedure
Establish Dox-inducible iPS/ES line
Time required: 27 days to obtain candidate lines.
-
1
Grow low passage iPSCs (between passages 10 to 40) to 70% confluence in a 6 well plate. iPSCs should be grown in dedicated tissue culture incubators (37 °C, 5% CO2, humidified). CRITICAL STEP: It is crucial that cell culture environments are free of mold, bacteria, and mycoplasma.
-
2
Prepare a matrigel-coated 6 well dish (see Box 2) and place it in the tissue culture incubator.
-
3
Lift cells as directed in Box 3. Critical step! To achieve highly efficient transfection, the cells have to be dissociated into single cells.
-
4 Coun
cells with a hemocytometer. Adjust cell density with PBS to 1 million cells per ml.
-
5
Prepare nucleofection solution: combine 82 μl of human stem cell nucleofector solution and 18 μl supplement 1 in a sterile 1.5 ml microfuge tube. Mix well.
-
6
Incubate solution at 37°C for 5 min.
-
7
Add 1 ml cell suspension to a sterile 1.5 ml eppendorf tube and centrifuge at room temperature (~20–23 °C) at 300 x g for 5 min in a bench top centrifuge.
-
8
Discard supernatant and resuspend cells in 100 μl of human stem cell nucleofector solution from Step 6.
-
9
Add to the cell suspension 1 μg Super piggyBac plasmid and 5 μg pPB-rtTA-hCas9-puro-PB plasmid.
-
10
Mix cells and DNA by gentle swirling. Transfer cells to a nucleofector cuvette using a 1 ml pipette tip. Put the cuvette into the nucleofector. Select Program B-016 and nucleofect cells by pressing button X.
-
11
Add 500 μl mTeSR1 medium with 10 μM Y-27632 to the nucleofection cuvette and aspirate the nucleofected cells from the cuvette using the provided plastic pipette.
-
12
Transfer cells drop-wise into one well of the matrigel-coated 6 well dish from Step 2 and incubate the cells at 37°C overnight.
-
13
The next day (Day 1 after nucleofection) change the medium to mTeSR1 and add puromyocin at final concentration at 1 μg/ml. Thereafter, change media with puromycin daily. Non-resistant cells will die, peaking at Day 4. When distinct colonies are visible to the naked eye (usually about Day 7), proceed to the next step.
TROUBLESHOOTING
-
14
Prepare a 10 cm matrigel-coated dish (Box 2) and place it in the tissue culture incubator.
-
15
Lift cells as described in Box 3.
-
16
Count cells with a hemocytometer. Plate 250 cells in the matrigel-coated 10 cm dish from Step 14 with 5 ml mTeSR1 media with 10 uM Y-27632.
TROUBLESHOOTING
-
17
Culture cells until individual clonal outgrowths are visible (about 12 days), changing media daily.
-
18
Pick 5–10 colonies and expand as directed in Box 4. The procedure described in Box 4 will yield iPSC clones in 24 well dishes, used in Step 19, and matching genomic DNA samples, analyzed in Steps 20–25 below.
-
19
When the iPSC clones growing in 24 well dishes reach 70% confluence, lift cells as described in Box 3. Freeze a sample of each iPSC-hCas9-PB clone as directed in Box 5 pending genotyping results, which are obtained by analyzing each clone’s genomic DNA sample as directed in Steps 20–25. PAUSE POINT: cells can remain frozen for several weeks while awaiting genotyping results.
Box 3. Lifting Cells.
The procedure below is written for one well of a six well dish. For different sized dishes, scale by the number of wells. For example, for a 24 well dish, use 1/4 the indicated volumes for each well.
Pretreat the culture by adding ROCK inhibitor Y-27632 to mTeSR1 media to 10 μM. Incubate overnight.
Aspirate cell culture media. Gently rinse the cells with 2 ml DPBS for each well of the six well plate.
Aspirate the DPBS, add 2 ml/well of versene, and put the culture back into the 37°C incubator until the cells become rounded up and loosely adherent, but not detached. Critical step! This requires 3–7 min and should be determined by observing under the microscope every 2–3 min. Sufficient time is needed for complete dissociation to single cells, but excessive time will lead to cell death.
Gently aspirate the versene. Add 1 ml mTeSR1 with Y-27632 (10 μM) and dislodge the cells by gently flowing mTeSR1 over them with a P1000 pipet. Critical step! The cells should be dislodged easily by the flow of medium over the cells. The cells should not require scraping to dislodge; if they do then rinse with DPBS, add more versene, and increase the time of incubation. CRITICAL STEP: Total versene incubation time should not exceed 15 min.
Collect the dislodged cells, gently triturate them into a single-cell suspension.
Box 4. Picking and expanding individual clonal outgrowths.
Treat clones that will be picked by changing media to mTeSR1 containing ROCK inhibitor Y-27632 at 10 μM. Incubate overnight.
Prepare a matrigel-coated 24 well plate (see Box 2) by putting it in the tissue culture incubator for 20 min – 2 hrs. Then aspirate the solution and replace with mTeSR1 with 10 μM Y-27632, 500 μl per well.
Put the dish containing the individual clones into the culture hood and pick colonies with a P10 pipette with filter tips and set at 10 μl. Pick up a clone by scratching it into small pieces. Transfer each clone to a separate well of the matrigel-coated 24 well plate from the previous Step. Use a different filter tip for each clone.
Change media daily with mTeSR1 without Y-27632.
After 4–5 days the cells within each well should reach about 40% confluence and will be ready to split. Culture cells overnight in mTeSR1 with 10 μM Y-27632, 500 μl per well.
Prepare a fresh 24-well plate coated with matrigel (Box 2) by placing it in the incubator for 20 min – 2 hr. Aspirate solution and replace with 125 μl mTeSR1.
Using the dish containing the cells from Step 5 of this Box, repeat Steps 2–5 of Box 3 (using 1/4 volumes).
Transfer 125 μl cell suspension into a well of the matrigel-coated 24-well plate from Step 6 of this Box. Add 125 μl mTeSR1 with Y-27632 (10 μM).
Transfer 125 μl cell suspension into a 1.5 ml eppendorf tube for genomic DNA extraction as described in Box 6.
Box 5. Freezing and thawing iPSCs.
These procedures for freezing and thawing iPSCs are written for a well of a 24 well plate. For different size plates, adjust volumes accordingly.
Freezing
-
1
When the culture reaches roughly 60–70% confluence, harvest cells following Steps 2–5 of Box 3, using 1/4 volumes to adjust for use of a 24 well plate, and using mFreSR medium to dislodge and resuspend cells.
-
2
Quantitate cell density using a hemocytometer and adjust to 0.5 million cells per ml.
-
3
Using a 1 ml pipet, transfer the 0.25 ml cell containing mFreSR medium into a labeled cryovial.
-
8
Put the vials into a Coolcell LX cell freezing box and place at −80 °C overnight. For long term storage, transfer the vials to a liquid nitrogen tank. Cells can be stored in this state for many years.
Thawing
One vial of cells cryopreserved as described above should be thawed into 1 – 2 wells of a 6-well plate. Cells should be thawed and plated as quickly as possible.
Prepare a 6 well dish coated with Matrigel (see Box 2).
Thaw the vial of cryopreserved cells in a 37 °C water bath. Gently and continuously agitate the cryovial. The cells should thaw within 1–2 minutes.
Transfer the contents of the cryovial into a 15 mL tube with 1 ml pipette.
Put 6 mL warm mTeSR1 into to the 15 mL tube.
Centrifuge cells at 300 x g for 5 minutes at room temperature.
Remove the supernatant and resuspend the cell pellet in 4 ml mTeSR1 with 10 μM Y-27632.
Aspirate the media from the Matrigel-coated dish from Step 1. CRITICAL STEP: Do not scratch the surface coating.
Transfer 2 ml of the cell suspension into each of two wells of the pre-coated 6-well.
Put the plate in a 37°C incubator. Distribute the cells evenly by gently rocking the plate front-to-back and side-to-side. The plate should not be disturbed for 24 hours.
Change the mTeSR1 medium daily until the cells are ready for splitting (typically in 4–5 days).
Quality Control of Dox-inducible iPSC/ESC clones
Time required: 2 days for genotyping. An additional 3–4 weeks will be needed for additional quality control of selected iPSC/ESC lines (Step 26).
-
20
To confirm transgene integration, prepare PCR genotyping reaction to analyze the genomic DNA samples from Step 18 as follows:
Genomic DNA from Step 18 50 ng hCas9F primer, 10 μM 1 μl hCas9R primer, 10 μM 1 μl ddH20 to 12.5 μl 2X KAPA HIFI Hot Start ReadyMix 12.5 μl
Final Volume 25 μl -
21
Run PCR program as follows:
1 95 °C 3 min 2 98 °C 20 sec 3 60 °C 15 sec 4 72 °C 15 sec Go to 2, 30 times 5 72 °C 1 min -
22
Add 4 μl DNA Loading Dye to the 25 μl PCR reaction and load on a 2% (w/v) agarose gel. Run at 7 V/cm for 30 min and visualize by staining with ethidium bromide (0.5 μg/ml). A 571 bp product in the puromycin-selected cells that is absent from the untransfected cells indicates successful stable transgene integration (see example in Fig. 3b).
-
23
Determine transgene copy number using quantitative PCR. The hCas9 amplicon is detected with FAM-labeled probe, and the genomic reference amplicon (EIF2C1) is detected with the HEX-labeled probe. Assemble the following reaction in triplicate technical replicates:
genomic DNA from Step 18 10 ng 20x hCas9 qPCR probe assay 1 μl 20x EIF2C1 qPCR probe assay 1 μl ddH20 to 10 μl 2X Taqman Real Time PCR Mix 10 μl
Final volume 20 μl -
24
Perform quantitative PCR, detecting both FAM and HEX, using the PCR program:
1 95 °C 10 min 2 95 °C 30 sec 3 60 °C 60 sec Go to 2, 40 times -
25
Calculate copy number per genome using the formula: 2 * 2^(CtEIF2C1-CtCas9). CRITICAL STEP: When interpreting the data, keep in mind that in our hands this assay underestimated by about 3-fold the actual transgene copy number, determined by whole genome sequencing and confirmed by targeted PCR amplification of each transgene. If digital droplet PCR is available, it may estimate transgene copy number slightly more accurately using the same qPCR probe assays (see Suppl. Fig. 3).
-
26
Using the genotyping results from Step 22 and the transgene copy number from Step 25, select those cell lines positive for the transgene and containing a low number of transgene copies (e.g. 1–5 copies) for further characterization. Thaw the chosen cell lines from Step 19 as directed in Box 5, and perform quality control for expression of pluripotency markers (qRTPCR and immunostaining for Oct4, Nanog, Sox2)20. Confirm normal karyotype by performing G-banded karyotyping through a cytogenetics testing service. Using qRTPCR22 and the hCas9 qPCR probe assay, confirm robust Cas9 upregulation by DOX (2 μg/ml). Confirm Cas9 exciseability as detailed in Steps 55–62.
-
27
Expand and freeze (Box 5) the best 2–3 lines for future use.
Genome editing using the stable iPSC-hCas9-PB cell line
Time required: 5 days
-
28
Prepare one of the iPSC-hCas9-PB stable cell lines from Step 27 for nucleofection as described in Steps 1–8.
-
29
To the cell suspension, add the following DNAs in a maximum volume of less than 20 μl:
gRNA expression construct 4–20 μg donor DNA 4 μg excision-only piggyBac transposase plasmid (optional)** 2 μg CRITICAL STEP We found that 4 μg gRNA expression construct is the minimal required for high (70–80%) transfection efficiency and over 20 μg DNA will cause excessive cell death. Generally 10 μg works best but increasing or decreasing the amount can improve efficiency.
CRITICAL STEP Addition of excision-only PB transposase plasmid is optional - for one step genome editing with concurrent removal of the hCas9-PB transgene, add 2 μg excision-only PB expression plasmid to the transfection mix. If further genome editing might be performed, then omit.
-
30
Repeat Steps 10–12, adding Dox at a final concentration of 2 μg/ml to the cells in the 6-well plate before overnight incubation.
-
31
The next day change the medium to mTeSR1 with Dox (2 μg/ml) but without Y-27632. Change daily with mTeSR1 containing Dox (2 μg/ml). Cells should not exceed 70% confluence; if they do, then lift cells (Box 3), replate, and continue Dox treatment.
-
32
On the fourth day after nucleofection, change to mTeSR1 containing 10 μM Y-27632 but without Dox. CRITICAL STEP Subsequent culture steps use media without Dox.
-
33
On the fifth day after nucleofection, prepare three matrigel-coated 10 cm dishes (Box 2).
-
34
Lift cells from Step 32 (as described in Box 3). Adjust cell density with DPBS to 100,000 cells/ml.
TROUBLESHOOTING
-
35
Seed the cells onto the matrigel-coated 10 cm dishes from Step 33, each containing 5 ml mTeSR1 medium with 10 μM Y-27632. Seed cells at densities of 5K,10K and 40K per dish. For culture of these cells, continue to Step 48. CRITICAL STEP: Be sure to distribute the cells evenly to avoid overlap of clones.
-
36
Extract genomic DNA from the remaining cells from Step 34, as directed in Box 6, and proceed to the Surveyor mutation detection assay (Step 37 below).
Box 6. Preparing Genomic DNA.
The protocol below is for preparing genomic DNA from a 6-well dish. The volumes can be scaled proportionately for other dish sizes. For example, use 1/4 the volumes for a well of a 24 well dish.
Add 250 μl lysis buffer to a cell pellet or to a well of a 6-well dish.
Incubate at 55 °C overnight.
Precipitate DNA by adding 250 μl isopropanol
Centrifuge for 30 minutes in a microfuge at maximum speed (20000 x g, room temperature). Aspirate supernatant. Wash with 1 ml 70% ethanol and repellet by centrifuging for 5 minutes. Aspirate supernatant and air dry for 5 min. Critical Step! The DNA pellet may be small and translucent. Be careful not to disturb the pellet when aspirating the supernatant. If DNA recovery is problematic, a coprecipitant such as Glycoblue may increase yield and help to visualize the pellet.
Resuspend DNA with 100–200 μl dH2O. Vortex vigorously. Allow several hours for the DNA to be resolubilized.
Surveyor Mutation Detection
Time required: 1 day
-
37
To prepare PCR amplicons of the target area using Surveyor mutation genotyping primers (see Reagent Setup), assemble the following reaction mix:
genomic DNA from Step 36 50–100 ng Forward primer, 10 μM 1 μl Reverse primer, 10 μM 1 μl ddH20 to 10 μl 2X Kapa Hifi Hotstart ReadyMix 10 μl
Final Volume 20 μl -
38
Run the following PCR program:
1 95 °C 3 min 2 98 °C 20 sec 3 60 °C 15 sec 4 72 °C 15 sec Go to 2, 30 times 5 72 °C 1 min -
39
Purify PCR products using the QiaQuick PCR purification kit, according to the manufacturer’s instructions.
-
40
Make 400 ng purified PCR product to 40 μl in 1x Taq PCR Buffer (GenScript).
-
41
Melt and anneal PCR products by placing in a thermocycler running the following program:
Step Temp Time 1 95 °C 10 min 2 95 °C to 85°C @ −2 °C/s 3 85 °C to 75 °c @ −0.3 °C/s 4 75 °C 1 min 5 75 °C to 65 °C @ −0.3 °C/s 6 65 °C 1 min 7 65 °C to 55 °C @ −0.3 C/s 8 55 °C 1 min 9 55 °C to 45 °C @ −0.3 C/s 10 45 °C 1 min 11 45 °C to 35 °C @ −0.3 C/s 12 35 °C 1 min 13 35 °C to 25 °C @ −0.3 C/s 14 25 °C 1 min -
42
Treat with Surveyor nuclease by adding:
0.15 M MgCl2 4 μl Surveyor Enhancer S 1 μl Surveyor Nuclease S 2 μl -
43
Mix well and incubate at 42 °C for 60 min.
-
44
Add 1/10 volume stop solution to terminate the reaction and 1/6 volume DNA Loading Dye.
-
45
Analyze 10 μl of surveyor nuclease digestion product by electrophoresis through the vertical 4–20% native polyacrylamide gel at 200 V for ~60 minutes.
-
46
Stain the gel with 0.5 μg/ml ethidium bromide in 1X TBE for 10 min. Wash the gel in water for 10 min.
-
47
Image the gel with a UV transilluminator. Detectable Surveyor nuclease cleavage fragments typically indicate Cas9 cutting efficiency that is sufficient to proceed with screening of individual clones. See Fig. 2a and Suppl. Fig. 1c for examples. Fig. 6 includes an example of inefficient gRNA cleavage. If detectable Surveyor nuclease cleavage fragments are visible, then one can proceed to analyzing single clonal outgrowths (see Step 48, below). If gRNA cleavage was inefficient, terminate the experiment and see Troubleshooting.
TROUBLESHOOTING
Fig. 6. Surveyor nuclease assay.

Examples of good, moderate, and poor gRNA-directed Cas9 cleavage followed by NHEJ, as determined by the surveyor assay. Arrows indicate surveyor nuclease cleavage products.
Picking and analyzing individual clonal outgrowths
Time required: 20 days
-
48
For the cells plated in Step 35, the day after plating change medium to mTeSR1 without Y-27632. Change media daily until individual clonal outgrowths are visible to the naked eye (approximately 12 days). Do not allow clones to become too big or to adhere to each other.
-
49
Pick and expand individual clones as directed in Box 4. The procedure described in Box 4 will yield iPSC clones in 24 well dishes, used in Step 50, and matching genomic DNA samples, analyzed in Steps 51–54 below.
-
50
When the iPSC clones growing in 24 well dishes reach 70% confluence, lift cells as described in Box 3. Freeze a sample of each clone as directed in Box 5 pending genotyping results, , which are obtained by analyzing each clone’s genomic DNA sample as directed in Steps 51–54. PAUSE POINT: cells can remain frozen for several weeks while awaiting genotyping results.
Genotype clones by Sanger sequencing
Time required: 3 days.
-
51
To evaluate individual clones for Cas9 genome editing, PCR amplify genomic DNA for each clone obtained in Step 50. Design primers so that putative changes are in the center of a 200–300 bp amplicon. The PCR primers should efficiently amplify genomic DNA to yield a single band. PCR reaction setup and PCR program are described in Steps 37–38.
-
52
Submit the PCR product for Sanger sequencing with appropriate sample processing for crude PCR products.
-
53
Analyze the Sanger sequencing chromatograms to detect desired sequence changes. Software such as PolyPeakParser22 may help to deconvolute overlapping, out of phase chromatograms, which frequently result from indel mutation on one or both alleles. It may be necessary to TA-clone PCR amplicons and then sequence individual bacterial colonies to definitively determine the sequence of each individual allele.
TROUBLESHOOTING
-
54
If piggyBac transposase was included in the transfection mix in Step 28, then also perform genotyping for the hCas9 transgene as described in Steps 20–22.
PiggyBac transposon removal
Time required: 20 days.
CRITICAL: If multiple genome editing steps are anticipated, it may be desirable to leave the piggyBac transposon in place, and then remove it at a final independent step.
-
55
Expand iPSC clones from Step 50 with desired genotype and still containing the hCas9 transgene as described in Box 4.
-
56
On the day prior to transfection, prepare a matrigel-coated 10 cm dish (Box 2).
-
57
On the day of transfection, remove the solution from the matrigel-coated 10 cm dish. Replace with 6 ml mTeSR1 medium with 10 μM Y-27632.
-
58
Prepare the iPSC clone from Step 55 for nucleofection, following Steps 1–8.
-
59
To the cell suspension, add 2 μg excision-only piggyBac transposase plasmid.
-
60
Repeat Steps 10 and 11.
-
61
Of the 500 μl suspension of nucleofected cells from Step 60, plate 30 μl onto the matrigel coated 10 cm dish from Step 56. Distribute cells evenly over the dish. Incubate the cells at 37°C overnight.
TROUBLESHOOTING
-
62
The next day change the medium to mTeSR1 without Y-27632 and change the medium daily.
-
63
Once the clones are big enough to see with the naked eye (about 12 days), pick 10 and expand (Box 4). The procedure in Box 4 will yield iPSC clones in a 24 well dish, used Step 64, and matching genomic DNA samples, analyzed in Step 65.
-
64
When the iPSC clones growing in 24 well dishes reach 70% confluence, lift cells as described in Box 3. Freeze a sample of each clone as directed in Box 5 pending genotyping results, which are obtained by analyzing genomic DNA from each clone from step 63 as directed in Step 65.
-
65
Genotype the genomic DNA of the clones from Step 63 for the hCas9 transgene as described in Steps 20–22.
TROUBLESHOOTING
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting Table.
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| Steps 16, 34, and 61 | Low iPSC number after nucleofection |
|
|
| Step 13 | No puromycin-resistant iPSC clones |
|
|
| Step 47 | No surveyor nuclease cleavage product (see Fig. 5 for example). |
|
|
| Step 53 | No HDR mutants obtained |
|
|
| Steps 65 | Low efficiency of piggyBac transposon excision |
|
|
Timing
Obtain Dox-inducible iPSC/ES clones (27 days)
Steps 3–12. Nucleofection with dox-inducible hCas9 plasmid and piggyBac. 2 hours.
Step 13. Outgrowth of puro-resistant cells. 7 days.
Steps 14–17. Clonal expansion of puro-resistant cells. 12 days.
Step 18. Pick individual puro-resistant clones. 2 hours.
Step 18. Outgrowth of puro-resistant clones, expand, split and prepare genomic DNA. 5 days.
Step 19. Grow clones to 70% confluence and freeze. 3 days.
Quality Control of Dox-inducible iPSC/ES clones
Steps 20–22. Confirm presence of hCas9 transgene. 1 day.
Steps 23–25. hCas9 transgene copy number. 1 day.
-
Step 26. Additional QC (can be done in parallel with each other):
cDNA preparation from cells. 4 hours.
qRTPCR for pluripotency markers. 4 hours.
qRTPCR to determine basal and induced hCas9 levels. 4 hours.
immunostaining and microscopy to visualize pluripotency proteins. 1 day.
send out G-banded karyotyping, 2 weeks.
confirm Cas9 exciseability. 16 days. Can be done in parallel with karyotyping.
Genome editing using stable iPSC-hCas9-PB line
Steps 28–30. Nucleofection with gRNA and HDR donor DNAs. 2 hours.
Steps 31–33. Outgrowth of nucleofected cells. 5 days.
Step 34–35. Lift and replate cells for clonal outgrowth. 1 hour.
Steps 36–47. Confirm efficient genomic DNA modification. 1 day.
Step 48. Wait for clonal outgrowths to grow up. 12 days.
Step 49. Pick individual clones into 24 well dishes, expand, split and prepare genomic DNA. 5 days.
Step 50. Grow clones to 70% confluence and freeze individual clones, 3 days.
Genotyping candidate clones
Steps 51–52. PCR amplify genomic DNA and submit amplicons for Sanger sequencing. 2 days.
Step 53. Analyze sequencing results to identify candidate clones. 1 day.
Step 54. PCR analysis of hCas9 excision (if performing one step editing with concurrent hCas9 excision). 4 hours, in parallel with Steps 51–53.
Piggybac transposon removal
Steps 55–60. Nucleofection with excision-only piggyBac transposase plasmid. 2 hours.
Steps 61–62. Outgrowth of single cell-derived clones. 12 days.
Step 63. Pick individual clones into 24 well dishes, expand, split, and prepare genomic DNA. 5 days.
Step 64. Grow clones to 70% confluence and freeze individual clones, 3 days.
Step 65. PCR analysis of hCas9 excision. 4 hours.
Anticipated Results
The first stage of this protocol is to integrate the piggyBac transposon containing the Dox-inducible Cas9 expression cassette into iPSCs, to yield iPSC-Cas9-PB. This step only needs to be done once to obtain a common parental line that is conducive for genome editing.
From iPSC-Cas9-PB cells, genome editing is highly efficient. As shown in Fig. 2 and Suppl. Fig. 1, we routinely obtain clones in which 19% have undergone HDR and 54% have undergone NHEJ. The surveyor nuclease assay is a useful predictor of efficient genome modification: if a robust nuclease cleavage product is observed, then analysis of 48–96 clones will likely yield the desired genome edited product. On the other hand, weak or absent nuclease cleavage product (Fig. 6) suggests that the experiment should be halted and steps should be taken to troubleshoot the genome editing efficiency (see Supplementary Table 1).
Supplementary Material
Acknowledgments
G.W. was supported by T32HL007572 and a Progenitor Cell Biology Consortium Jump Start award. W.T.P was funded by U01 HL100401 and R01 HL128694 and the Barth Syndrome Foundation. LY, DG and XRV were funded by NIH Centers of Excellence in Genomic Sciences grant P50 HG005550
Footnotes
Author Contributions
G.W. and L.Y. developed the protocol. L.Y. and D.G. analyzed sequencing data. G.W. and W.T.P. wrote the manuscript with input from the other authors. W.T.P. and G.M.C. supervised the project. L.Y.Y. performed experiments. D.Z. contributed to iPSC culture and differentiation. Y.H. contributed genome editing data. X.R.V. contributed to development of the Dox-inducible Cas9 expression plasmid.
Competing Financial Interests
L.Y. and G.M.C. are inventors on a patent filed by Harvard University on Cas9 genome editing using the technology described in this protocol.
Editorial Summary
This protocol uses a doxycycline-inducible Cas9 transgene carried on a piggyBac transposon for robust, highly efficient and scarless genome editing of human iPSCs, enabling a parental line to be engineered to harbor many separate mutations.
Tweet
Highly efficient, scarless hiPSC genome editing using doxycycline-inducible Cas9 transgene on a piggyBac transposon
References
- 1.Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim HS, Bernitz JM, Lee DF, Lemischka IR. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev. 2014;23:2673–2686. doi: 10.1089/scd.2014.0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang G, McCain ML, Yang L, He A, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616–623. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang L, Grishin D, Wang G, Aach J, et al. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat Commun. 2014;5:5507. doi: 10.1038/ncomms6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, Güell M, Niu D, George H, et al. Genome-wide inactivation of porcine endogenous retroviruses (PERVs) Science. 2015;350:1101–1104. doi: 10.1126/science.aad1191. [DOI] [PubMed] [Google Scholar]
- 6.Gupta RM, Musunuru K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J Clin Invest. 2014;124:4154–4161. doi: 10.1172/JCI72992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mali P, Aach J, Stranges PB, Esvelt KM, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cong L, Ran FA, Cox D, Lin S, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Guell M, Byrne S, Yang JL, et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013;41:9049–9061. doi: 10.1093/nar/gkt555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li K, Wang G, Andersen T, Zhou P, Pu WT. Optimization of Genome Engineering Approaches with the CRISPR/Cas9 System. PLoS One. 2014;9:e105779. doi: 10.1371/journal.pone.0105779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding Q, Regan SN, Xia Y, Oostrom LA, et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12:393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.González F, Zhu Z, Shi ZD, Lelli K, et al. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bione S, D’Adamo P, Maestrini E, Gedeon AK, et al. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet. 1996;12:385–389. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez IL. Barth syndrome: TAZ gene mutations, mRNAs, and evolution. Am J Med Genet A. 2005;134:409–414. doi: 10.1002/ajmg.a.30661. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Burnight ER, Cooney AL, Malani N, et al. piggyBac transposase tools for genome engineering. Proceedings of the National Academy of Sciences. 2013;110:E2279–E2287. doi: 10.1073/pnas.1305987110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiba Y, Fernandes S, Zhu WZ, Filice D, et al. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012;489:322–325. doi: 10.1038/nature11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veres A, Gosis BS, Ding Q, Collins R, et al. Low Incidence of Off-Target Mutations in Individual CRISPR-Cas9 and TALEN Targeted Human Stem Cell Clones Detected by Whole-Genome Sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki K, Yu C, Qu J, Li M, et al. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell. 2014;15:31–36. doi: 10.1016/j.stem.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohnuki M, Takahashi K, Yamanaka S. Generation and characterization of human induced pluripotent stem cells. Current protocols in stem cell biology. 2009:4A–42. doi: 10.1002/9780470151808.sc04a02s9. [DOI] [PubMed] [Google Scholar]
- 21.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-throughput real-time quantitative reverse transcription PCR. Current protocols in molecular biology. 2006:15–18. doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
- 22.Hill JT, Demarest BL, Bisgrove BW, Su YC, et al. Poly peak parser: Method and software for identification of unknown indels using sanger sequencing of polymerase chain reaction products. Dev Dyn. 2014;243:1632–1636. doi: 10.1002/dvdy.24183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graham DB, Root DE. Resources for the design of CRISPR gene editing experiments. Genome Biol. 2015;16:260. doi: 10.1186/s13059-015-0823-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L, Yang JL, Byrne S, Pan J, Church GM. CRISPR/Cas9-Directed Genome Editing of Cultured Cells. Curr Protoc Mol Biol. 2014;107:31.1.1–31.117. doi: 10.1002/0471142727.mb3101s107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.