

Abstract
G9a-like protein (GLP) and G9a are highly homologous protein lysine methyltransferases (PKMTs) sharing approximately 80% sequence identity in their catalytic domains. GLP and G9a form a heterodimer complex and catalyze mono- and dimethylation of histone H3 lysine 9 and nonhistone substrates. Although they are closely related, GLP and G9a possess distinct physiological and pathophysiological functions. Thus, GLP or G9a selective small-molecule inhibitors are useful tools to dissect their distinct biological functions. We previously reported potent and selective G9a/GLP dual inhibitors including UNC0638 and UNC0642. Here we report the discovery of potent and selective GLP inhibitors including 4 (MS0124) and 18 (MS012), which are >30-fold and 140-fold selective for GLP over G9a and other methyltransferases, respectively. The cocrystal structures of GLP and G9a in the complex with either 4 or 18 displayed virtually identical binding modes and interactions, highlighting the challenges in structure-based design of selective inhibitors for either enzyme.
Graphical Abstract

INTRODUCTION
Protein lysine methyltransferases (PKMTs) catalyze the transfer of the methyl group from the cofactor S-5′-adenosyl-L-methionine (SAM) to the ε-amino group of the targeted lysine residue in histone and nonhistone substrates, leading to mono-, di-, or trimethylation.1–3 Lysine methylation does not change the charge state of the residue. Instead, this modification increases the bulkiness of the amino group and alters the hydrophobicity and hydrogen bond formation potential.2 As a result, lysine methylation has dramatic effects on the interactions of the modified protein with other proteins, thus its physiological function.2 Depending on the methylation state and location, histone lysine methylation can lead to either activation or repression of gene expression and transcription.3–7 In general, methylation of histone H3 lysine 4, 36, and 79 (H3K4, H3K36, and H3K79) is associated with transcription activation and methylation of histone H3 lysine 9 and 27 (H3K9 and H3K27) is typically a hallmark of transcription repression.8–11
GLP (also known as euchromatic histone-lysine N-methyltransferase 1 (EHMT1) or lysine methyltransferase 1D (KMT1D)) and G9a (also known as EHMT2 or KMT1C) are two closely related PKMTs with approximately 80% sequence identity in their respective suppressor of variegation 3–9 enhancer-of-zeste trithorax (SET) domains.12,13 It has been shown that the heterodimeric complex of G9a and GLP is the main functional methyltransferase in mouse embryonic stem cells, depositing mono- and dimethylated H3K9 (H3K9me1 and H3K9me2).14,15 Besides H3K9, other histone substrates, such as histone H1,16,17 H3K27,18–20 and H3K56,21 have been reported. G9a and/or GLP can also methylate a variety of nonhistone proteins,22 including the tumor suppressor p53,23 sirtuin 1 (SIRT1),24 reptin,25 myogenic differentiation 1 (MyoD),26 chromodomain Y-like protein (CDYL1),27 widely interspaced zinc finger motifs protein (WIZ),27 and G9a.28 Furthermore, G9a and/or GLP have been reported to play roles29 in germ cell development and meiosis,30 embryo development,14,15 tumor cell growth and metathesis,23,31–33 immune response,34,35 cocaine-induced plasticity,36 cognition and adaptive behavior,4,37 and provirus silencing.38 Dysregulation of G9a and/or GLP has been implicated in many human diseases such as cancer, inflammatory diseases, and neurogenerative disorder.4,6,32,37,39,40
Although G9a and GLP share high sequence homology and many biological activities, they do possess distinct physiological and pathophysiological functions.41 For example, the ankyrin repeat domain of G9a mainly associates with H3K9me2, whereas the ankyrin domain of GLP preferentially associates with H3K9me1.42 Mutations at the ankyrin domain of G9a and GLP result in drastic phenotype differences in mice.42 In addition, G9a and GLP often display distinct tissue-specific expression profiles. Different from G9a, GLP is highly expressed in brown adipose tissue and controls brown adipose cell fate and energy homeostasis.43 Moreover, G9a and GLP have opposite roles during early steps of skeletal muscle terminal differentiation. GLP, but not G9a, stabilizes MyoD, which is the major player in triggering muscle terminal differentiation, by down-regulating proteasome subunit-encoding gene expression.44
Potent and selective small-molecule inhibitors of G9a/GLP are useful tools for investigating biological functions of these proteins and testing the therapeutic hypotheses associated with them. Over the past decade, a number of G9a/GLP inhibitors have been reported.12,13,45–56 Compound 1 (BIX01294) was the first selective dual G9a/GLP inhibitor (Figure 1).12 It was discovered via high throughput screening and is competitive with the peptide substrate and noncompetitive with the cofactor SAM. On the basis of the crystal structure of the GLP–1 complex,13 our group and others have discovered more potent G9a/GLP inhibitors including UNC0224,45 UNC0321,46 and 2 (E72).47 Further optimization of this chemical series led to the discovery of potent, selective, and cell-active G9a/GLP dual inhibitors 3 (UNC0638)48,49,57 and UNC0642,50 which has sufficient in vivo pharmacokinetic properties and is suitable for animal studies (Figure 1). Among all of these inhibitors, compound 1 is the only one that has some selectivity for GLP over G9a (about 3–5-fold).13,49 Because G9a and GLP are not functionally redundant and have been shown to have distinct biological functions, G9a or GLP selective inhibitors will be valuable tools to study distinct functions of these two closely related PKMTs. Here, we describe the design, synthesis, and biochemical evaluation of new compounds based on the quinazoline scaffold shared by compounds 1–3. Our structure–activity relationship (SAR) studies resulted in the discovery of potent and selective GLP inhibitors including 18 (MS012), which is remarkably selective for GLP over G9a (>140-fold) in biochemical assays.
Figure 1.

Structures of selected G9a/GLP inhibitors.
RESULTS AND DISCUSSION
Discovery of 4 (MS0124) as a GLP Selective Inhibitor
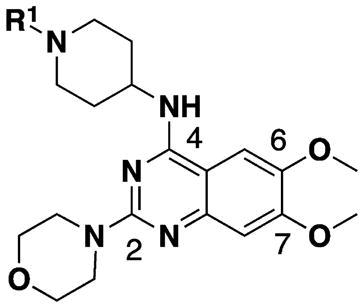
To identify selective inhibitors for GLP or G9a, we screened our quinazoline compound collection against these two enzymes using radioactivity based scintillation proximity biochemical assays, which measure the potency of test compounds at inhibiting the transfer of the methyl group from 3H-SAM to the peptide substrates.50 We were pleased to find compound 4 was 34-fold more selective for GLP (IC50 = 13± 4 nM) over G9a (IC50 = 440 ± 63 nM) (Table 1). We were also able to obtain cocrystal structures of both GLP and G9a in complex with 4 in the presence of SAM at resolutions of 2.6 and 1.95 Å, respectively (Figure 2 and Supporting Information, Table 1). These two cocrystal structures are extremely similar, with a root-mean-square deviation (RMSD) of 0.31 Å. Compound 4 occupies the peptide substrate binding site with the same binding mode for GLP and G9a. The interactions between compound 4 and these two enzymes are conserved. For example, the secondary amino group at the C4 position forms a direct hydrogen bond with the side chain of GLP Asp1171 and the equivalent G9a Asp1083. In addition, the N1 nitrogen on the pyrimidine ring forms another direct hydrogen with the side chain of GLP Asp1176 and G9a Asp1088. Moreover, the oxygen atoms of both 6- and 7-methoxy groups interact with a water molecule in both structures.
Table 1.
SAR of the N-Capping Group of the Piperidine Moietya
| |||
|---|---|---|---|
| Compound | R1 | IC50 (nM) | |
| GLP | G9a | ||
| 4 |
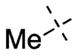
|
13 ± 4 | 440 ± 63 |
| 7 |
|
26 ± 8 | 323 ± 51 |
| 8 |
|
11 ± 1 | 224 ± 63 |
| 9 |
|
15 ± 4 | 262 ± 40 |
| 10 |
|
12 ± 1 | 313 ± 133 |
IC50 determination experiments were performed at substrate and cofactor concentrations equal to the respective Km values for each enzyme. IC50 determination experiments were performed in triplicate and the values are presented as mean ± SD.
Figure 2.

X-ray cocrystal structures of (A) GLP (green) (PDB 5TUZ) and (B) G9a (blue) (PDB 5TUY) in complex with 4 in the presence of SAM. Water molecule is illustrated as a red sphere. Main interactions are shown in yellow dashed lines. (C) Overlay of (A) and (B).
To improve potency and selectivity of 4, we conducted structure–activity relationship (SAR) studies at the following four regions: (1) N-capping group of the piperidine moiety, (2) 6-methoxy moiety, (3) 7-methoxy moiety, and (4) 2-morpholine moiety (Tables 1–3).
Table 3.
SAR of the 2-Morpholine Moietya
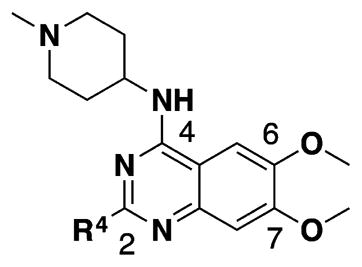
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R4 | IC50 (nM) | Compound | R4 | IC50 (nM) | ||
| GLP | G9a | GLP | G9a | ||||
| 11 |
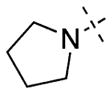
|
12 ± 1 | 313 ± 133 | 16 |
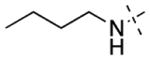
|
35 ± 7 | 436 ± 141 |
| 12 |
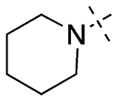
|
12 ± 1 | 31 ± 9 | 17 |
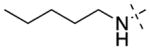
|
63 ± 5 | 341 ± 26 |
| 13 |
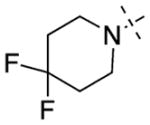
|
97 ± 6 | 1060 ± 180 | 18 |
|
7 ± 2 | 992 ± 337 |
| 14 |
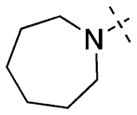
|
16 ± 3 | 190 ± 55 | 19 |
|
44 ± 12 | 756 ± 218 |
| 15 |
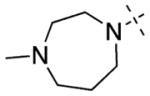
|
4 ± 2 | 9 ± 3 | 20 |
|
35 ± 13 | 407 ± 136 |
| 53 |
|
34 ± 9 | 438 ± 135 | 21 |
|
28 ± 7 | 306 ± 75 |
| 54 |
|
341 ± 18 | 1910 ± 560 | 22 |

|
18 ± 3 | 311 ± 37 |
IC50 determination experiments were performed at substrate and cofactor concentrations equal to the respective Km values for each enzyme. IC50 determination experiments were performed in triplicate and the values are presented as Mean ± SD.
Synthesis
We previously developed a two-step synthetic sequence to explore the 2-amino and 4-amino regions of the 6,7-dimethoxyquinazoline scaffolds.45 Using this efficient synthesis, we converted the commercially available 2,4-dichloro-6,7-dimethoxyquinazoline 5 to compounds 4 and 7–22 by two consecutive chloro displacement reactions (Scheme 1 and Tables 1 and 3).
Scheme 1. Synthesis of 4 and 7–22a.

aReagents and conditions: (a) 1-alkylpiperidin-4-ylamine, K2CO3, DMF, rt, 80–90%; (b) R′ amines, 4 N HCl in dixoane, i-PrOH, microwave, 160 °C, 75–85%.
To investigate the SAR of 6- and 7-methoxy moieties, we synthesized analogues with ethoxy, isopropoxy, fluoro, and methyl groups at C6 and C7 positions (Scheme 2 and Table 2). We developed four synthetic routes to prepare 4,5-disubtituted-2-cyanoaniline intermediates 29, 30, 35, and 36. Briefly, etherification of commercially available phenols, 23 and 24, followed by nitration and subsequent nitro reduction produced 2-cyanoanilines (29 and 30) bearing different alkoxy groups (Scheme 2A). The fluoro and methyl substituted intermediates, 35 and 36, were synthesized from anilines 31 and 32 through bromination and subsequent cyanation reactions (Scheme 2B). The 4,5-disubstituted-2-cyanoaniline intermediates 29, 30, 35, and 36 were then reacted with methyl chloroformate to yield carbamates 37. After saponification of the cyano group, cyclization reaction afforded quinazolinediones 38. Chlorination and subsequent two consecutive chloro substitutions provided quinazolines 41–48 (Scheme 2C and Table 2).
Scheme 2. Synthesis of 41–48a.

aReagents and conditions: (a) EtI or i-PrBr, K2CO3, acetone, reflux; (b) HNO3, Ac2O, 0 °C to rt, 81–94% over 2 steps; (c) Fe dust, NH4OAc, H2O, reflux, 60–70%; (d) Bu4NBr3, ethyl acetate, 0 °C to rt, 80–90%; (e) Zn(CN)2, Pd(pph3)4, DMF, microwave, 130 °C, 50–72%; (f) Br2, Ac2O, 50 °C, 64%; then HCl, EtOH, 95%; (g) Br2, K2CO3, DCM, –15 °C, 70%; (h) CuCN, DMF, sealed tube, 160 °C, 56%; (i) ClCO2Me, DIEA, DCM/DMF, 0°C to rt; (j) H2O2, NaOH, H2O/MeOH, reflux; (k) PhNEt2, POCl3, reflux, 30–40% over 3 steps; (l) 1-methylpiperidin-4-amine, K2CO3, DMF, rt; (m) morpholine, 4 N HCl in dixoane, i-PrOH, microwave, 160 °C, 75–85% over 2 steps.
Table 2.
SAR of the 6- and 7-Methoxy Moietiesa
| ||||
|---|---|---|---|---|
| Compound | R2 | R3 | IC50 (nM) | |
| GLP | G9a | |||
| 4 |
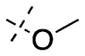
|
|
13 ± 4 | 440 ± 63 |
| 41 |
|
>5000 | >6000 | |
| 42 |
|
>6000 | >6000 | |
| 43 |
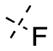
|
1230 ± 340 | 3690 ± 1780 | |
| 44 |
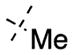
|
808 ± 378 | 4070 ± 950 | |
| 45 |
|
|
>1000 | >5000 |
| 46 |
|
>5000 | >5000 | |
| 47 |
|
>5000 | >5000 | |
| 48 |
|
29 ± 12 | 1150 ± 47 | |
IC50 determination experiments were performed at substrate and cofactor concentrations equal to the respective Km values for each enzyme. IC50 determination experiments were performed in triplicate and the values are presented as mean ± SD.
Quinazoline analogues with alkyl groups at the C2 position (53 and 54) were synthesized from aniline 49. Amide coupling of 49 with acid chlorides provided intermediates 50, which were then converted to 51 through the saponification and subsequent cyclization reaction. Chlorination followed by chloro displacement provided 2-alkyl analogues 53 and 54 (Scheme 3 and Table 3).
Scheme 3. Synthesis of 53 and 54a.

aReagents and conditions: (a) acid chloride, DIEA, DCM/DMF, 0 °C to rt, 90–95%; (b) H2O2, NaOH, H2O/MeOH, reflux, 46–55% over 2 steps; (c) PhNEt2, POCl3, reflux, 46–55% over 2 steps; (d) 1-methylpiperidin-4-amine, DIEA, i-PrOH, microwave, 160 °C, 80–90%.
SAR in GLP and G9a Biochemical Assays
All of the synthesized compounds were evaluated in the GLP and G9a radioactivity based scintillation proximity biochemical assays. IC50 values of the compounds are summarized in Tables 1–3.
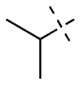
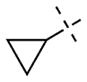
For the N-capping group of the piperidine moiety, we found that a variety of alkyl groups were well tolerated for both GLP and G9 (Table 1). Replacing the methyl group (4) with the ethyl (7), n-propyl (8), i-propyl (9), or cyclopropyl (10) group did not significantly change the potency for either GLP or G9a. These SAR results are consistent with the crystal structures of GLP and G9a in complex with 4, which show the N-capping group is solvent exposed. Because the methyl group in 4 provides slightly better selectivity and is the smallest substituent, we chose the methyl group as the N-capping group for the following SAR studies.
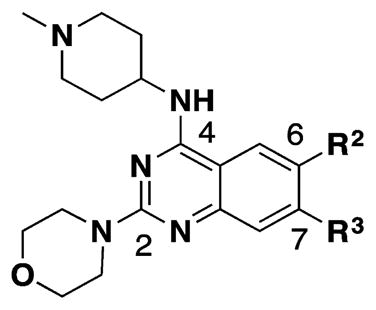
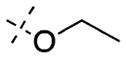
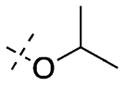
In contrast to the N-capping groups, the 6-methoxy moiety was much more sensitive to structural changes (Table 2). Displacing the methoxy group with the ethoxy (41) or isopropoxy (42) group totally diminished their activities against both GLP and G9a (IC50 > 5000 nM). From the cocrystal structures of G9a and GLP in complex with 4, the 6-methoxy group occupies a shallow binding groove. A larger group such as ethoxy or isopropoxy likely clashes with the binding groove, thus decreasing the potency. The removal of the oxygen atom from the methoxy group and installation of a smaller group also led to less potent analogues. In addition, GLP was more sensitive to these structural changes than G9a. For example, 6-fluoro group (43) decreased the potency for GLP (IC50 = 1230 ± 340 nM) by 95-fold and for G9a (IC50 = 3690 ± 1780 nM) by 8-fold. Similarly, 6-methyl group (44) led to a 62-fold potency drop for GLP (IC50 = 808 ± 378 nM) and a 9-fold potency decrease for G9a (IC50 = 4070 ± 950 nM). These SAR results confirmed the importance of the interactions between the water molecule and 6-methoxy group in the cocrystal structures.
The SAR of the C7 substituents followed the similar trend as the 6-substituents (Table 2). The ethoxy (45) or isopropoxy (46) group abolished the activities against both GLP and G9a. From the cocrystal structures, the 7-methoxy group is located at the neck of the lysine binding channel and also interacts with the water molecule. In our previous report, the pyrrolidin-1-yl-propoxy group at this position greatly increased G9a potency by forming an addition hydrogen bond with Leu1086 and cation–π interaction with Tyr1154.49 The drastic potency loss of the ethoxy and isopropoxy analogues highlights the importance of the interactions between the basic amino group and Leu1086 and Tyr1154. We also found that, while the fluoro group (47) completely abolished the activities against both GLP and G9a, the methyl group (48) was tolerated with only about 2–3-fold potency loss for GLP (IC50 = 29 ± 12 nM) and G9a (IC50 = 1150 ± 47 nM).
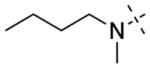
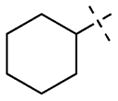
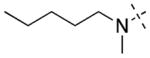
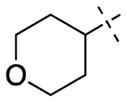
Having established initial SAR for the N-capping group, and 6- and 7-methoxy regions, we next explored the 2-morpholine moiety using cyclic amines (11–15), cyclic alkyl groups (53 and 54), and acyclic amines (16–22). We first compared the pyrrolidinyl (11), piperidinyl (12), and azepanyl (14) analogues and found the effect of the ring size on the potency for GLP was minimal with IC50 values of 12 ± 1, 12 ± 1, and 16 ± 3 nM, respectively. However, the ring size was more important for modulating G9a potency. Among the three groups, the five-membered pyrrolidinyl group (11) led to the weakest G9a inhibition (IC50 = 313 ± 133 nM) and the six-membered piperidinyl group (12) resulted in the most potent G9a inhibitor (IC50 = 31 ± 9 nM). Substitutions on the cyclic amino groups also exhibited a drastic impact on the potency for both GLP and G9a. Compared with the piperidinyl analogue 12, the 4,4-difluoro substituted piperidinyl group (13) decreased potency for GLP by 8-fold and for G9a by 34-fold. Compared with the azepanyl analogue 14, the methylhomopiperazinyl analogue 15 increased potency for GLP by 4-fold and G9a by 21-fold. Replacing the 2-cycloamino group with a 2-cycloalkyl group led to much less potent compounds for both GLP and G9a. For example, compound 53 was 3-fold less potent for GLP and 14-fold less potent for G9a compared with 12. In addition, compound 54 was 26-fold less potent for GLP and 4-fold less potent for G9a compared with 4. These SAR results indicate the important role of the 2-amino group for maintaining high potency for both GLP and G9a. We next explored a number of acyclic alkyl amines (16–22) at the 2-position. For the secondary amine analogues 16–19, the length of the alkyl chain had the most profound effect on the potency for GLP over G9a. We were delighted to find that compound 18, which contains a 2-hexylamino group, was >140-fold selective for GLP over G9a, representing the most selective GLP inhibitor to date. The tertiary amine analogues 20–22, which possess an extra N-methyl substitute, did not improve potency or selectivity for GLP over G9a.
Characterization of 4 and 18 in Selectivity, Biophysical, and Mechanism of Action Studies (MOA)
To further assess the selectivity of 4 and 18, we tested them against 29 protein lysine methyltransferases (PKMTs), protein arginine methyltransferases (PRMTs), DNA methyltransferases (DNMTs), and RNA methyltransferases (RNMTs). We found that 4 did not inhibit any of these methyltransferases at 10 μM (Figure 3A). While 18 did not inhibit any of these methyltransferase at 1 μM, it displayed modest inhibition (57%) of the N6-adenosine-methyltransferase subunit METTL3 and 14 complex (METTL3–14) at 10 μM (Figure 3B).
Figure 3.

Selectivity of 4 and 18 against methyltransferases. Selectivity of 4 (A) and 18 (B) against a panel of 29 PKMTs, PRMTs, DNMTs, and RNMTs was determined at two compound concentrations of 1 μM (blue bars) and 10 μM (red bars).
We next confirmed bindings of 4 and 18 to GLP and G9a using isothermal titration calorimetry (ITC) in the presence of SAM (Figure 4). The binding of 4 and 18 to GLP and G9a is enthalpy driven. Compound 4 displayed higher binding affinity to GLP (Kd = 40 ± 5 nM) than G9a (Kd = 445 ± 40 nM) (Figure 4A,B). Similarly, compound 18 also exhibited stronger binding affinity to GLP (Kd = 46 ± 15 nM) than G9a (Kd = 610 ± 68 nM) (Figure 4C,D). Thus, in ITC biophysical assays, 4 and 18 were 11-fold and 13-fold selective for GLP over G9a, respectively.
Figure 4.

Binding confirmation of 4 and 18. Isothermal titration calorimetry (ITC) was used to confirm binding of 4 to (A) GLP with a Kd of 40 ± 5 nM and to (B) G9a with a Kd of 445 ± 40 nM. Similarly, ITC was used to confirm the binding of 18 to (C) GLP with a Kd of 46 ± 15 nM and to (D) G9a with a Kd of 610 ± 68 nM. ITC experiments for GLP and G9a were performed in triplicate.
To assess MOA of 4 and 18, we evaluated the effect of peptide and SAM concentrations on IC50 values of 4 and 18 against GLP. As illustrated in parts A and C of Figure 5, increasing the peptide substrate concentration significantly increased the IC50 values of 4 and 18, indicating that both compounds are peptide competitive. As illustrated in parts B and D of Figure 5, increasing the SAM concentration decreased the IC50 values of 4 and 18, indicating that both compounds are SAM uncompetitive.
Figure 5.

Mechanism of GLP inhibition by 4 and 18. Mechanism of inhibition of GLP activity by 4 and 18 was explored by IC50 determination at various peptide (A and C, respectively) or SAM (B and D, respectively) concentrations while the concentration of the other substrate was kept constant. Increase in peptide concentration in assays resulted in significant increase in IC50 values, indicating that both compounds are peptide competitive (A,C). Decrease in IC50 values upon increasing SAM concentration indicates that both compounds are SAM uncompetitive inhibitors (B,D).
Cocrystal Structures of 18
We solved the ternary crystal structures of GLP and G9a in complex with 18 in the presence of the cofactor SAM (Figure 6A–C, and Supporting Information, Table 2). Similar to 4, compound 18 conserved three key hydrogen bonds with GLP and G9a: (1) the secondary amino group at the 4-position interacts with GLP Asp1171 and G9a Asp1083; (2) the N1 nitrogen on the quinazoline ring interacts with GLP Asp1176 and G9a Asp1088; (3) the oxygen atoms of both 6- and 7-methoxy groups interact weakly with a water molecule in both GLP and G9a structures. Interestingly, the 2-hexylamino group of 18 points to a different direction compared to the 2-(3′-dimethylamino)propylamino group of compound 2 in the cocrystal structure of GLP–SAH–2 (Figure 6D).47 The salt bridge47 formed between the 3′-dimethylamino group of 2 and Asp1131 likely leads to the 2-(3′-dimethylamino)propylamino side chain adapting to the different geometry. Notably, the crystal structures of GLP and G9a in the complex with 4 or 18 revealed the virtually identical inhibitor binding modes and ligand–protein interactions and did not provide insight for the structural basis of the high selectivity for GLP over G9a. We are currently conducting additional studies to understand the structural basis of the observed high selectivity and will report our progress in future publications.
Figure 6.

X-ray cocrystal structures of GLP and G9a in complex with 18. (A) Structure of GLP–SAM–18 (green) (PDB 5TTG). (B) Structure of G9a–SAM–18 (blue) (PDB 5TTF). Water molecule is illustrated as a red sphere. Main interactions are shown in yellow dashed lines. (C) Overlay of (A) and (B). (D) Overlay of (A) and GLP–SAH–2 (purple) (PDB 3MO5).
Lastly, we assessed membrane permeability of compound 18 in a Caco-2 cell permeability assay and found that 18 had poor membrane permeability (5.0 nm/s) and high efflux ratio (80). Thus, 18 is not suitable for cellular studies. Future work will attempt to improve membrane permeability and reduce efflux while maintaining the high selectivity for GLP over G9a.
CONCLUSIONS
By testing our quinazoline compound collection, we identified compound 4 as a GLP selective inhibitor. We next conducted SAR studies and explored four regions of 4, which led to the discovery of compound 18, which is >140-fold selective for GLP over G9a. We confirmed the bindings of 4 and 18 to GLP and G9a by ITC. Both 4 and 18 are also selective for GLP over a broad range of other PKMTs, PRMTs, DNMTs, and RNMTs. We also obtained high-resolution X-ray cocrystal structures of GLP and G9a in complex with either 4 or 18, which further confirmed the bindings of these inhibitors to GLP and G9a and revealed that the inhibitors bind to GLP and G9a in virtually identical binding modes. This work will help develop high-quality chemical probes of GLP.
EXPERIMENTAL SECTION
Chemistry General Procedures
HPLC spectra for all compounds were acquired using an Agilent 1200 series system with DAD detector. Chromatography was performed on a 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 mL/min. The gradient program was as follows: 1% B (0–1 min), 1–99% B (1–4 min), and 99% B (4–8 min). High-resolution mass spectra (HRMS) data were acquired in positive ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Nuclear magnetic resonance (NMR) spectra were acquired on a Bruker DRX-600 spectrometer (600 MHz 1H, 150 MHz 13C) or a Varian Mercury spectrometer (400 MHz 1H, 100 MHz 13C). Chemical shifts are reported in ppm (δ). Preparative HPLC was performed on Agilent Prep 1200 series with UV detector set to 254 nm. Samples were injected into a Phenomenex Luna 750 mm × 30 mm, 5 μm, C18 column at room temperature. The flow rate was 40 mL/min. A linear gradient was used with 10% (or 50%) of MeOH (A) in H2O (with 0.1% TFA) (B) to 100% of MeOH (A). HPLC was used to establish the purity of target compounds. All final compounds had >95% purity using the HPLC methods described above.
6,7-Dimethoxy-N-(1-methylpiperidin-4-yl)-2-morpholinoquinazolin-4-amine (4)
To a solution of 2,4-dichloro-6,7-dimethoxyquinazoline (50 mg, 0.19 mmol) in DMF (2 mL) were added K2CO3 (55 mg, 0.40 mmol) and 4-methylpiperidin-1-amine (0.05 mL, 0.40 mmol). After being stirred for 2 h, the reaction was quenched with water and extracted with DCM (3 × 5 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was dissolved in i-PrOH (1 mL) and treated with HCl (0.1 mL, 4 M in dioxane, 0.4 mmol) and morpholine (0.03 mL, 0.34 mmol). The solution was heated in a microwave reactor at 160 °C for 15 min before being concentrated under reduced pressure. The resulting residue was purified by preparative HPLC to yield the title compound (60 mg, 82% over 2 steps). 1H NMR (600 MHz, CDCl3) δ 6.92 (s, 1H), 6.79 (s, 1H), 5.21 (d, J = 7.2 Hz, 1H), 4.17–4.12 (m, 1H), 3.96 (s, 3H), 3.95 (s, 3H), 3.83–3.79 (m, 8H), 2.90 (d, J = 10.8 Hz, 2H), 2.33 (s, 3H), 2.21–2.15 (m, 4H), 1.71–1.61 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 158.95, 158.32, 154.45, 149.05, 145.68, 106.11, 103.39, 100.69, 67.10, 56.32, 55.98, 54.80, 47.75, 46.20, 44.67, 32.13. HRMS(ESI-TOF) m/z: [M + H]+ calcd for C20H30N5O3 388.2343, found 388.2382.
N-(1-Ethylpiperidin-4-yl)-6,7-dimethoxy-2-morpholinoquinazolin-4-amine (7)
The title compound (75% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.87 (s, 1H), 6.73 (s, 1H), 5.13 (d, J = 7.6 Hz, 1H), 4.08–4.05 (m, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 3.78–3.73 (m, 8H), 2.93 (d, J = 12.0 Hz, 2H), 2.41 (q, J = 6.8 Hz, 2H), 2.14–2.08 (m, 4H), 1.63–1.54 (m, 2H), 1.07 (q, J = 7.2 Hz, 3H). MS (ESI) m/z 402.3 [M + H]+.
6,7-Dimethoxy-2-morpholino-N-(1-propylpiperidin-4-yl)-quinazolin-4-amine (8)
The title compound (76% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.89 (s, 1H), 6.74 (s, 1H), 5.12 (d, J = 7.2 Hz, 1H), 4.13–4.10 (m, 1H), 3.93 (s, 3H), 3.91 (s, 3H), 3.81–3.74 (m, 8H), 2.92 (d, J = 11.6 Hz, 2H), 2.30 (q, J = 6.8 Hz, 2H), 2.15–2.11 (m, 4H), 1.61–1.47 (m, 4H), 0.90 (q, J = 7.2 Hz, 3H). MS (ESI) m/z 416.1 [M + H]+.
N-(1-Isopropylpiperidin-4-yl)-6,7-dimethoxy-2-morpholinoquinazolin-4-amine (9)
The title compound (85% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.88 (s, 1H), 6.78 (s, 1H), 5.24 (d, J = 7.2 Hz, 1H), 4.12–4.03 (m, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.81–3.74 (m, 8H), 2.91 (d, J = 12.0 Hz, 2H), 2.81–2.75 (m, 1H), 2.33 (t, J = 10.8 Hz, 2H), 2.16–2.14 (m, 2H), 1.67–1.58 (m, 2H), 1.06 (d, J = 6.8 Hz, 6H). MS (ESI) m/z 416.3 [M + H]+.
N-(1-Cyclopropylpiperidin-4-yl)-6,7-dimethoxy-2-morpholinoquinazolin-4-amine (10)
The title compound (77% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.91 (s, 1H), 6.70 (s, 1H), 5.01 (d, J = 6.8 Hz, 1H), 4.18–4.08 (m, 1H), 3.95 (s, 3H), 3.92 (s, 3H), 3.83–3.77 (m, 8H), 3.04 (d, J = 12.4 Hz, 2H), 2.39 (td, J = 11.6, 3.6 Hz, 2H), 2.15–2.11 (m, 2H), 1.66–1.60 (m, 1H), 1.54 (qd, J = 11.2, 4.0 Hz, 2H), 0.49–0.45 (m, 2H), 0.44–0.40 (m, 2H). MS (ESI) m/z 414.3 [M + H]+.
6,7-Dimethoxy-N-(1-methylpiperidin-4-yl)-2-(pyrrolidin-1-yl)-quinazolin-4-amine (11)
The title compound (80% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.92 (s, 1H), 6.78 (s, 1H), 5.12 (d, J = 6.8 Hz, 1H), 4.19–4.09 (m, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.64–3.60 (m, 4H), 2.85 (d, J = 12.0 Hz, 2H), 2.30 (s, 3H), 2.18–2.13 (m, 4H), 1.96–1.93 (m, 4H), 1.64–1.55 (m, 2H). MS (ESI) m/z 372.3 [M + H]+.
6,7-Dimethoxy-N-(1-methylpiperidin-4-yl)-2-(piperidin-1-yl)-quinazolin-4-amine (12)
The title compound (80% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, MeOH-d4) δ 7.64 (s, 1H), 7.11 (s, 1H), 4.58–4.46 (m, 1H), 3.94 (s, 3H), 3.90 (s, 3H), 3.87–3.81 (m, 4H), 3.63 (d, J = 12.8 Hz, 2H), 3.26–3.19 (m, 1H), 2.89 (s, 3H), 2.34 (d, J = 12.9 Hz, 2H), 2.10–1.96 (m, 2H), 1.82–1.66 (m, 7H). MS (ESI) m/z 386.3 [M + H]+.
2-(4,4-Difluoropiperidin-1-yl)-6,7-dimethoxy-N-(1-methylpiperidin-4-yl)quinazolin-4-amine (13)
The title compound (84% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.89 (s, 1H), 6.71 (s, 1H), 5.01 (d, J = 7.2 Hz, 1H), 4.15–4.05 (m, 1H), 3.99 (t, J = 6.0 Hz, 4H), 3.96 (s, 3H), 3.95 (s, 3H), 2.89–2.86 (m, 2H), 2.33 (s, 3H), 2.22–2.13 (m, 4H), 2.06–1.96 (m, 4H), 1.69–1.59 (m, 2H). MS (ESI) m/z 422.3 [M + H]+.
2-(Azepan-1-yl)-6,7-dimethoxy-N-(1-methylpiperidin-4-yl)-quinazolin-4-amine (14)
The title compound (79% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.88 (s, 1H), 6.72 (s, 1H), 4.97 (d, J = 6.4 Hz, 1H), 4.12–4.05 (m, 1H), 3.93 (s, 3H), 3.91 (s, 3H), 3.78 (t, J = 6.0 Hz, 4H), 2.88–2.85 (m, 2H), 2.33 (s, 3H), 2.17–2.13 (m, 4H), 1.82–1.74 (m, 4H), 1.64–1.53 (m, 6H). MS (ESI) m/z 400.3 [M + H]+.
6,7-Dimethoxy-2-(4-methyl-1,4-diazepan-1-yl)-N-(1-methylpiperidin-4-yl)quinazolin-4-amine (15)
The title compound (78% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, MeOH-d4) δ 7.38 (s, 1H), 6.89 (s, 1H), 4.15–4.07 (m, 1H), 3.93–3.89 (m, 2H), 3.88 (s, 3H), 3.87 (s, 3H), 3.82 (t, J = 6.4 Hz, 2H), 2.94 (d, J = 12.1 Hz, 2H), 2.76–2.70 (m, 2H), 2.60–2.57 (m, 2H), 2.34 (s, 3H), 2.30 (s, 3H), 2.17 (td, J = 12.1, 2.2 Hz, 2H), 2.12–2.04 (m, 2H), 2.04–1.96 (m, 2H). MS (ESI) m/z 415.3 [M + H]+.
N2-Hexyl-6,7-dimethoxy-N4-(1-methylpiperidin-4-yl)quinazoline-2,4-diamine (16)
The title compound (85% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.84 (s, 1H), 6.82 (s, 1H), 5.32 (d, J = 6.8 Hz, 1H), 4.79 (br, 1H), 4.13–4.11 (m, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.45-3.40 (m, 2H), 2.83 (d, J = 11.6 Hz, 2H), 2.28 (s, 3H), 2.15-2.10 (m, 4H), 1.62–1.57 (m, 4H), 1.45–1.37 (m, 2H). 0.93 (t, J = 7.2 Hz, 3H). MS (ESI) m/z 374.3 [M + H]+.
6,7-Dimethoxy-N4-(1-methylpiperidin-4-yl)-N2-pentylquinazoline-2,4-diamine (17)
The title compound (76% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.83 (s, 1H), 6.82 (s, 1H), 5.40 (d, J = 5.2 Hz, 1H), 4.96 (br, 1H), 4.16–4.07 (m, 1H), 3.87 (s, 3H), 3.84 (s, 3H), 3.39 (q, J = 6.8 Hz, 2H), 2.84–2.81 (m, 2H), 2.26 (s, 3H), 2.14–2.08 (m, 4H), 1.63–1.54 (m, 4H), 1.34–1.31 (m, 4H), 0.86 (t, J = 7.2 Hz, 3H). MS (ESI) m/z 388.2 [M + H]+.
N2-Hexyl-6,7-dimethoxy-N4-(1-methylpiperidin-4-yl)quinazoline-2,4-diamine (18)
The title compound (81% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.86 (s, 1H), 6.76 (s, 1H), 5.14 (d, J = 7.2 Hz, 1H), 4.77 (br, 1H), 4.15–4.12 (m, 1H), 3.92 (s, 3H), 3.90 (s, 3H), 3.46–3.40 (q, J = 6.8 Hz, 2H), 2.50 (d, J = 12.0 Hz, 2H), 2.30 (s, 3H), 2.18–2.12 (m, 4H), 1.60–1.56 (m, 4H), 1.32–1.28 (m, 6H), 0.88 (t, J = 7.2 Hz, 3H). MS (ESI): 402 [M + H]+. 13C NMR (151 MHz, CDCl3) δ 158.68, 158.39, 154.77, 145.62, 104.35, 103.15, 101.36 (2C), 56.48, 56.12, 54.81, 47.97, 46.27, 41.58, 32.19, 31.68, 30.01, 26.79, 22.66, 14.10. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H36N5O2 402.2864, found 402.2896.
N2-Heptyl-6,7-dimethoxy-N4-(1-methylpiperidin-4-yl)-quinazoline-2,4-diamine (19)
The title compound (75% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.84 (s, 1H), 6.82 (s, 1H), 5.46 (d, J = 6.7 Hz, 1H), 4.95–4.80 (m, 1H), 4.21-4.03 (m, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 3.39 (dd, J = 12.9, 6.9 Hz, 2H), 2.81 (d, J = 11.9 Hz, 2H), 2.25 (s, 3H), 2.17–1.98 (m, 4H), 1.67–1.48 (m, 4H), 1.43–1.14 (m, 8H), 0.83 (t, J = 6.9 Hz, 3H). MS (ESI) m/z 416.2 [M + H]+.
N2-Butyl-6,7-dimethoxy-N2-methyl-N4-(1-methylpiperidin-4-yl)-quinazoline-2,4-diamine (20)
The title compound (83% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.90 (s, 1H), 6.74 (s, 1H), 5.02 (d, J = 7.0 Hz, 1H), 4.10 (dt, J = 11.0, 5.3 Hz, 1H), 3.93 (s, 3H), 3.91 (s, 3H), 3.79–3.55 (t, J = 7.2 Hz, 2H), 3.18 (s, 3H), 2.86 (t, J = 10.2 Hz, 2H), 2.31 (s, 3H), 2.16–2.11 (m, 4H), 1.72–1.48 (m, 4H), 1.44–1.27 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). MS (ESI) m/z 388.3 [M + H]+.
6,7-Dimethoxy-N2-methyl-N4-(1-methylpiperidin-4-yl)-N2-pentyl-quinazoline-2,4-diamine (21)
The title compound (79% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.88 (s, 1H), 6.77 (s, 1H), 5.10 (d, J = 6.8 Hz, 1H), 4.13–4.03 (m, 1H), 3.90 (s, 3H), 3.87 (s, 3H), 3.62 (t, J = 7.2 Hz, 2H), 3.17 (s, 3H), 2.85 (d, J = 12.0 Hz, 2H), 2.29 (s, 3H), 2.15–2.09 (m, 4H), 1.64–1.54 (m, 4H), 1.37–1.23 (m, 4H), 0.88 (t, J = 7.2 Hz, 3H). MS (ESI) m/z 402.3 [M + H]+.
N2-Hexyl-6,7-dimethoxy-N2-methyl-N4-(1-methylpiperidin-4-yl)-quinazoline-2,4-diamine (22)
The title compound (82% over 2 steps) was prepared according to synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.88 (s, 1H), 6.78 (s, 1H), 5.13 (d, J = 6.8 Hz, 1H), 4.13–4.03 (m, 1H), 3.90 (s, 3H), 3.87 (s, 3H), 3.62 (t, J = 7.2 Hz, 2H), 3.17 (s, 3H), 2.85 (d, J = 12.0 Hz, 2H), 2.29 (s, 3H), 2.15–2.09 (m, 4H), 1.64–1.54 (m, 4H), 1.37–1.23 (m, 6H), 0.88 (t, J = 7.2 Hz, 3H). MS (ESI) m/z 416.3 [M + H]+.
2-Amino-5-ethoxy-4-methoxybenzonitrile (29a)
The title compound (50% over 3 steps) was prepared according to synthetic procedure for 30a. 1H NMR (600 MHz, CDCl3) δ 6.78 (s, 1H), 6.23 (s, 1H), 4.30–4.25 (br, 2H), 3.96 (q, J = 7.1 Hz, 2H), 3.81 (s, 3H), 1.40 (t, J = 7.0 Hz, 3H).
2-Amino-5-isopropoxy-4-methoxybenzonitrile (29b)
The title compound (56% over 3 steps) was prepared according to synthetic procedure for 30a. 1H NMR (600 MHz, CDCl3) δ 6.88 (s, 1H), 6.88 (s, 1H), 6.24 (s, 1H), 4.33–4.29 (m, 1H), 4.26–4.22 (br, 2H), 3.83 (s, 3H), 1.32 (d, J = 6.1 Hz, 6H).
2-Amino-4-ethoxy-5-methoxybenzonitrile (30a)
To a solution of 4-hydroxy-3-methoxybenzonitrile (1.49 g, 10 mmol) in acetone (50 mL) were added ethyl bromide (1.4 mL, 19 mmol), followed by K2CO3 (2.77 g, 20 mmol). The mixture was refluxed overnight. After removal of the solvent, the residue was suspended in water (50 mL) and extracted with ethyl acetate (3 × 50 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to provide intermediate 26a. To a solution of intermediate 26a in Ac2O (50 mL) was added HNO3 (3.6 mL, 39 mmol) slowly at 0 °C. The solution was stirred at rt overnight, during which time solid was precipitated out. The reaction mixture was poured into ice water and filtered. The solid was collected and dried to yield the intermediate 28a (2.09 g, 94%) as yellow solid. To a suspension of 28a (2.09 g, 9.4 mmol) in a mixture of water (50 mL) and ethyl acetate (50 mL) were added iron powder (2.10 g, 37.6 mmol) and NH4OAc (4.43 g, 57.4 mmol). The resulting mixture was heated under reflux for 2 h. After cooling to rt, the mixture was filtered and the filtrate was extracted with ethyl acetate. The organic layer was dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield the title compound (1.06 g, 60%) as brown solid. 1H NMR (400 MHz, CDCl3) δ 6.84 (s, 1H), 6.17 (s, 1H), 5.80–5.50 (br, 2H), 4.06 (q, J = 7.0 Hz, 2H), 3.80 (s, 3H), 1.46 (t, J = 7.0 Hz, 3H).
2-Amino-4-isopropoxy-5-methoxybenzonitrile (30b)
The title compound (55% over 3 steps) was prepared according to synthetic procedure for 30a. 1H NMR (400 MHz, CDCl3) δ 6.86 (s, 1H), 6.19 (s, 1H), 5.72–5.55 (br, 2H), 4.58–4.52 (m, 1H), 3.79 (s, 3H), 1.39 (d, J = 6.1 Hz, 6H).
2-Bromo-5-methoxy-4-methylaniline (33a)
To a solution of 3-methoxy-4-methylaniline (420 mg, 3.1 mmol) in ethyl acetate (15 mL) was added Bu3NBr3 (1.56 g, 3.2 mmol) at 0 °C. After being stirred at rt overnight, the reaction was quenched with saturated aqueous NaHCO3 solution (10 mL). The organic layer was separated and dried with anhydrous Na2SO4. After removal of the solvent, the resulting residue was purified by silica gel chromatograph to yield the desire product (585 mg, 90%). 1H NMR (600 MHz, CDCl3) δ 7.15 (s, 1H), 6.31 (s, 1H), 4.00–3.90 (br, 2H), 3.79 (s, 3H), 2.11 (s, 3H).
2-Bromo-4-methoxy-5-methylaniline (34a)
To a solution of 3-methyl-4-methoxyaniline (600 mg, 4.3 mmol) in acetic acid (10 mL) was added acetic anhydride (0.42 mL, 4.4 mmol) dropwise at rt. The reaction was heated at 50 °C, at which temperature bromine (0.23 mL, 4.4 mmol) was added. After being stirred for 2 h at 50 °C, the reaction was cooled to rt and poured into ice–water. The resulting precipitate was filtered, washed with water, and dissolved in ethyl acetate. The solution was washed with saturated sodium chloride aqueous solution, dried with sodium sulfate, and evaporated to dryness to give 2-bromo-4-methoxy-5-methylacetanilide as a crude product. This intermediate was dissolved in ethanol (10 mL) and treated with concentrated hydrochloric acid (2.6 mL). The mixture was refluxed for 2 h before being concentrated. The residue was treated with aqueous sodium hydroxide (1.5 M, 10 mL) to precipitate the crude product. After filtration, the solid was washed with water and dried under reduced pressure to give the title compound (743 mg, 80% over 2 steps). 1H NMR (600 MHz, CDCl3) δ 6.90 (s, 1H), 6.64 (s, 1H), 3.77 (s, 3H), 3.75–3.70 (br, 2H), 2.14 (s, 3H).
2-Bromo-5-fluoro-4-methoxyaniline (34b)
To a suspension of 3-fluoro-4-methoxyaniline (5.06 g, 35.9 mmol) in DCM (160 mL) was added a solution of Br2 (1.85 mL, 36.0 mmol) in DCM (20 mL) dropwise at −15 °C. After being stirred for 30 min, the reaction was quenched with water (100 mL). The organic layer was dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatograph to yield the desired compound (5.50 g, 70%). 1H NMR (400 MHz, CDCl3) δ 7.02 (d, J = 8.5 Hz, 1H), 6.55 (d, J = 12.3 Hz, 1H), 3.88–3.80 (br, 2H), 3.79 (s, 3H).
2-Amino-4-methoxy-5-methylbenzonitrile (35a)
To a solution of 33a (97 mg, 0.46 mmol) in DMF (2 mL) was added Zn(CN)2 (111 mg, 0.94 mmol) and Pd(PPh3)4 (54 mg, 0.04 mmol). The suspension was heated in a microwave reactor at 0 °C for 15 min. After cooling to rt, the reaction was diluted with DCM (4 mL) and ammonium hydroxide solution (2 mL). The resulting mixture was extracted with DCM (3 × 4 mL). The combined organic layer was dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel chromatograph to yield the desired product (53 mg, 72%). 1H NMR (400 MHz, CDCl3) δ 7.07 (s, 1H), 6.14 (s, 1H), 4.32–4.20 (br, 2H), 3.78 (s, 3H), 2.04 (s, 3H).
2-Amino-5-fluoro-4-methoxybenzonitrile (35b)
The title compound (50% over 2 steps) was prepared according to the synthetic procedure for compound 35a. 1H NMR (400 MHz, CDCl3) δ 7.05 (d, J = 10.5 Hz, 1H), 6.24 (d, J = 7.1 Hz, 2H), 4.38–4.17 (br, 2H), 3.86 (s, 3H).
2-Amino-5-methoxy-4-methylbenzonitrile (36a)
The title compound (75%) was prepared from 34a according to the synthetic procedure for compound 35a. 1H NMR (600 MHz, CDCl3) δ 6.74 (s, 1H), 6.58 (s, 1H), 4.16–4.06 (br, 2H), 3.76 (s, 3H), 2.19 (s, 3H).
2-Amino-4-fluoro-5-methoxybenzonitrile (36b)
To a solution of 34b (127 mg, 0.58 mmol) in DMF (160 mL) was added CuCN (126 mg, 1.42 mmol). After being stirred for 18 h in sealed tube at 120 °C, the reaction was cooled to rt, poured into ammonium hydroxide solution (3 mL), and extracted with DCM (3 × 3 mL). The combined organic layers were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatograph to yield the desired compound (54 mg, 56%). 1H NMR (400 MHz, CDCl3) δ 6.92 (d, J = 8.7 Hz, 1H), 6.49 (d, J = 12.2 Hz, 1H), 4.30–4.16 (br, 2H), 3.79 (s, 3H).
2,4-Dichloro-7-ethoxy-6-methoxyquinazoline (39a)
To a solution of 30a (1.06 g, 5.5 mmol) in DCM (12 mL) and DMF (12 mL) were added DIEA (1.8 mL, 10.9 mmol) and methyl chloroformate (0.62 mL, 8.2 mmol) at 0 °C. After being stirred for 2 h, the reaction solution was poured into water and extracted with DCM. The organic layer was dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield 37a as brown solid. To a solution of 37a in EtOH (40 mL) was added H2O2 (35 mL) and NaOH (278 mg, 6.9 mmol). The mixture was refluxed for 2 h before being cooled to rt. The precipitated solid was collected and dried to yield the intermediate 38a. A suspension of 38a in PhNEt2 (1.5 mL, 1.4 mmol) and POCl3 (10 mL) was refluxed for 4 h before being cooled to rt. After removal of the solvent, cold water was added, followed by saturated aqueous NaHCO3 solution. The mixture was extracted with ethyl acetate (3 × 30 mL). The combined organic layers were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel chromatograph to yield the title compound (450 mg, 30%). 1H NMR (600 MHz, CDCl3) δ 7.38 (s, 1H), 7.29 (s, 1H), 4.31 (q, J = 7.0 Hz, 2H), 4.09 (s, 3H), 1.60 (t, J = 7.2 Hz, 3H).
2,4-Dichloro-7-isopropoxy-6-methoxyquinazoline (39b)
The title compound (40%) was prepared according to the synthetic procedure for 39a. 1H NMR (600 MHz, CDCl3) δ 7.37 (s, 1H), 7.29 (s, 1H), 4.83–4.79 (m, 1H), 4.07 (s, 3H), 1.52 (d, J = 6.1 Hz, 6H).
2,4-Dichloro-6-ethoxy-7-methoxyquinazoline (39c)
The title compound (38%) was prepared according to the synthetic procedure for 39a. 1H NMR (400 MHz, CDCl3) δ 7.27 (s, 1H), 7.26 (s, 1H), 4.28 (q, J = 7.0 Hz, 2H), 4.05 (s, 3H), 1.58 (t, J = 7.0 Hz, 3H).
2,4-Dichloro-6-isopropoxy-7-methoxyquinazoline (39d)
The title compound (40%) was prepared according to the synthetic procedure for 39a. 1H NMR (400 MHz, CDCl3) δ 7.27 (s, 1H), 7.26 (s, 1H), 4.83–4.77 (m, 1H), 4.03 (s, 3H), 1.49 (d, J = 6.1 Hz, 6H).
2,4-Dichloro-6-methoxy-7-methylquinazoline (39e)
The title compound (39%) was prepared according to the synthetic procedure for 39a. 1H NMR (400 MHz, CDCl3) δ 7.30 (s, 1H), 7.24 (s, 1H), 4.01 (s, 3H), 2.43 (s, 3H).
2,4-Dichloro-7-fluoro-6-methoxyquinazoline (39f)
The title compound (31%) was prepared according to the synthetic procedure for 39a. 1H NMR (600 MHz, CDCl3) δ 7.67 (d, J = 10.8 Hz, 1H), 7.55 (d, J = 8.4 Hz, 1H), 4.10 (s, 3H).
2,4-Dichloro-6-fluoro-7-methoxyquinazoline (39g)
The title compound (30%) was prepared according to the synthetic procedure for 39a. 1H NMR (600 MHz, CDCl3) 7.78 (dd, J = 10.3, 0.9 Hz, 1H), 7.33 (d, J = 7.4 Hz, 1H), 4.07 (s, 3H).
2,4-Dichloro-7-methoxy-6-methylquinazoline (39h)
The title compound (34%) was prepared according to the synthetic procedure for 39a. 1H NMR (600 MHz, CDCl3) δ 7.95 (s, 1H), 7.24 (s, 1H), 4.03 (s, 3H), 2.43 (s, 3H).
6-Ethoxy-7-methoxy-N-(1-methylpiperidin-4-yl)-2-morpholinoquinazolin-4-amine (41)
The title compound (79%) was prepared from 39a according to the synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.87 (s, 1H), 6.77 (s, 1H), 5.09 (d, J = 7.6 Hz, 1H), 4.10–4.05 (m, 3H), 3.879 (s, 3H), 3.80–3.73 (m, 8H), 2.84–2.82 (m, 2H), 2.28 (s, 3H), 2.17–2.10 (m, 4H), 1.64–1.54 (m, 2H), 1.44 (t, J = 6.8 Hz, 3H). MS (ESI) m/z 402.3 [M + H]+.
6-Isopropoxy-7-methoxy-N-(1-methylpiperidin-4-yl)-2-morpholinoquinazolin-4-amine (42)
The title compound (77%) was prepared from 39b according to the synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.88 (s, 1H), 6.86 (s, 1H), 5.09 (d, J = 7.6 Hz, 1H), 4.52–4.46 (m, 1H), 4.12–4.03 (m, 1H), 3.87 (s, 3H), 3.79–3.72 (m, 8H), 2.84–2.81 (m, 2H), 2.28 (s, 3H), 2.17–2.08 (m, 4H), 1.64–1.54 (m, 2H), 1.32 (d, J = 6.4 Hz, 6H). MS (ESI) m/z 416.3 [M + H]+.
6-Fluoro-7-methoxy-N-(1-methylpiperidin-4-yl)-2-morpholinoquinazolin-4-amine (43)
The title compound (85%) was prepared from 39c according to the synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 7.13 (d, J = 11.6 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 5.04 (d, J = 7.2 Hz, 1H), 4.11–4.02 (m, 1H), 3.91 (s, 3H), 3.81–3.76 (m, 8H), 2.83–2.80 (m, 2H), 2.29 (s, 3H), 2.19–2.07 (m, 4H), 1.64–1.55 (m, 2H). MS (ESI) m/z 376.2 [M + H]+.
7-Methoxy-6-methyl-N-(1-methylpiperidin-4-yl)-2-morpholinoquinazolin-4-amine (44)
The title compound (76%) was prepared from 39d according to the synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 7.18 (s, 1H), 6.79 (s, 1H), 5.15 (d, J = 7.2 Hz, 1H), 4.12–4.03 (m, 1H), 3.86 (s, 3H), 3.81–3.76 (m, 8H), 2.83–2.80 (m, 2H), 2.29 (s, 3H), 2.23 (s, 3H), 2.18–2.08 (m, 4H), 1.64–1.554 (m, 2H). MS (ESI): 372.3 [M + H]+.
7-Ethoxy-6-methoxy-N-(1-methylpiperidin-4-yl)-2-morpholinoquinazolin-4-amine (45)
The title compound (78%) was prepared from 39e according to the synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.85 (s, 1H), 6.77 (s, 1H), 5.17 (d, J = 7.6 Hz, 1H), 4.12 (q, J = 7.2 Hz, 2H), 4.09–4.03 (m, 1H), 3.87 (s, 3H), 3.78–3.74 (m, 8H), 2.84–2.81 (m, 2H), 2.27 (s, 3H), 2.16–2.09 (m, 4H), 1.63–1.53 (m, 2H), 1.07 (t, J = 6.8 Hz, 3H). MS (ESI) m/z 402.3 [M + H]+.
7-Isopropoxy-6-methoxy-N-(1-methylpiperidin-4-yl)-2-morpholinoquinazolin-4-amine (46)
The title compound (81%) was prepared from 39f according to the synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 6.85 (s, 1H), 6.79 (s, 1H), 5.20 (d, J = 6.8 Hz, 1H), 4.69–4.60 (m, 1H), 4.12–4.03 (m, 1H), 3.85 (s, 3H), 3.79–3.72 (m, 8H), 2.84–2.81 (m, 2H), 2.26 (s, 3H), 2.15–2.08 (m, 4H), 1.62–1.53 (m, 2H), 1.36 (d, J = 6.0 Hz, 6H). MS (ESI) m/z 416.3 [M + H]+.
7-Fluoro-6-methoxy-N-(1-methylpiperidin-4-yl)-2-morpholinoquinazolin-4-amine (47)
The title compound (85%) was prepared from 39g according to the synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 12.8 Hz, 1H), 6.90 (d, J = 8.8 Hz, 1H), 5.28 (d, J = 7.6 Hz, 1H), 4.19–4.10 (m, 1H), 3.95 (s, 3H), 3.83–3.74 (m, 8H), 2.97–2.94 (m, 2H), 2.38 (s, 3H), 2.56 (t, J = 11.2 Hz, 2H), 2.18–2.15 (m, 2H), 1.79–1.69 (m, 2H). MS (ESI) m/z 376.2 [M + H]+.
6-Methoxy-7-methyl-N-(1-methylpiperidin-4-yl)-2-morpholinoquinazolin-4-amine (48)
The title compound (75%) was prepared from 39h according to the synthetic procedure for 4. 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 1H), 6.65 (s, 1H), 5.19 (d, J = 7.2 Hz, 1H), 4.13–4.05 (m, 1H), 3.83 (s, 3H), 3.79–3.71 (m, 8H), 2.84–2.82 (m, 2H), 2.28 (s, 3H), 2.24 (s, 3H), 2.16–2.11 (m, 4H), 1.64–1.55 (m, 2H). MS (ESI) m/z 372.3 [M + H]+.
N-(2-Cyano-4,5-dimethoxyphenyl)tetrahydro-2H-pyran-4-carboxamide (50a)
To a solution of 2-amino-4,5-dimethoxybenzonitrile (372 mg, 2.1 mmol) in DCM (10 mL) were added DIEA (0.55 mL, 3.3 mmol) and tetrahydro-2H-pyran-4-carbonyl chloride (374 mg, 2.5 mmol) at 0 °C. After being stirred for 2 h, the reaction was concentrated, and the resulting residue was purified by silica gel chromatography to yield the title compound (577 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 8.49 (s, 1H), 6.92 (s, 1H), 5.92–5.51 (br, 1H), 4.07 (t, J = 3.5 Hz, 1H), 4.04 (t, J = 3.5 Hz, 1H), 4.00 (dd, J = 7.3, 1.2 Hz, 1H), 3.96 (s, 3H), 3.89 (s, 3H), 3.52–3.41 (m, 2H), 1.96–1.85 (m, 4H).
4-Chloro-6,7-dimethoxy-2-(tetrahydro-2H-pyran-4-yl)quinolone (52a)
To a solution of 50a (405 mg, 1.4 mmol) in EtOH (10 mL) were added H2O2 (10 mL) and NaOH (160 mg, 4.0 mmol). The resulting mixture was heated under reflux for 2 h before being cooled to rt. The precipitated solid was collected and dried under vacuum to yield intermediate 51a as a yellow solid. A mixture of 51a, PhNEt2 (1.5 mL, 1.4 mmol), and POCl3 (10 mL) was refluxed for 4 h before being cooled to rt. After removal of the solvent, cold water was added, followed by saturated aqueous NaHCO3 solution. The mixture was extracted with ethyl acetate (3 × 30 mL). The combined organic layers were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel chromatograph to yield the title compound (240 mg, 55%). 1H NMR (600 MHz, CDCl3) δ 7.40 (s, 1H), 7.33 (s, 1H), 4.14 (dd, J = 11.4, 3.2 Hz, 2H), 4.09 (s, 3H), 4.09 (s, 3H), 3.61 (t, J = 11.0 Hz, 2H), 3.34–3.11 (m, 1H), 2.11 (m 2H), 2.03 (m, 2H).
4-Chloro-2-cyclohexyl-6,7-dimethoxyquinoline (52b)
The title compound (41% over 3 steps) was prepared according to synthetic procedure for 52a. 1H NMR (600 MHz, CDCl3) δ 7.39 (s, 1H), 7.32 (s, 1H), 4.08 (s, 6H), 2.97 (tt, J = 11.9, 3.4 Hz, 1H), 2.16–2.01 (m, 2H), 1.92–1.90 (m, 2H), 1.83–1.67 (m, 3H), 1.52–1.41 (m, 2H), 1.40–1.30 (m, 1H).
2-Cyclohexyl-6,7-dimethoxy-N-(1-methylpiperidin-4-yl)-quinazolin-4-amine (53)
A mixture of 52a (60 mg, 0.2 mmol), 4-methylpiperidin-1-amine (0.05 mL, 0.40 mmol), and DIEA (0.06 mL, 0.36 mmol) in i-PrOH (1 mL) was heated by microwave irradiation at 160 °C for 15 min. After concentration, the crude product was purified by preparative HPLC to yield the title compound (69 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 7.13 (s, 1H), 6.86 (s, 1H), 5.31 (d, J = 6.8 Hz, 1H), 4.25–4.16 (m, 1H), 3.90 (s, 3H), 3.89 (s, 3H), 2.86–2.83 (m, 2H), 2.70 (tt, J = 11.6, 3.6 Hz, 1H), 2.29 (s, 3H) 2.21–2.12 (m, 4H), 1.97–1.94 (m, 2H), 1.82–1.79 (m, 2H), 1.72–1.54 (m, 5H), 1.42–1.22 (m, 3H). MS (ESI) m/z 385.3 [M + H]+.
6,7-Dimethoxy-N-(1-methylpiperidin-4-yl)-2-(tetrahydro-2H-pyran-4-yl)quinazolin-4-amine (54)
The title compound (80%) was prepared according to synthetic procedure for 53. 1H NMR (400 MHz, CDCl3) δ 7.12 (s, 1H), 6.83 (s, 1H), 5.26 (d, J = 7.6 Hz, 1H), 4.25–4.16 (m, 1H), 4.05 (dd, J = 11.2, 4.4 Hz, 2H), 3.92 (s, 6H), 3.52 (td, J = 12.0, 1.6 Hz, 2H), 2.93 (tt, J = 12.0, 4.0 Hz, 1H), 2.86–2.83 (m, 2H), 2.29 (s, 3H) 2.20–2.12 (m, 4H), 2.04 (qd, J = 12.0, 4.4 Hz, 2H), 1.87–1.84 (m, 2H), 1.59 (qd, J = 12.0, 4.0 Hz, 2H). MS (ESI) m/z 387.3 [M + H]+.
GLP and G9a IC50 Determination
Methyltransferase activity assays for G9a and GLP were performed by monitoring the incorporation of tritium-labeled methyl group to lysine 9 of H3 (1–25) peptide using scintillation proximity assay (SPA). The enzymatic reactions were performed at 23 °C with 20 min incubation of 10 μL of reaction mixture in 25 mM potassium phosphate pH 8.0, 1 mM EDTA, 2 mM MgCl2, 0.01% Triton X-100 containing 8 μM of cold SAM, and 2 μM of 3H-SAM (catalogue no. NET155 V250UC; PerkinElmer; www.perkinelmer.com), 1 μM of biotinylated H3 (1–25), 5 nM G9a or GLP, and compound titrations from 1.5 nM to 25 μM. To stop the reactions, 10 μL of 7.5 M guanidine hydrochloride was added, followed by 60 μL of buffer (20 mM Tris, pH 8.0), mixed, and transferred to a 384-well streptavidin coated Flash-plate (PerkinElmer, http://www.perkinelmer.ca). After mixing, the mixtures in Flash-plate were incubated for 2 h, and the CPM counts were measured using Topcount plate reader (PerkinElmer, www.perkinelmer.com). The CPM counts in the absence of compound for each data set were defined as 100% activity. In the absence of the enzyme, the CPM counts in each data set were defined as background (0%). All enzymatic reactions were performed in triplicate, and IC50 values were determined by fitting the data to Four Parameter Logistic equation using GraphPad Prism 7 software.
Selectivity Assays
Effect of compound on methyltransferase activity of SUV39H1, SUV39H2, SETDB1, SETD8, SUV420H1, SUV420H2, SETD7, MLL1 trimeric complex, MLL3 pentameric complex, EZH2 trimeric complex, PRMT1, PRMT3, PRMT4, PRMT5/MEP50 complex, PRMT6, PRMT7, PRMT8, PRMT9, PRDM9, SETD2, BCDIN3D, METTL3-14, SMYD2, SMYD3, and DNMT1 was assessed by monitoring the incorporation of tritium-labeled methyl group to lysine or arginine residues of peptide substrates using scintillation proximity assay (SPA). Assays were performed in a 10 μL reaction mixture containing 3H-SAM (catalogue no. NET155 V250UC; PerkinElmer; www.perkinelmer.com) and substrate at concentrations close to Km values for each enzyme. Two concentrations (1 μM and 10 μM) of compound were used in all selectivity assays. To stop the enzymatic reactions, 10 μL of 7.5 M guanidine hydrochloride was added, followed by 180 μL of buffer (20 mM Tris, pH 8.0), mixed, and then transferred to a 96-well FlashPlate (catalogue no. SMP103; PerkinElmer; www.perkinelmer.com). After mixing, the reaction mixtures in Flash plates were incubated for 1 h and the CPM were measured using Topcount plate reader (PerkinElmer, www.perkinelmer.com). The CPM counts in the absence of compound for each data set were defined as 100% activity. In the absence of the enzyme, the CPM counts in each data set were defined as background (0%).
For DNMT1, the dsDNA substrate was prepared by annealing two complementary strands (biotinylated forward strand B-GAGCCC-GTAAGCCCGTTCAGGTCG and reverse strand CGACCT-GAACGGGCTTACGGGCTC), synthesized by Eurofins MWG Operon.
For DOT1L, ASH1L, DNMT3A/3L, and DNMT3B/3L, a filter-based assay was used. In this assay, 10 μL of reaction mixtures were incubated at RT for 1 h, and 50 μL of 10% TCA was added, mixed, and transferred to filter-plates (Millipore; catalogue no. MSFBN6B10; www.millipore.com). Plates were centrifuged at 2000 rpm (Allegra X-15R - Beckman Coulter, Inc.) for 2 min followed by two additional 10% TCA washes and one ethanol wash (180 μL) followed by centrifugation. Plates were dried, and 100 μL of MicroO (MicroScint-O; catalogue no. 6013611, PerkinElmer; www.perkinelmer.com) were added to each well, centrifuged, and removed. Then 70 μL of MicroO was added again and CPM was measured using a Topcount plate reader.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) measurements were made at 25 °C on a MicroCal ITC200 Instrument (Malvern Instruments). Co-concentrated G9a–SAM and GLP–SAM (protein/SAM molar ratio of 1:5) were diluted at 35 μM in ITC buffer [50 mM Tris (pH 8.0), 150 mM NaCl] supplemented with 1% DMSO. Compounds 4 and 18 were prepared in DMSO at 50 mM and diluted to 0.5 mM in ITC buffer with a final DMSO concentration of 1%. Binding constants were calculated by fitting the data using the ITC data analysis module in Origin 7.0 (OriginLab Corp.).
Mechanism of Action (MOA) Studies
IC50 values were determined for compound 4 and 18 at various concentrations of SAM (3.1, 6.2,12.5, 25, 50, and 100 μM), 5 nM of GLP, 5 μM of biotinylated H3 (1–25) peptide, or at various concentrations of biotinylated H3 (1–25) peptide substrate (0.31, 0.62, 1.2, 2.5, 5, and 10 μM), 5 nM of GLP, and 50 μM SAM. The reaction mixtures were incubated for 15 min at 23 °C. To stop the enzymatic reactions, an equal volume of 7.5 M guanidine hydrochloride was added, followed by 180 μL of buffer (20 mM Tris, pH 8.0), mixed, and then transferred to a 96-well FlashPlate (catalogue no. SMP103; PerkinElmer; www.perkinelmer.com). After mixing, the reaction mixtures in Flash plates were incubated for 2 h and the CPM were measured using a Topcount plate reader (PerkinElmer, www.perkinelmer.com). The CPM counts in the absence of compound for each data set were defined as 100% activity. In the absence of the enzyme, the CPM counts in each data set were defined as background (0%). The IC50 values were determined using GraphPad Prism 7 software.
Protein Expression, Purification, Crystallization, Data Collection, and Structure Determination for Compound 4
The recombinant proteins GLP catalytic SET domain (982–1266) and G9a catalytic SET domain (913–1193), from plasmids given by Alexander Plotnikov, were overexpressed in Escherichia coli BL21 (DE3) codon plus RIL strain (Stratagene) by addition of 0.3 mM isopropyl-1-thio-D-galactopyranoside with a supplement of 1 mM zinc sulfate and incubated overnight at 15 °C. Harvested cells were resuspended in 50 mM sodium phosphate buffer, pH 7.5, supplemented with 0.5 mM sodium chloride and 5% glycerol, and lysed using a microfluidizer (Microfluidics) at 10000 psi. After clarification of the crude extract by high-speed centrifugation, the lysate was loaded onto a 5 mL HiTrap chelating column (GE Healthcare) charged with Ni2+. The column was washed, and the protein was eluted with 50 mM Tris buffer, pH 8.0, 250 mM sodium chloride, 250 mM imidazole, and supplemented with 0.5 mM of TCEP. The protein was next purified on a Superdex200 column (26/600; GE Healthcare) equilibrated with 50 mM Tris-HCl buffer, pH 8.0, and 150 mM sodium chloride, and the elution fractions were pooled and supplemented with 0.5 mM of TCEP. All purification steps were performed at 4 °C and in the presence of a protease inhibitor AEBSF (Goldbio). The size and the purity of the recombinant G9a and GLP proteins were checked by SDS-PAGE and mass spectroscopy.
Purified GLP and G9a were mixed in the presence of S-adenosyl-L-methionine (Sigma) and compound 4 (protein/SAM/4 molar ratio of 1:5:10), coconcentrated to about 20 mg/mL and crystallized using the hanging drop vapor diffusion method at 17 °C by mixing equal volume of the protein solution with the reservoir solution. The GLP–SAM–4 ternary complex was crystallized in 20% PEG 20,000, 2% (v/v) 1,4-dioxane, 0.1 M bicine (pH 9.0). The G9a–SAM–4 ternary complex was crystallized in 20% (w/v) PEG 3350, 0.2 M NaF, 0.1 M Bis-Tris propane (pH 6.5). GLP–SAM–4 crystals were soaked in the corresponding mother liquor supplemented with 20% ethylene glycol, and the G9a–SAM–4 complex crystals in a mixture of parafin 70% and silicon 30% as cryoprotectant before flash freezing in liquid nitrogen.
X-ray diffraction data were collected at 100 K at NE-CAT beamline 24-ID-E of Advanced Photon Source (APS) at Argonne National Laboratory. The data were processed with MOSFLM, SCALA, and other programs from the CCP4 suite.58 The structures of the G9a and GLP complexes were solved by molecular replacement using the PHASER59 and the atomic model of the G9a and GLP SET domains (PDB 3K5K and 3HNA, respectively). The locations of the bound molecules were determined from a Fo–Fc difference electron density map. REFMAC60 and phenix.refine61,62 were used for structure refinement. Graphic program COOT63 was used for model building and visualization. The overall assessment of model quality was performed using MOLPROBITY.64
Protein Expression, Purification, Crystallization, Data Collection, and Structure Determination for Compound 18
Human G9a and GLP catalytic domains were cloned, expressed, and purified as previously described.65 Purified G9a and GLP proteins were premixed with compound 18 at 1:2 molar ratio of protein:compound. The protein–compound complexes were crystallized using sitting drop vapor diffusion method at 20 °C by mixing an equal volume of the protein solution with the reservoir solution containing 25% PEG3350, 0.2 M NaCl, 0.1 M Bis-Tris pH 6.5 (for G9a), and 20% 2-propanol, 20% PEG 4 K, 0.1 M Na citrate, pH 5.6 (for GLP).
X-ray diffraction data for G9a–18 and GLP–18 were both collected at 100 K at beamline 19ID of Advanced Photon Source (APS), Argonne National Laboratory. Both data sets were processed using the HKL-3000 suite.66 The structures of G9a and GLP with 18 were solved by molecular replacement using MOLREP,67 with PDBs 3K5K and 4I51 as search template, respectively. REFMAC60 was used for structure refinement. Geometry restraints for the compound refinement were prepared with by GRADE Developed at Global Phasing Ltd. (O. S. Smart, T. O. Womack, A. Sharff, C. Flensburg, P. Keller, W. Paciorek, C. Vonrhein, and G. Bricogne (2011) grade v1.102. Cambridge, United Kingdom, Global Phasing Ltd.) Graphics program COOT63 was used for model building and visualization. MOLPRO-BITY64 was used for structure validation. Crystal diffraction data and refinement statistics for the structure are displayed in Table 1.
Supplementary Material
Acknowledgments
We thank Albina Bolotokova for compound management and Taraneh Hajian for protein purification. The research described here was supported by the grant R01GM103893 (to J.J.) from the U.S. National Institutes of Health. The SGC is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust. We thank the staff at the 24-ID-E beamline of Advanced Photon Source (APS) at Argonne National Laboratory for facilitating x-ray data collection.
ABBREVIATIONS USED
- GLP
G9a-like protein 1
- PKMT
protein lysine methyltransferase
- PRMT
protein arginine methyltransferase
- H3K9me1
monomethylation of lysine 9 on histone H3
- H3K9me2
dimethylation of lysine 9 on histone H3
- SAM
S-5′-adenosyl-L-methionine
- SAH
S-5′-adenosyl-L-homocysteine
- SAR
structure–activity relationship
- DNMT
DNA methyltransferase
- RNMT
RNA methyltransferases
- SET
suppressor of variegation 3–9 enhancer-of-zeste trithorax
- SIRT1
sirtuin 1
- MyoD
myogenic differentiation 1
- CDYL1
chromodomain Y-like protein
- WIZ
widely interspaced zinc finger motifs protein
- RMSD
root-mean-square deviation
- METTL3–14
N6-adenosine-methyltransferase subunit METTL3 and 14 complex
- ITC
isothermal titration calorimetry
Footnotes
Accession Codes
The structure of GLP in complex with 4 has been deposited under PDB ID 5TUZ. The structure of G9a in complex with 4 has been deposited under PDB ID 5TUY. The structure of GLP in complex with 18 has been deposited under PDB ID 5TTG. The structure of G9a in complex with 18 has been deposited under PDB ID 5TTF. Authors will release the atomic coordinates and experimental data upon article publication.
ORCID
Jian Jin: 0000-0002-2387-3862
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.6b01645.
1H and 13C NMR spectra of compound 4 and 18; crystallography data and refinement statistics (PDF) Molecular formula strings (CSV)
References
- 1.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 2.Kaniskan HU, Jin J. Chemical probes of histone lysine methyltransferases. ACS Chem Biol. 2015;10:40–50. doi: 10.1021/cb500785t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaniskan HU, Konze KD, Jin J. Selective inhibitors of protein methyltransferases. J Med Chem. 2015;58:1596–1629. doi: 10.1021/jm501234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benevento M, van de Molengraft M, van Westen R, van Bokhoven H, Kasri NN. The role of chromatin repressive marks in cognition and disease: A focus on the repressive complex GLP/G9a. Neurobiol Learn Mem. 2015;124:88–96. doi: 10.1016/j.nlm.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 5.Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mozzetta C, Pontis J, Ait-Si-Ali S. Functional crosstalk between lysine methyltransferases on histone substrates: the case of G9A/GLP and polycomb repressive complex 2. Antioxid Redox Signaling. 2015;22:1365–1381. doi: 10.1089/ars.2014.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mozzetta C, Boyarchuk E, Pontis J, Ait-Si-Ali S. Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat Rev Mol Cell Biol. 2015;16:499–513. doi: 10.1038/nrm4029. [DOI] [PubMed] [Google Scholar]
- 8.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z, Pugh BF. High-resolution genome-wide mapping of the primary structure of chromatin. Cell. 2011;144:175–186. doi: 10.1016/j.cell.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, Snyder JP, Bedford MT, Cheng X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Mol Biol. 2009;16:312–317. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–1791. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005;19:815–826. doi: 10.1101/gad.1284005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the jumonji domain-containing JMJD2/KDM4 proteins. J Biol Chem. 2009;284:8395–8405. doi: 10.1074/jbc.M807818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss T, Hergeth S, Zeissler U, Izzo A, Tropberger P, Zee BM, Dundr M, Garcia BA, Daujat S, Schneider R. Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenet Chromatin. 2010;3:7. doi: 10.1186/1756-8935-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patnaik D, Chin HG, Esteve PO, Benner J, Jacobsen SE, Pradhan S. Substrate specificity and kinetic mechanism of mammalian G9a histone H3 methyltransferase. J Biol Chem. 2004;279:53248–53258. doi: 10.1074/jbc.M409604200. [DOI] [PubMed] [Google Scholar]
- 19.Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. SET domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem. 2001;276:25309–25317. doi: 10.1074/jbc.M101914200. [DOI] [PubMed] [Google Scholar]
- 20.Wu H, Chen XZ, Xiong J, Li YF, Li H, Ding XJ, Liu S, Chen S, Gao SR, Zhu B. Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell Res. 2011;21:365–367. doi: 10.1038/cr.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu YX, Song CY, Zhang QY, DiMaggio PA, Garcia BA, York A, Carey MF, Grunstein M. Histone H3 lysine 56 methylation regulates DNA replication through its interaction with PCNA. Mol Cell. 2012;46:7–17. doi: 10.1016/j.molcel.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biggar KK, Li SSC. Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol. 2015;16:5–17. doi: 10.1038/nrm3915. [DOI] [PubMed] [Google Scholar]
- 23.Huang J, Dorsey J, Chuikov S, Perez-Burgos L, Zhang XY, Jenuwein T, Reinberg D, Berger SL. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J Biol Chem. 2010;285:18122–18122. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore KE, Carlson SM, Camp ND, Cheung P, James RG, Chua KF, Wolf-Yadlin A, Gozani O. A general molecular affinity strategy for global detection and proteomic analysis of lysine methylation. Mol Cell. 2013;50:444–456. doi: 10.1016/j.molcel.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JS, Kim Y, Kim IS, Kim B, Choi HJ, Lee JM, Shin HJR, Kim JH, Kim JY, Seo SB, Lee H, Binda O, Gozani O, Semenza GL, Kim M, Kim KI, Hwang D, Baek SH. Negative regulation of hypoxic responses via induced reptin methylation. Mol Cell. 2010;39:71–85. doi: 10.1016/j.molcel.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ling BMT, Bharathy N, Chung TK, Kok WK, Li S, Tan YH, Rao VK, Gopinadhan S, Sartorelli V, Walsh MJ, Taneja R. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc Natl Acad Sci U S A. 2012;109:841–846. doi: 10.1073/pnas.1111628109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, Komatsu Y, Shinkai Y, Cheng XD, Jeltsch A. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol. 2008;4:344–346. doi: 10.1038/nchembio.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sampath SC, Marazzi I, Yap KL, Sampath SC, Krutchinsky AN, Mecklenbrauker I, Viale A, Rudensky E, Zhou MM, Chait BT, Tarakhovsky A. Methylation of a histone mimic within the histone methyltransferase G9a regulates protein complex assembly. Mol Cell. 2007;27:596–608. doi: 10.1016/j.molcel.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 29.Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tachibana M, Nozaki M, Takeda N, Shinkai Y. Functional dynamics of H3K9 methylation during meiotic prophase progression. EMBO J. 2007;26:3346–3359. doi: 10.1038/sj.emboj.7601767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kondo Y, Shen L, Suzuki S, Kurokawa T, Masuko K, Tanaka Y, Kato H, Mizuno Y, Yokoe M, Sugauchi F, Hirashima N, Orito E, Osada H, Ueda R, Guo Y, Chen XL, Issa JPJ, Sekido Y. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol Res. 2007;37:974–983. doi: 10.1111/j.1872-034X.2007.00141.x. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe H, Soejima K, Yasuda H, Kawada I, Nakachi I, Yoda S, Naoki K, Ishizaka A. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008;8:15. doi: 10.1186/1475-2867-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kondo Y, Shen L, Ahmed S, Boumber Y, Sekido Y, Haddad BR, Issa JPJ. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS One. 2008;3:e2037. doi: 10.1371/journal.pone.0002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas LR, Miyashita H, Cobb RM, Pierce S, Tachibana M, Hobeika E, Reth M, Shinkai Y, Oltz EM. Functional analysis of histone methyltransferase G9a in B and T lymphocytes. J Immunol. 2008;181:485–493. doi: 10.4049/jimmunol.181.1.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehnertz B, Northrop JP, Antignano F, Burrows K, Hadidi S, Mullaly SC, Rossi FMV, Zaph C. Activating and inhibitory functions for the histone lysine methyltransferase G9a in T helper cell differentiation and function. J Exp Med. 2010;207:915–922. doi: 10.1084/jem.20100363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maze I, Covington HE, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren YH, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaefer A, Sampath SC, Intrator A, Min A, Gertler TS, Surmeier DJ, Tarakhovsky A, Greengard P. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron. 2009;64:678–691. doi: 10.1016/j.neuron.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leung DC, Dong KB, Maksakova IA, Goyal P, Appanah R, Lee S, Tachibana M, Shinkai Y, Lehnertz B, Mager DL, Rossi F, Lorincz MC. Lysine methyltransferase G9a is required for de novo DNA methylation and the establishment, but not the maintenance, of proviral silencing. Proc Natl Acad Sci U S A. 2011;108:5718–5723. doi: 10.1073/pnas.1014660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleefstra T, Smidt M, Banning MJG, Oudakker AR, Van Esch H, de Brouwer APM, Nillesen W, Sistermans EA, Hamel BCJ, de Bruijn D, Fryns JP, Yntema HG, Brunner HG, de Vries BBA, van Bokhoven H. Disruption of the gene euchromatin histone methyl transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J Med Genet. 2005;42:299–306. doi: 10.1136/jmg.2004.028464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antignano F, Zaph C. Regulation of CD4 T-cell differentiation and inflammation by repressive histone methylation. Immunol Cell Biol. 2015;93:245–252. doi: 10.1038/icb.2014.115. [DOI] [PubMed] [Google Scholar]
- 41.Kramer JM. Regulation of cell differentiation and function by the euchromatin histone methyltranserfases G9a and GLP. Biochem Cell Biol. 2016;94:26–32. doi: 10.1139/bcb-2015-0017. [DOI] [PubMed] [Google Scholar]
- 42.Liu N, Zhang ZQ, Wu H, Jiang YH, Meng LJ, Xiong J, Zhao ZD, Zhou XH, Li J, Li H, Zheng Y, Chen S, Cai T, Gao SR, Zhu B. Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes Dev. 2015;29:379–393. doi: 10.1101/gad.254425.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohno H, Shinoda K, Ohyama K, Sharp LZ, Kajimura S. EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature. 2013;504:163–167. doi: 10.1038/nature12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Battisti V, Pontis J, Boyarchuk E, Fritsch L, Robin P, Ait-Si-Ali S, Joliot V. Unexpected distinct roles of the related histone H3 lysine 9 methyltransferases G9a and G9a-like protein in myoblasts. J Mol Biol. 2016;428:2329–2343. doi: 10.1016/j.jmb.2016.03.029. [DOI] [PubMed] [Google Scholar]
- 45.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wasney GA, Dong AP, Barsyte D, Kozieradzki I, Senisterra G, Chau I, Siarheyeva A, Kireev DB, Jadhav A, Herold JM, Frye SV, Arrowsmith CH, Brown PJ, Simeonov A, Vedadi M, Jin J. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J Med Chem. 2009;52:7950–7953. doi: 10.1021/jm901543m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wigle TJ, Wasney GA, Dong AP, Senisterra G, Chau I, Siarheyeva A, Norris JL, Kireev DB, Jadhav A, Herold JM, Janzen WP, Arrowsmith CH, Frye SV, Brown PJ, Simeonov A, Vedadi M, Jin JA. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J Med Chem. 2010;53:5844–5857. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang YQ, Ganesh T, Horton JR, Spannhoff A, Liu J, Sun AM, Zhang X, Bedford MT, Shinkai YC, Snyder JP, Cheng XD. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J Mol Biol. 2010;400:1–7. doi: 10.1016/j.jmb.2010.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu F, Barsyte-Lovejoy D, Allali-Hassani A, He YL, Herold JM, Chen X, Yates CM, Frye SV, Brown PJ, Huang J, Vedadi M, Arrowsmith CH, Jin J. Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. J Med Chem. 2011;54:6139–6150. doi: 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, DiMaggio PA, Wasney GA, Siarheyeva A, Dong AP, Tempel W, Wang SC, Chen X, Chau I, Mangano TJ, Huang XP, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7:648–648. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu F, Barsyte-Lovejoy D, Li FL, Xiong Y, Korboukh V, Huang XP, Allali-Hassani A, Janzen WP, Roth BL, Frye SV, Arrowsmith CH, Brown PJ, Vedadi M, Jin J. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem. 2013;56:8931–8942. doi: 10.1021/jm401480r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Konze KD, Pattenden SG, Liu F, Barsyte-Lovejoy D, Li FL, Simon JM, Davis IJ, Vedadi M, Jin J. A chemical tool for in vitro and in vivo precipitation of lysine methyltransferase G9a. Chem Med Chem. 2014;9:549–553. doi: 10.1002/cmdc.201300450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sweis RF, Pliushchev M, Brown PJ, Guo J, Li FL, Maag D, Petros AM, Soni NB, Tse C, Vedadi M, Michaelides MR, Chiang GG, Pappano WN. Discovery and development of potent and selective inhibitors of histone methyltransferase G9a. ACS Med Chem Lett. 2014;5:205–209. doi: 10.1021/ml400496h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan Y, Wang Q, Paulk J, Kubicek S, Kemp MM, Adams DJ, Shamji AF, Wagner BK, Schreiber SL. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem Biol. 2012;7:1152–1157. doi: 10.1021/cb300139y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Devkota K, Lohse B, Liu Q, Wang MW, Staerk D, Berthelsen J, Clausen RP. Analogues of the natural product sinefungin as inhibitors of EHMT1 and EHMT2. ACS Med Chem Lett. 2014;5:293–297. doi: 10.1021/ml4002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kondengaden SM, Luo LF, Huang K, Zhu MY, Zang LL, Bataba E, Wang RL, Luo C, Wang BH, Li KK, Wang PG. Discovery of novel small molecule inhibitors of lysine methyltransferase G9a and their mechanism in leukemia cell lines. Eur J Med Chem. 2016;122:382–393. doi: 10.1016/j.ejmech.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 56.Chen WL, Wang ZH, Feng TT, Li DD, Wang CH, Xu XL, Zhang XJ, You QD, Guo XK. Discovery, design and synthesis of 6H-anthra[1,9-cd]isoxazol-6-one scaffold as G9a inhibitor through a combination of shape-based virtual screening and structure-based molecular modification. Bioorg Med Chem. 2016;24:6102–6108. doi: 10.1016/j.bmc.2016.09.071. [DOI] [PubMed] [Google Scholar]
- 57.Lehnertz B, Pabst C, Su L, Miller M, Liu F, Yi L, Zhang R, Krosl J, Yung E, Kirschner J, Rosten P, Underhill TM, Jin J, Hebert J, Sauvageau G, Humphries RK, Rossi FM. The methyltransferase G9a regulates HoxA9-dependent transcription in AML. Genes Dev. 2014;28:317–327. doi: 10.1101/gad.236794.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bailey S. The CCP4 suite - programs for protein crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 59.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr, Sect D: Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 61.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr, Sect D: Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davis IW, Murray LW, Richardson JS, Richardson DC. MolProbity: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu H, Min JR, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, Allali-Hassani A, Campagna-Slater V, Vedadi M, Arrowsmith CH, Plotnikov AN, Schapira M. Structural biology of human H3K9 methyltransferases. PLoS One. 2010;5:e8570. doi: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 67.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.