

Abstract
Clinically isolated syndrome (CIS) is a term that describes the first clinical onset of potential multiple sclerosis (MS). The term CIS is typically applied to young adults with episodes of acute or subacute onset, which reaches a peak quite rapidly within 2–3 weeks. In 85% of young adults who develop MS, onset occurs with an acute, CIS of the optic nerves, brainstem, or spinal cord. When clinically silent brain lesions are seen on MRI, the likelihood of developing MS is high. Because no single clinical feature or diagnostic test is sufficient for the diagnosis of CIS, diagnostic criteria have included a combination of both clinical and paraclinical studies. Diagnostic criteria from the International Panel of McDonald and colleagues incorporate MRI evidence of dissemination in time and space to allow a diagnosis of definite MS in patients with CIS. As CIS is typically the earliest clinical expression of MS, research on patients with CIS may provide new insights into early pathological changes and pathogenetic mechanisms that might affect the course of the disorder. With recent improvements in diagnosis and the advent of disease-modifying treatments for MS, there has been growing interest and research in patients with CIS.
Keywords: Clinically isolated syndrome, multiple sclerosis, diagnostic criteria
CLINICALLY ISOLATED SYNDROME AND ITS RELATION TO MULTIPLE SCLEROSIS
Clinically isolated syndrome (CIS) refers to a single clinical attack of central nervous system (CNS) inflammatory demyelinating symptoms that are suggestive of multiple sclerosis (MS). CIS presentations can be monofocal or multifocal and typically involve the optic nerve, brainstem, cerebellum, spinal cord, or cerebral hemispheres. Although CIS may represent the first manifestation of definite multiple sclerosis (DMS), some patients may not develop a second clinical relapse. To be termed as CIS, the episode should last for at least 24 h and should occur in the absence of fever or infection and with no clinical features of encephalopathy (1,2,3,4).
The course of MS after CIS is variable: after 15–20 years, a third of the patients have a benign course with minimal or no disability, while half will develop secondary progressive MS with increasing disability. Current predictions of the long-term course at disease onset are unreliable (1). The onset of MS in 85% of young adults (aged 20–45 years) is with a subacute CIS of the optic nerves, brainstem, or spinal cord. While multifocal brain lesions are present on a magnetic resonance imaging (MRI) in many patients with CIS, some patients have additional abnormalities on quantitative MRI in otherwise normal-appearing white and gray matter, suggesting an extensive pathological process (5). Although patients usually recover from their presenting episode, CIS is often the first manifestation of MS. The most notable predictors for developing MS are clinically silent MRI lesions and cerebrospinal fluid (CSF) oligoclonal bands (OCBs), while weak or uncertain risk factors include vitamin D deficiency, Epstein–Barr virus infection, smoking, HLA genes, and miscellaneous immunological abnormalities (1).
A review of a large database of patients with CISs found that 21% presented with optic neuritis, 46% with long-tract signs and symptoms, 10% with a brainstem syndrome, and 23% with multifocal abnormalities. Therefore, the syndrome was isolated in space in only 77% of the presentations, although all presentations were isolated in time. The presentation of MS affects disease course and prognosis (Table 1) (4).
Table 1.
Features of CIS and early multiple sclerosis reported to affect prognosis (4)
| Good prognosis | Poor prognosis |
| Optic neuritis | “Multifocal” CIS |
| Isolated sensory symptoms | Efferent systems affected |
| Long interval to second relapse | High relapse rate in the first 2–5 years |
| No disability after 5 years | Substantial disability after 5 years |
| Normal MRI | Abnormal MRI with a large lesion load |
CIS: clinically isolated syndrome; MRI: magnetic resonance imaging
DIAGNOSIS
When patients present with CIS, clinicians are faced with many questions. Is CIS due to a disorder other than MS? What is the likelihood that the person will develop MS? If MS does develop, is it possible to ascertain the likelihood of disability? Should the patient be assessed with MRI, CSF analysis, or other tests? What should patients be told? Should patients be treated to assist recovery from CIS or to delay the development of MS? (4).
DIFFERENTIAL DIAGNOSIS
The ability of making an accurate diagnosis as early as possible is important for patient management, counseling, and optimal therapy (Figure 1) (6). The diagnosis of patients with CIS-MS, another neurological disorder, or CIS only is a clinical decision and should be made by a neurologist experienced in the relevant differential diagnosis. Certain inflammatory or infectious disorders (e.g., systemic lupus erythematosus and neuroborreliosis) that are commonly part of the differential diagnosis can be investigated through blood and CSF analyses (4).
Figure 1.
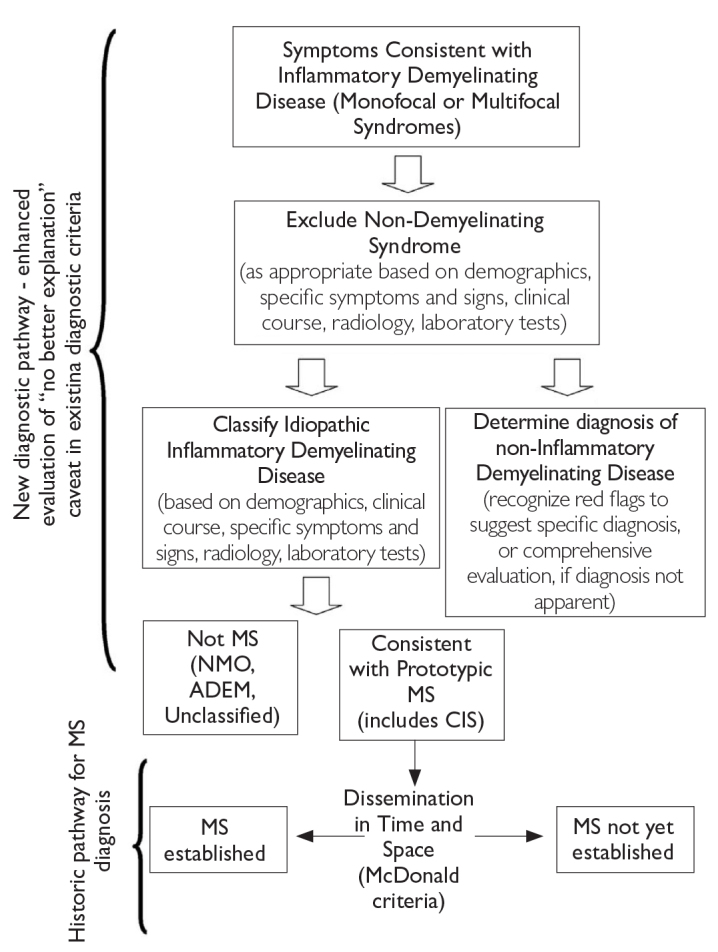
Steps in MS differential diagnosis (6)
MS: multiple sclerosis
Table 2 categorizes CIS presentations of patients eventually diagnosed as having MS into (i) those that are typical of patients eventually diagnosed as having MS, (ii) those that are less common but nevertheless may be an initial presentation in patients eventually diagnosed with MSor may signal another disease, and (iii) those that are atypical, suggesting an alternative diagnosis (6).
Table 2.
Clinical features of CIS and likelihood of signaling an MS diagnosis (6)
| CIS features typically seen in MS | Less common CIS features that may be seen in MS | Atypical CIS features not expected in MS |
|---|---|---|
|
| ||
| Optic nerve | ||
| Unilateral optic neuritis | Bilateral simultaneous optic neuritis | Progressive optic neuropathy |
| Pain on eye movement | No pain | Severe, continuous orbital pain |
| Partial and mainly central visual blurring | No light perception | Persistent complete loss of vision |
| Normal disc or mild disc swelling | Moderate to severe disc swelling with no hemorrhages Uveitis (mild, posterior) | Neuroretinitis (optic disc swelling with macular star) Uveitis (severe, anterior) |
|
| ||
| Brain stem/cerebellum | ||
| Bilateral internuclear ophthalmoplegia | Unilateral internuclear ophthalmoplegia, facial palsy, facial myokmia | Complete external ophthmalmoplegia; vertical gaze palsies |
| Ataxia and multidirectional nystagmus | Deafness | Vascular territory syndrome, e.g., lateral medullary |
| Sixth nerve palsy | One-and-a-half syndrome | Third nerve palsy |
| Facial numbness | Trigeminal neuralgia Paroxysmal tonic spasms |
Progressive trigeminal sensory neuropathy Focal dystonia, torticollis |
|
| ||
| Spinal cord | ||
| Partial myelopathy | Complete transverse myelitis | Anterior spinal artery territory lesion (sparing posterior columns only) |
| Lhermitte’s symptom Deafferented hand | Radiculopathy, areflexia Segmental loss of pain and temperature sensation | Cauda equina syndrome |
| Numbness | Partial Brown–Sequard syndrome (sparing posterior colums) | Sharp sensory level to all modalities and localized spinal pain |
| Urinary urgency, incontinence, erectile dysfunction | Fecal incontinence | Complete Brown–Sequard syndrome |
| Progressive spastic paraplegia (asymmetrical) | Progressive spastic paraplegia (symmetrical) | Acute urinary retention Progressive sensory ataxia (posterior columns) |
|
| ||
| Celebral hemispheres | ||
| Mild subcortical cognitive impairment | Epilepsy | Encephalopathy (obtundation, confusion, drowsiness)a |
| Hemiparesis | Hemianopia | Cortical blindness |
Although encephalopathy is required for ADEM, it may also be seen at presentation and/or during the course of MS.
CIS: clinically isolated syndrome; MS: multiple sclerosis
Magnetic resonance imaging is a crucial investigation for the differentiation of brainstem and spinal cord CIS owing to inflammatory demyelination from other structural lesions (e.g., brainstem vascular malformation and cord compression) (6). MRI can also influence a decision as to whether a presentation is due to monofocal or multifocal lesions and the likelihood for the ultimate diagnosis of MS (Table 3) (6).
Table 3.
Clinically isolated syndromes in the differential diagnosis of MS (6)
| Type 1 CIS: clinically monofocal, at least one asymptomatic MRI lesion |
| Type 2 CIS: clinically multifocal, at least one asymptomatic MRI lesion |
| Type 3 CIS: clinically monofocal, may appear normal; no symptomatic MRI lesion |
| Type 4 CIS: clinically multifocal, may appear normal; no symptomatic MRI lesion |
| Type 5 CIS: no clinical presentation to suggest demyelinating disease, but MRI is suggestive |
Note: symptomatic lesion should appear typical for demyelination; they may be located in the brain or cord, although they are more often occur in the brain; current evidence on the prognostic value of asymptomatic lesion mainly comes from brain imaging. CIS: clinically isolated syndrome; MS: multiple sclerosis; MRI: magnetic resonance imaging
The three most common CIS syndromes seen at presentation in MS diagnosis include those affecting the optic nerve, brain stem, and spinal cord (6). In patients with suspected optic neuritis, there are many clinical features that suggest a cause other than inflammatory demyelination (4). Figures 2, 3, and 4 show clinical aspects that are typical for demyelination as seen in MS and atypical features that should trigger a consideration of other diagnoses (1,6).
Figure 2.
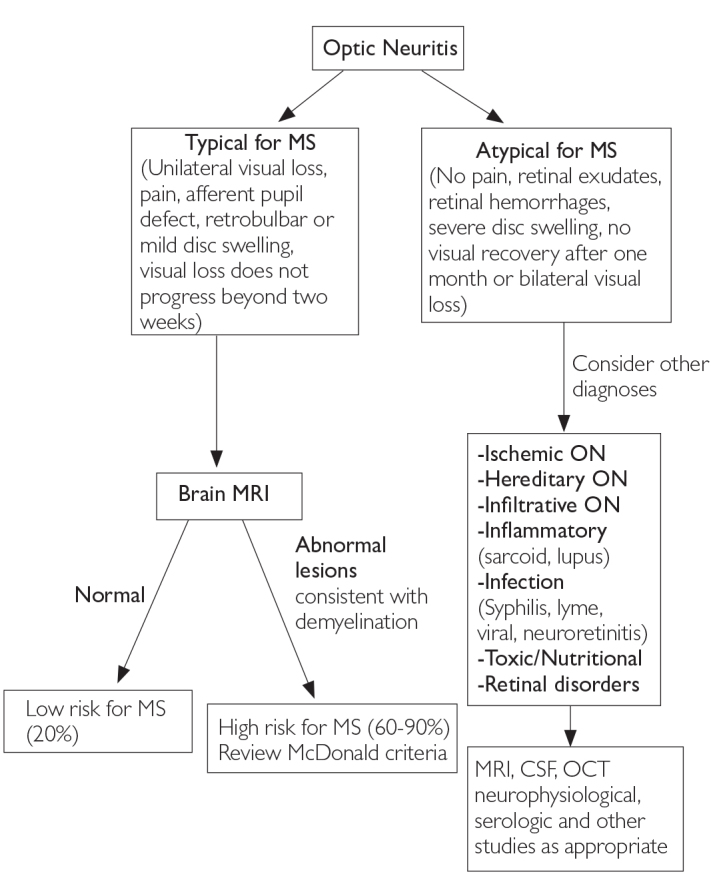
Differential diagnosis upon presentation with demyelinating optic neuritis (6)
MRI: magnetic resonance imaging; MS: multiple sclerosis; CSF: cerebrospinal fluid
Figure 3.
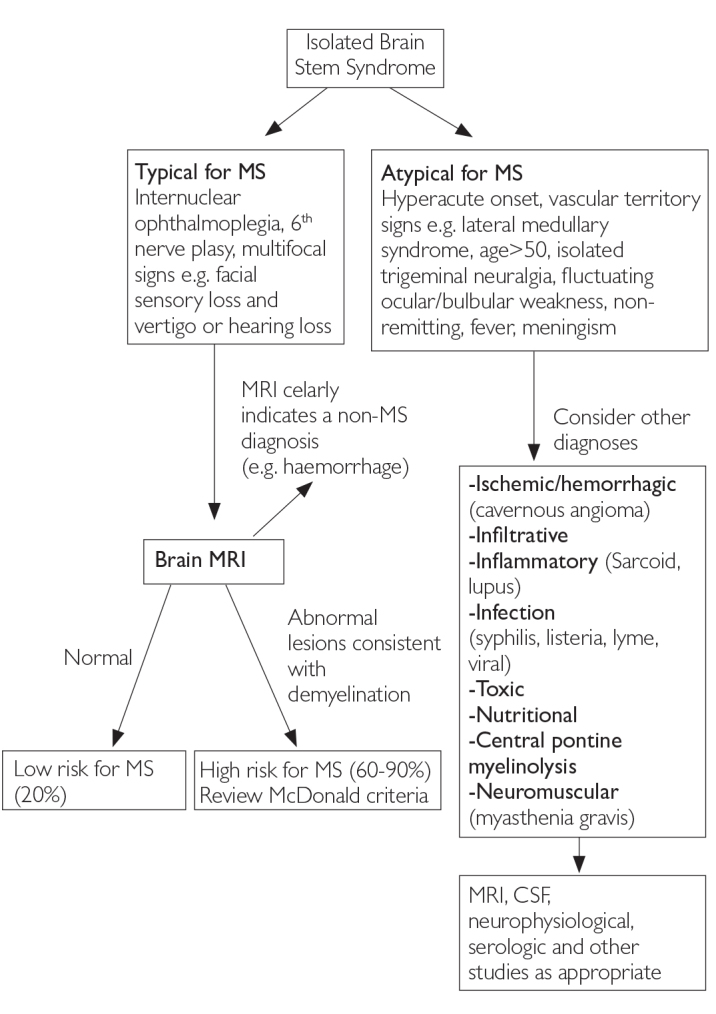
Differential diagnosis upon presentation with demyelinating brain stem syndrome (6)
MRI: magnetic resonance imaging; MS: multiple sclerosis; CSF: cerebrospinal fluid
Figure 4.
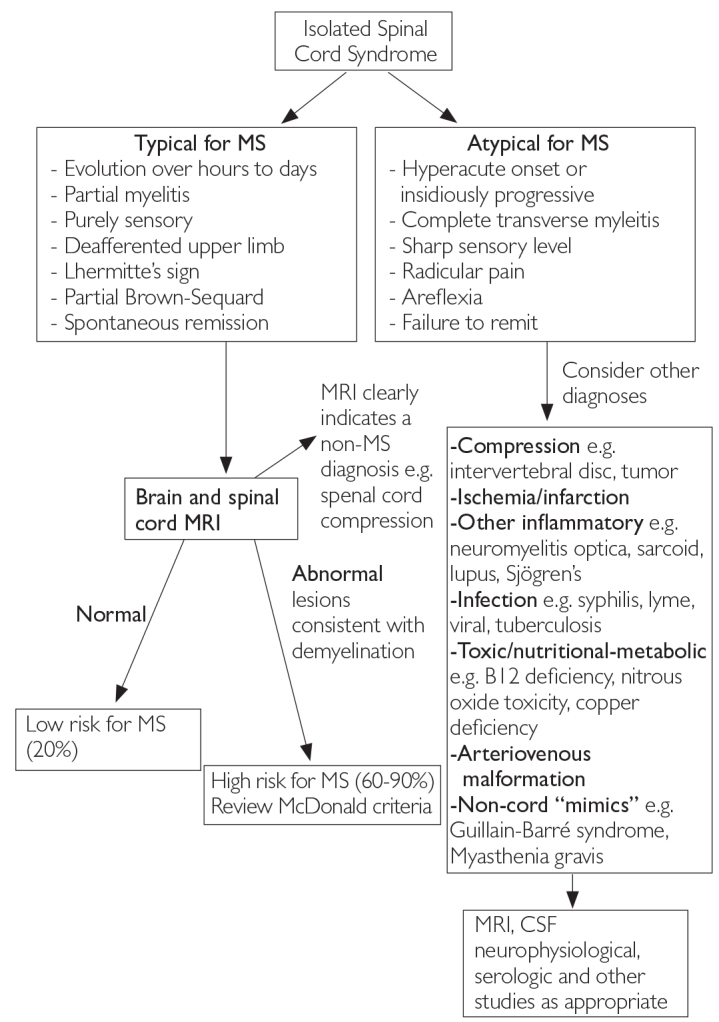
Differential diagnosis upon presentation with demyelinating spinal cord syndrome (6)
MRI: magnetic resonance imaging; MS: multiple sclerosis; CSF: cerebrospinal fluid
CSF
Although CSF OCBs increase the risk of CIS developing into MS, they add little to MRI-assigned risk. Tintore et al. (7) recorded a conversion to MS in 59% of all patients with CIS with more than 10 brain lesions and in 64% of patients who had more than 10 brain lesions and CSF OCBs. For MS diagnosis or prediction, many neurologists think that MRI alone is sufficient. However, CSF examination helps predict the conversion to MS in patients with a negative MRI or with MRI showing few lesions (i.e., MRI that does not meet the McDonald criteria for dissemination in space). In patients with negative MRI, the presence of OCBs increases the risk for developing MS from 4% to 23%. Therefore, the development of MS is unlikely in patients with CIS showing few or no MRI lesions and no CSF OCBs (1).
Typical CSF findings in MS include a slightly elevated leukocyte count (5–50 cells per μL compared with a normal range of <5 cells per μl), the presence of activated B cells or plasma cells on cytological analysis, elevated immunoglobulin G (IgG) synthesis, OCBs, and an increased synthesis of intrathecally produced immunoglobulins against measles, rubella, and varicella zoster viruses (Table 4). CSF albumin levels and the CSF albumin:serum ratio (an accurate measure of the integrity of the blood–CSF barrier) are usually normal but can be elevated in some patients with MS. Normal CSF findings are present in up to 10% of patients with MS (8).
Table 4.
Diagnostic CSF markers in clinically definite MS (8)
| Marker | Detection method | Normal range | MS range | Prevalence in patients with MS (%) |
|---|---|---|---|---|
|
| ||||
| Oligoclonal lgG bands | Isoelectric focusing and immunoblot | ≤1 band | Unique in CSF >1 band | 90–100 |
|
| ||||
| Albumin CSF:serum ratio | Nephelometry | Age dependent <5–10×10−3 | <10×10−3 | 85–90 |
|
| ||||
| Activated B lymphocytes or plasma cells | Cytochemical staining | <0.1% | lgG type predominant | 80 |
|
| ||||
| lgG index (linear formula) | Nephelometry and calculation | ≤0.7 | >.7 | 50–75 |
| lgG synthesis rate (linear formula) | <3.5 mg per day | >6.0 mg per day | ||
| lgG local synthesis (linear formula) | 0 g/L | >.0 g/L | ||
|
| ||||
| Leukocyte count | Funchs–Rosenthal chamber, panoptical staining and light microscopy | <5.0 cells per μL | 5.0–50.0 cells per μL | 50–60 |
|
| ||||
| MRZ reaction (optional) | Quantitative immunoassay and antibody index | Antibody index <1.5 | Antibody index >1.4 for two or more virus-specific antibodies | 70–90 |
CSF: cerebrospinal fluid; MRZ: measles rubella zoster; MS: multiple sclerosis
Although a typical constellation of CSF findings can support the diagnosis of MS, it is by no means pathognomonic for this disease. However, deviations from typical CSF findings can aid in making a correct diagnosis, which is important for the management of patients (8).
DIAGNOSTIC CRITERIA
Diagnostic criteria for MS have evolved over the past 50 years. Although successive versions have differed in emphasis, all have required the dissemination of disease in space and time documented by either clinical, paraclinical, or laboratory criteria. Additionally, MS diagnostic criteria have emphasized that an alternative explanation for the clinical presentation must be considered and excluded before the diagnosis of MS can be made (6).
POSER CRITERIA
The main reason for establishing these criteria is to restrict therapeutic trials and other research protocols to patients with DMS, while the category of probable is designed for the purpose of prospectively evaluating new diagnostic methods (9). In 1983, the diagnostic criteria of Poser et al. (9) were published. Originally, they were developed as guidelines for use in research protocols, but they also became widely applied in clinical practice. Two major groups were defined, “definite” and “probable” MS, each with two subgroups: “clinically definite” and “laboratory supported.” The classification included paraclinical evidence of CNS lesions, e.g., by means of evoked potentials and tissue imaging procedures. Laboratory support was defined as OCBs or increased IgG production in CSF. The diagnosis of “clinically definite” MS according to the Poser criteria requires the occurrence of at least two attacks and the clinical evidence of two separate lesions or two attacks with clinical evidence of one lesion and paraclinical evidence of another separate lesion (Table 5) (10).
Table 5.
Poser criteria for multiple sclerosis (8)
| New Diagnostic Criteria for Multiple Sclerosis | |||||
|---|---|---|---|---|---|
|
| |||||
| Category | Attacks | Clinical evidence | Paraclinical evidence | CSF OB/lgG | |
|
| |||||
| A. Clinically definite | |||||
| CDMS A1 | 2 | 2 | |||
| CDMS A2 | 2 | 1 | and | 1 | |
|
| |||||
| B. Laboratory-supported definite | |||||
| LSDMS B1 | 2 | 1 | or | 1 | + |
| LSDMS B2 | 1 | 2 | + | ||
| LSDMS B3 | 1 | 1 | and | 1 | + |
|
| |||||
| C. Clinically probable | |||||
| CPMS C1 | 2 | 1 | |||
| CPMS C2 | 1 | 2 | |||
| CPMS C3 | 1 | 1 | and | 1 | |
|
| |||||
| D. Labarotory supported | |||||
| Probable | |||||
| LSPMS D1 | 2 | + | |||
OB/lgG=oligoconal bands or increased lgG. CDMS: clinically definite MS. Needs two attacks and some clinical or paraclinical evidences; LSDMS: laboratory-supported definite MS, showing oligoclonal bands and clinical or paraclinical evidences; CPMS: clinically probable MS, with less restrict combinations; LSPMS: laboratory-supported probable MS. Only two attacks are enough to enter this category; CSF: cerebrospinal fluid
MCDONALD CRITERIA: 2001 AND 2005 REVISIONS
In 2001, diagnostic criteria from the International Panel of McDonald and colleagues incorporated the MRI evidence of dissemination in time and space to allow a diagnosis of MS in patients with CISs. From clinical and MRI examinations prospectively performed at baseline and at 3 months, 1 year, and 3 years of follow-up, the frequency of developing MS was ascertained by the application of both the new McDonald criteria and the Poser criteria for clinically DMS (CDMS) (8). The key difference between the revised McDonald criteria and the previous Poser criteria is that MS could now be readily diagnosed in individuals who have had a single attack suggestive of the disease. This could allow an earlier diagnosis and initiation of treatment (10).
Dalton et al. (2) showed that the use of McDonald criteria more than doubled the rate of the diagnosis of MS within a year of presentation with CIS. The high specificity, positive predictive value, and accuracy of the criteria for CDMS support their clinical relevance (2).
In the years since their original presentation, several publications have appeared that support the utility of the McDonald criteria: Retrospective analyses of extent datasets have shown that the criteria could reliably signal the development of CDMS earlier than prior criteria and that they had a reasonably high level of specificity and sensitivity compared with those of prior criteria. Additional published studies have explored potential modifications of the original criteria, with particular emphasis on determining the dissemination of lesions in time and space (the core concept in MS diagnosis), incorporating different types of imaging criteria to the diagnostic scheme, and assessing the value of CSF analysis, particularly for the diagnosis of primary progressive MS (PPMS) (11).
Although Dalton et al. (12) demonstrated that a new T2 lesion at 3 months is a reliable marker of dissemination in time, there was a median of 5 weeks from the onset of symptoms to the baseline MRI scan in their studies. The International Panel on Diagnosis of MS (the Panel) believed that T2 lesions could be useful for demonstrating dissemination in time more rapidly than over the 3-month period required in the original McDonald criteria, but it agreed that T2 lesions occurring in the first few weeks after the onset of the first clinical episode should not be considered a separate, new event. Practically any new T2 lesion occurring at any time point after a so-called reference scan performed at least 30 days after the onset of the initial clinical event is useful in fullfilling imaging diagnostic criteria for dissemination in time. This revision would simplify and clarify the prior criteria, allow for a more rapid diagnosis, and provide more flexibility in imaging criteria, while still carrying an unequivocal proof of dissemination in time (Table 6) (11).
Table 6.
Magnetic resonance imaging criteria to demonstrate dissemination of lesions in time (11)
| Original McDonald criteria | 2005 revisions |
|---|---|
|
|
The original McDonald criteria set out specific criteria that needed to be fulfilled largely based on brain MRI scan outcomes to demonstrate a diagnostically relevant brain abnormality. These criteria, from the work of Barkhof et al. (13) as modified by Tintore et al. (7), include evidence for three of the following four outcomes: one gadolinium-enhancing lesion or nine T2-hyperintense lesions if there is no gadolinium-enhancing lesion, at least one infratentorial lesion, at least one juxtacortical lesion, or at least three periventricular lesions (Table 7). As noted, the Panel concluded that although it is biologically plausible to liberalize these requirements for “positive” MRI indicating MS-like brain abnormality, the lack of prospective data to test the specificity and sensitivity of any such liberalized criteria make it unwise at this point to change these criteria (11).
Table 7.
Magnetic resonance imaging criteria to demonstrate brain abnormality and demonstration of dissemination in space (11)
| Original McDonald criteria | 2005 revisions |
|---|---|
Three of the following:
|
Three of the following:
|
It was recommended in the original McDonald criteria that “one spinal cord lesion can be substituted for one brain lesion,” a statement that in retrospect is confusing and provides insufficient guidance for the use of spinal cord imaging in the diagnostic workup. At its Amsterdam meeting, the Panel also reached a consensus on the revisions and guidance related to spinal cord lesions (11).
The Panel continues to believe that a positive CSF finding (preferably based on isoelectric focusing evidence of oligoclonal IgG bands with immunofixation, demonstrating that bands are different from those in serum or an increased IgG index or both) increases the “comfort level” for the diagnosis of MS in individuals with an insidious progression of the disease from the onset. However, such CSF findings are not specific and may be commonly detected in patients with progressive myelopathies of other causes, particularly those associated with infection (e.g., retrovirus). Depending on the strength of other diagnostic criteria, a positive CSF finding is no longer a requirement for the diagnosis of PPMS (Table 8) (11).
Table 8.
Diagnosis of multiple sclerosis in disease with progression from onset (11)
| Original McDonald criteria | 2005 revisions |
|---|---|
|
|
The 2005 Revisions of the MS diagnostic criteria (Table 9) retain the core features of the original McDonald criteria: emphasis on objective clinical findings, dependence on the evidence of the dissemination of lesions in time and space, use of supportive and confirmatory paraclinical examinations to speed the process and help eliminate false-negative and -positive diagnoses, a focus on specificity rather than sensitivity, and a need to eliminate better explanations for the diagnosis (11).
Table 9.
The 2005 revisions to the McDonald diagnostic criteria for multiple sclerosis (11)
| Clinical presentation | Additional data needed for MS diagnosis |
|---|---|
| Two or more attcaksa; objective clinical evidence of two or more lesions Two or more attcaksa; objective clinical evidence of one lesion One attacka; objective clinical evidence of two or more lesions One attacka; objective clinical evidence of one lesion (monosymptomatic presentation; clinically isolated syndrome) Insidious neurological progression suggestive of MS |
Noneb Dissemination in space, demonstrated by:
Two of the following: |
If criteria indicated are fulfilled and there is no better explanation for the clinical presentation, the diagnosis is MS; if suspicious, but the criteria are not completely met, the diagnosis is “possible MS”; if another diagnosis arises during the evaluation that better explains the entire clinical presentation, then the diagnosis is “not MS.”
An attack is defined as an episode of neurological disturbance for which causative lesions are likely to be inflammatory and demyelinating in nature. There should be subjective report (backed up by objective findings) or objective observation that the event lasts for at least 24 hours.
No additional tests are required; however, if tests (MRI, CSF) are undertaken and are negative, extreme caution needs to be taken before making a diagnosis of MS. Alternative diagnoses must be considered. There must be no better explanation for the clinical picture and some objective evidence to support a diagnosis of MS.
MRI demonstration of space dissemination must fulfill the criteria in Table 10.
Positive CSF determined by oligoclonal bands detected by established methods (isoelectric focusing) different from any such bands in serum, or by an increased IgG index.
MRI demonstration of time dissemination must fulfill the criteria in Table 9.
Abnormal VEP of the type seen in MS.
MS: multiple sclerosis; MRI: magnetic resonance imaging; CSF: cerebrospinal fluid; VEP: visual-evoked potential.
MCDONALD CRITERIA: 2010 REVISIONS
New evidence and consensus has led to a further revision of the McDonald criteria for the diagnosis of MS. In May 2010 in Dublin, Ireland, the Panel met for a third time to examine the requirements for demonstrating DIS and DIT and to focus on the application of the McDonald criteria in pediatric, Asian, and Latin American populations. The use of imaging for the demonstration of the dissemination of CNS lesions in space and time has been simplified, and in some circumstances, the dissemination in space and time can be established by a single scan. These revisions simplify the criteria, preserve their diagnostic sensitivity and specificity, address their applicability across populations, and may allow earlier diagnosis and their more uniform and widespread use (Table 10) (15).
Table 10.
The 2010 McDonald criteria for diagnosis of MS (15)
| Clinical presentation | Additional data needed for MS diagnosis |
|---|---|
| ≥2 attacksa; objective clinical evidence of ≥2 lesions or objective clinical evidence of 1 lesion with reasonable historical evidence of a prior attackb | Nonec |
| ≥2 attacksa; objective clinical evidence of 1 lesion | Dissemination in space, demonstrated by: ≥ 1 T2 lesion in at least 2 of 4 MS-typical regions of the CNS (periventricular, juxtacortical, infratentorial, or spinal cord)d; or Await a further clinical attacka implicating a different CNS site |
| 1 attacka; objective clinical evidence of ≥ 2 lesions | Dissemination in time, demonstrated by: Simultaneous presence of asymptomatic gadolinium-enhancing and nonenhancing lesions at any time; or A new T2 and/or gadolinium-enhancing lesion(s) on follow-up MRI, irrespective of its timing with reference to a baseline scan; or Await a second clinical attacka |
| 1 attacka; objective clinical evidence of 1 lesion (clinically isolated syndrome) | Dissemination in space and time, demonstrated by: For DIS: ≥1 T2 lesion in at least 2 of 4 MS-typical regions of the CNS (periventricular, juxtacortical, infratentorial or spinal cord)d; or Await a second clinical attacka implicating a different CNS site; and For DIT: Simultaneous presence of asymptomatic gadolinium-enhancing and nonenhancing lesions at any time; or A new T2 and/or gadolinium-enhancing lesion(s) on follow-up MRI, irrespective of its timing with reference to a baseline scan; or Await a second clinical attacka |
| Insidious neurological progression suggestive of MS (PPMS) | 1 year of disease progression (retrospectively or prospectively determined) plus 2 of 3 of the following criteriad:
|
If the criteria are fulfilled and there is no better explanation for the clinical presentation, the diagnosis is “MS”; if suspicious, but the criteria are not completely met, the diagnosis is “possible MS”; if another diagnosis arises during the evaluation that better explains the clinical presentation, then the diagnosis is “not MS.”
An attack (relapse; exacerbation) is defined as patient-reported or objectively observed events typical of an acute inflammatory demyelinating event in the CNS, current or historical, with duration of at least 24 hours, in the absence of fever or infection. It should be documented by contemporaneous neurological examination, but some historical events with symptoms and evolution characteristic for MS, but for which no objective neurological findings are documented, can provide reasonable evidence of a prior demyelinating event. Reports of paroxysmal symptoms (historical or current) should, however, consist of multiple episodes occurring over not less than 24 hours. Before a definite diagnosis of MS can be made, at least 1 attack must be corroborated by findings on neurological examination, visual-evoked potential response in patients reporting prior visual disturbance, or MRI consistent with demyelination in the area of the CNS implicated in the historical report of neurological symptoms.
Clinical diagnosis based on objective clinical findings for 2 attacks is most secure. Reasonable historical evidence for 1 past attack, in the absence of documented objective neurological findings, can include historical events with symptoms and evolution characteristics for a prior inflammatory demyelinating event; at least 1 attack, however, must be supported by objective findings.
No additional tests are required. However, it is desirable that any diagnosis of MS be made with access to imaging based on these criteria. If imaging or other tests (for instance, CSF) are undertaken and are negative, extreme caution needs to be taken before making a diagnosis of MS, and alternative diagnoses must be considered. There must be no better explanation for the clinical presentation, and objective evidence must be present to support a diagnosis of MS.
Gadolinium-enhancing lesions are not required; symptomatic lesions are excluded from consideration in subjects with brainstem or spinal cord syndromes.
MS: multiple sclerosis; CNS: central nervous system; MRI: magnetic resonance imaging; DIS: dissemination in space; DIT: dissemination in time; PPMS: primary progressive multiple sclerosis; CSF: cerebrospinal fluid; IgG: immunoglobulin G.
In the past versions of the McDonald criteria, DIS demonstrated by MRI was based on the Barkhof/Tintore (13,14) criteria. Despite having good sensitivity and specificity, these criteria have been difficult to consistently apply by nonimaging specialists. The European MAGNIMS multicenter collaborative research network, which studies MRI in MS, compared the Barkhof/Tintore (13,14) criteria for DIS with simplified criteria developed by Swanton et al. (16,17).
In the MAGNIMS work, DIS can be demonstrated with at least one T2 lesion in at least two of four locations considered characteristic for MS and as specified in the original McDonald criteria (juxtacortical, periventricular, infratentorial, and spinal cord), with lesions within the symptomatic region excluded in patients with brainstem or spinal cord syndromes. In 282 patients with CIS, the Swanton-based DIS criteria were shown to be simpler and slightly more sensitive than the original McDonald criteria for DIS, without compromising specificity and accuracy. The Panel accepted these MAGNIMS DIS criteria, which can simplify the diagnostic process for MS while preserving specificity and improving sensitivity (15, 16).
More recently, the MAGNIMS group confirmed earlier studies by showing that in patients with typical CIS, a single brain MRI study that demonstrates DIS and both asymptomatic gadolinium-enhancing and nonenhancing lesions is highly specific for predicting the early development of CDMS and reliably substitutes prior imaging criteria for DIT. After reviewing these data, the Panel accepted that the presence of both gadolinium-enhancing and nonenhancing lesions on the baseline MRI can substitute for a follow-up scan to confirm DIT, as long as it can be reliably determined that the gadolinium-enhancing lesion is not due to a non-MS pathology (15).
MANAGEMENT OF CIS
Many CIS episodes are mild and are resolved without therapeutic intervention. Clinical features that favor treatment include severe visual loss, pain in optic neuritis, or both, and pronounced motor dysfunction, ataxia, or vertigo in spinal cord and brainstem syndromes (1,6).
Corticosteroids are used when the symptoms are functionally disabling or when patients do not spontaneously improve (6). High-dose intravenous methylprednisolone in acute optic neuritis shortens the duration of visual deficits but not the visual outcome after 1 year. Oral high-dose methylprednisolone (1 g per day for 3–5 days) is probably an acceptable alternative to intravenous methylprednisolone (1 g per day for 3–5 days) (1). Patients treated with intravenous methylprednisolone and oral prednisone have a lower risk of developing CDMS over the next 2 years than those given a placebo (9% vs. 17%, respectively). However, after the third year of follow-up, the treatment and placebo groups’ risks of conversion to CDMS did not differ (6).
β-interferon and glatiramer acetate extend the time to the next relapse of CDMS. Patients treated with intramuscular β-interferon-1a (30 μg) once a week had a 37% conversion to CDMS after 2 years compared with a 50% conversion of those who received a placebo. Weekly subcutaneous β-interferon-1a (22 μg) was associated with a 35% conversion to MS after 2 years compared with a 50% conversion in those given a placebo. Subcutaneous β-interferon-1b (250 mg) on alternate days reduced the conversion to CDMS from 55% (in placebo) to 35% after 2 years. Daily subcutaneous glatiramer acetate (30 mg) was associated with a 35% conversion to CDMS compared with a 50% conversion in the placebo group. These treatments also reduce new MRI lesion formation and might slow down brain atrophy (1).
In addition to the suppression of inflammation, the challenge exists for treatments to prevent neurodegeneration as CIS evolves to MS and beyond. Ultimately, a combination of anti-inflammatory and neuroprotective treatments might be needed to prevent long-term disability. Strategies for neuroprotection include sodium-channel blockers, statins, glutamate antagonists, and remyelination with stem cells and other molecular targets that promote repair. However, no effective neuroprotective agent has yet been identified for MS (1).
DIAGNOSIS AND TREATMENT OF INDIVIDUAL PATIENTS
According to Miller et al. (4), most clinicians who see patients with CIS or suspected early MS agree that it is important to diagnose early and having done so, to weigh-up the potential benefits, risks, and uncertainties of disease-modifying treatment, while ensuring that the patient is kept fully informed and participates in the decision-making (4).
CIS MANAGEMENT IN CLINICAL TRIALS
Three large clinical trials have shown that IFN beta treatments are effective in patients with CIS [Controlled High-Risk Avonex® Multiple Sclerosis Prevention Study (CHAMPS), Early Treatment of MS (ETOMS) and Betaferon® in Newly Emerging Multiple Sclerosis for Initial Treatment (BENEFIT)] (Table 11) (18). The PreCISe trial studied GA in patients with CIS and has also shown superior efficacy to placebos, although the full results are still pending, and thus will not be considered further here, except to note that arguments made in favor of early IFN beta therapy are likely to apply equally to GA (Table 12) (19–24).
Table 11.
Comparison of efficacy results from the early treatment studies (18)
| Study | Treatment | Patients: drug/placebo | Multifocal presentationa | Patients with ≥1 Gd+ lesion on initial MRIa | Conversion to CDMS (2 years) | |
|---|---|---|---|---|---|---|
| IFNβ Placebo | ||||||
| CHAMPS | IFNβ-1a IM, qw | 193/190 | 0% | 28% | 20%b | 35%b |
| ETOMS | IFNβ-1a sc, tiw | 154/154 | 39.0% | 58% | 34% | 45% |
| BENEFIT | IFNβ-1b sc, eod | 292/176 | 47.4% | 42% | 28% | 45% |
Data for all patients (drug and placebo groups):
Patients converting to CDMS at 25 months.
CDMS: clinically definite MS; eod: every other day; Gd+: gadolinium-positive; IFNβ: interferon beta, IM: intramuscular; qw: weekly; sc: subcutaneous; tiw: twice weekly; CHAMPS: Controlled High-Risk Avonex® Multiple Sclerosis Prevention Study; ETOMS: Early Treatment of MS; BENEFIT: Betaferon® in Newly Emerging Multiple Sclerosis for Initial Treatment.
Table 12.
Comparison of the REFLEX study and other large trials of disease-modifying drugs in patients with a first clinical demyelinating event (19–24)
| Active treatment | Study design | Conversion to CDMS versus placebo | Notes | |
|---|---|---|---|---|
| Comi et al. (19) (REFLEX) |
Subcutaneous interferon beta 1-a (44 μg three times a week or once a week) | Primary endpoint, McDonald MS (2005 criteria); secondary endpoint, CDMS | Three times a week, adjusted HR 0.48 (95% CI 0.31–0.73 p<0.0004) once a week, adjusted HR 0.53 (95% CI 0.35–0.79; p=0.0023) | Monofocal presentation, 54%; steroid treatment, 71% |
| Kapsos et al. (20) (BENEFIT) |
Subcutaneous interferon beta 1-b (250 μg every other day) | Primary endpoint, CDMS; secondary endpoint, McDonald MS (2001 criteria) | Adjusted HR 0.50 (95% CI 0.36–0.70; p<0.0001) | Similar conversion to McDonald MS in placebo group; similar risk reduction for McDonald MS and CDMS; monofocal presentation, 53%; steroid treatment, 71% |
| Jacobs et al. (21) and O’Connor et al. (22) (CHAMPS) |
Intramuscular interferon beta-1a (30 μg once a week) | Primary endpoint, CDMS | Rate ratio 0.56 (95% CI 0.38–0.81; p=0.002); adjusted rate ratio 0.49 (95% CI 0.33–0.73; p<0.001) | Monofocal presentation 70%; steroid treatment, 100% |
| Comi et al. (23) (PRECISE) |
Subcutaneous glatiramer acetate (20 mg per day) | Primary endpoint, CDMS | HR 0.55 (95% CI 0.40–0.77; p=0.0005) | Monofocal presentation, 100%; steroid treatment, 61% |
| Comi et al. (24) (ETOMS) |
Subcutaneous interferon beta 1-a (22 μg once a week) | Primary endpoint, CDMS | Odds ratio 0.61 (95% CI 0.37–0.99: p=0.045) | Monofocal presentation, 61%; steroid treatment, 64% |
CDMS: clinically definite multiple sclerosis; REFLEX: REbif FLEXible dosing in early MS; HR: hazard ratio; MS: multiple sclerosis
As the CIS stage represents the first opportunity for treatment in most patients, many neurologists advocate starting therapy at this point. Of the available treatments, only 30 mg of IFNb-1a by weekly intramuscular (im) injection and 250 mg of IFNb-1b by subcutaneous (sc) injection every other day are currently licensed for the treatment of patients with a CIS suggestive of MS. Nevertheless, low-dose 22 mg of IFNb-1a by sc injection once weekly has also been demonstrated to be effective in CIS, and a recent placebo-controlled trial of 20 mg of GA by daily sc administration was stopped early at the 3-year interim analysis due to the superiority of the treatment arm. Thus, in reality, the use of any of these agents is appropriate in the clinical context of a CIS (18).
The CHAMPS study enrolled 383 patients with CIS with monofocal disease randomized to two groups: 193 were given once-weekly 30 mg of IFNb-1a, im, and 190 were given weekly placebo injections. The development of CDMS was defined by the occurrence of a second attack or an increase of at least 1.5 points on the EDSS. The 30 mg IFNb-1a im treated group demonstrated a 44% reduction in the 3-year cumulative probability of developing CDMS (rate ratio 0.56, 95% confidence interval 0.38 to 0.8; p=0.002) (18,25). At 18 months, treatment with 30 mg of IFNb-1a im was associated with a significant reduction of new T2 lesions, gadolinium enhanced lesions, and T2 lesion volume. Among the placebo-treated patients, 82% had developed a new subclinical MRI signal abnormality by the eighteenth month after study entry (25).
In the ETOMS trial, 309 patients with CIS were randomized to once-weekly injections of 22 mg of IFNb-1a sc (n=154) or a placebo (n=155). A significantly higher proportion of patients in the placebo group than the IFNb-1a group (45% vs. 34%, respectively; rate ratio 0.65; p=0.047) converted from CIS to CDMS (as defined by the occurrence of a second clinical attack) over the 2-year course of the study (19,20). The time when 30% of patients had converted to CDMS was 569 days in the interferon group and 252 in the placebo group (p=0.034). The annual relapse rates were 0.33 and 0.43 (p=0.045). The number of new T2-weighted MRI lesions and the increase in lesion burden were significantly lower with active treatment. The ETOMS group concluded that interferon beta-1a treatment at an early stage of DMS had significant positive effects on clinical and MRI outcomes (24).
The BENEFIT study randomized 468 patients with CIS to two groups receiving injections of either 250 μg of IFNb-1b sc (n=292), or a placebo (n=176) every other day. As in the other two studies, the IFNb group showed a significantly lower rate of conversion from CIS to CDMS, as defined by a second event or an increase of at least 1.5 points on the EDSS. According to proportional hazards regression, the 2-year cumulative probability for CDMS was 28% in the IFNb-1b group and 45% in the placebo group (rate ratio 0.5; p<0.0001). In addition to defining CDMS by the Poser criteria, the BENEFIT study also employed the new international criteria to define progression to MS. In this analysis, again, treatment with IFNb-1b was associated with a reduction in the progression to International MS within 2 years, compared with placebo (rate ratio 0.54; p<0.00001) (18,24,25). The BENEFIT group concluded that interferon beta-1b 250 μg subcutaneously every other day delayed the conversion to CDMS and should be considered as a therapeutic option in patients presenting with a first clinical event suggestive of MS (20).
The PRECISE study randomized 481 patients presenting with CIS with unifocal manifestation, and two or more T2-weighted brain lesions measuring 6 mm or more. Patients were randomly assigned to receive either subcutaneous glatiramer acetate 20 mg per day (n=243) or a placebo (n=238) for up to 36 months, unless they converted to CDMS. The randomization scheme used SAS-based blocks stratified by center and patients, and all the personnel were masked to treatment assignment. The primary endpoint was the time to develop CDMS, based on a second clinical attack. Glatiramer acetate reduced the risk of developing CDMS by 45% compared with placebo (hazard ratio 0.55, 95% CI 0.40–0.77; p=0.0005). The time for 25% of patients to convert to the clinically definite disease was prolonged by 115%, from 336 days for the placebo group to 722 days for the glatiramer acetate group. The most common adverse events in the glatiramer acetate group were injection-site reactions (135 [56%] glatiramer acetate vs. 56 [24%] placebo) and immediate post-injection reactions (47 [19%] vs. 12 [5%]). The PRECISE study group concluded that early treatment with glatiramer acetate is efficacious in delaying the conversion to CDMS in patients presenting with CIS and brain lesions detected by MRI (23).
In the Early Treatment of MS (ETOMS) study, subcutaneous interferon beta-1a at 22 μg once a week was effective in delaying MS. However, until now the effect of subcutaneous interferon beta-1a at 44 μg three times a week—the licensed dosing regimen for MS—has not been assessed in people with a clinically isolated event. In REbif FLEXible dosing in early MS [REFLEX] study, which was a 2-year, double-blind phase 3 study, the effects of two dosing frequencies of a serum-free formulation of subcutaneous interferon beta-1a at 44 μg and a placebo on the time to develop MS, as defined in the 2005 version of the McDonald criteria, were compared. This study is the first comparison of the efficacy of two dosing frequencies of subcutaneous interferon beta-1a. Participants were randomly assigned in a 1:1:1 ratio by use of a centralized interactive voice response system to receive the serum-free formulation of subcutaneous interferon beta-1a at 44 μg three times a week or once a week (plus placebo twice a week for masking), or placebo three times a week for up to 24 months. The primary endpoint was the time to a diagnosis of MS as defined by the 2005 McDonald criteria and the main secondary endpoint was the time to develop CDMS as defined by the Poser criteria. 517 patients were randomly assigned (171 to subcutaneous interferon beta-1a three times a week, 175 to subcutaneous interferon beta-1a once a week, and 171 to placebo) and 515 were treated. The 2-year cumulative probability of McDonald MS was significantly lower in patients treated with subcutaneous interferon beta-1a (three times a week 62.5%, p<0.0001, hazard ratio [HR] 0.49 [95% CI 0.38–0.64]; once a week 75.5%, p=0.008, HR 0.69 [0.54–0.87]) versus placebo (85.8%). The 2-year rates of conversion to CDMS were lower for both interferon beta-1a dosing regimens (three times a week 20.6%, p=0.0004, HR 0.48 [0.31–0.73]; once a week 21.6%, p=0.0023, HR 0.53 [0.35–0.79]) than for placebo (37.5%). Thus, the three times a week and once a week regimens similarly delayed the occurrence of a clinical relapse, but the three times a week regimen had a more pronounced effect on McDonald MS than did the once a week regimen (19).
Comi et al. (24) indicated that the long-term differences between interferon beta-1a three times a week and interferon beta-1a once a week, in terms of the effect on MRI-related disease activity and adverse-event profile, warrants further study and is the focus of the ongoing preplanned, dose-frequency-blinded, 3-year extension of this study (REFLEXION-REFLEX extensION study) (19).
CONCLUSION
Clinically isolated syndrome describes a single, first-occurrence attack caused by inflammation/demyelination at one or more locations in CNS (1). Patients with CIS (and abnormal MRI scans) are at a high risk of developing CDMS (18). Hence, when an individual presents with CIS, the challenge for the clinician is to determine the likelihood of the patient having CDMS. The ability to make an accurate diagnosis as early as possible is important for patient management, counseling, and optimal therapy (6,26). Diagnosis in patients with CIS is a clinical decision and should be made by a neurologist experienced in the relevant diagnosis (4).
Diagnostic criteria for MS have evolved over the past 50 years (6). These criteria provide neurologists with a systematic approach for MS diagnosis (heavily based on MRI findings) for a range of clinical presentations, including CIS, RRMS, and primary progressive MS (18). Although successive versions have differed in emphasis, all require the dissemination of the disease in space and time documented by either clinical, paraclinical, or laboratory criteria (6). Recently, the International Panel on Diagnosis of Multiple Sclerosis has proposed new MRI criteria for the diagnosis of MS in patients with CIS. These criteria were developed in Western Caucasian populations and then studied in a Spanish cohort, and similar results were obtained with regard to sensitivity and specificity, hence reinforcing their usefulness in MS diagnosis. Currently, the international validation of the 2010 criteria is moving forward, and new information about their sensitivity and specificity has been provided for several MS populations (27).
Patients with CIS with MRI-detected brain damage similar to that seen in MS are in the high-risk category for a second neurologic event. This group may benefit from early treatment with immunomodulators to delay the diagnosis of CDMS (28). IFNb treatment is particularly effective in the early stages of MS, when the levels of inflammatory activity are highest, and has been shown to be effective and well tolerated in the treatment of patients with CIS in clinical trials (18). Much has been learned in the past 10 years about CIS and its relation to MS. Robust and practical new diagnostic criteria aid earlier MS diagnosis in patients with typical CIS, and immunomodulatory treatments favorably modify the early clinical course (over 2–5 years) in those at a high risk of MS (1).
Footnotes
Conflict of Interest: No conflict of interest was declared by the authors.
REFERENCES
- 1.Miller DH, Chard DT, Ciccarelli O. Clinically isolated syndromes. Lancet Neurol. 2012;11:157–169. doi: 10.1016/S1474-4422(11)70274-5. http://dx.doi.org/10.1016/S1474-4422(11)70274-5. [DOI] [PubMed] [Google Scholar]
- 2.Dalton CM, Brex PA, Miszkiel KA, Hickman SJ, MacManus DG, Plant GT, Thompson AJ, Miller DH. Application of the New McDonald criteria to patients with clinically isolated syndromes suggestive of multiple sclerosis. Ann Neurol. 2002;52:47–53. doi: 10.1002/ana.10240. http://dx.doi.org/10.1002/ana.10240. [DOI] [PubMed] [Google Scholar]
- 3.Platten M, Lanz T, Bendszus M, Diem R. Clinically isolated syndrome. Der Nervenarzt. 2013;84:1247–1259. doi: 10.1007/s00115-013-3845-1. http://dx.doi.org/10.1007/s00115-013-3845-1. [DOI] [PubMed] [Google Scholar]
- 4.Miller D, Barkhof F, Montalban X, Thompson A, Filippi M. Clinically isolated syndromes suggestive of multiple sclerosis, part I: natural history, pathogenesis, diagnosis, and prognosis. Lancet Neurol. 2005;4:281–288. doi: 10.1016/S1474-4422(05)70071-5. http://dx.doi.org/10.1016/S1474-4422(05)70071-5. [DOI] [PubMed] [Google Scholar]
- 5.Miller D, Barkhof F, Montalban X, Thompson A, Filippi M. Clinically isolated syndromes suggestive of multiple sclerosis, part 2: non-conventional MRI, recovery processes, and management. Lancet Neurol. 2005;4:341–348. doi: 10.1016/S1474-4422(05)70095-8. http://dx.doi.org/10.1016/S1474-4422(05)70071-5. [DOI] [PubMed] [Google Scholar]
- 6.Miller D, Barkhof F, Montalban X, Thompson A, Filippi M. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler. 2008;14:1157–1174. doi: 10.1177/1352458508096878. http://dx.doi.org/10.1177/1352458508096878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tintoré M, Rovira A, Río J, Tur C, Pelayo R, Nos C, Téllez N, Perkal H, Comabella M, Sastre-Garriga J, Montalban X. Do oligoclonal bands add information to MRI in first attacks of multiple sclerosis? Neurology. 2008;70:1079–1083. doi: 10.1212/01.wnl.0000280576.73609.c6. http://dx.doi.org/10.1212/01.wnl.0000280576.73609.c6. [DOI] [PubMed] [Google Scholar]
- 8.Stangel M, Fredrikson S, Meinl E, Petzold A, Stüve O, Tumani H. The utility of cerebrospinal fluid analysis in patients with multiple sclerosis. Nat Rev Neurol. 2013;9:267–276. doi: 10.1038/nrneurol.2013.41. http://dx.doi.org/10.1038/nrneurol.2013.41. [DOI] [PubMed] [Google Scholar]
- 9.Poser CM, Paty DW, Scheinberg L, McDonald WI, Davis FA, Ebers GC, Johnson KP, Sibley WA, Silberberg DH, Tourtellotte WW. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13:227–231. doi: 10.1002/ana.410130302. http://dx.doi.org/10.1002/ana.410130302. [DOI] [PubMed] [Google Scholar]
- 10.Fangerau T, Schimrigk S, Haupts M, Kaeder M, Ahle G, Brune N, Klinkenberg K, Kotterba S, Möhring M, Sindern E. Diagnosis of multiple sclerosis: comparison of the Poser criteria and the new McDonald criteria. Acta Neurol Scand. 2004;109:385–389. doi: 10.1111/j.1600-0404.2004.00246.x. http://dx.doi.org/10.1111/j.1600-0404.2004.00246.x. [DOI] [PubMed] [Google Scholar]
- 11.Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, Lublin FD, Metz LM, McFarland HF, O’Connor PW, Sandberg-Wollheim M, Thompson AJ, Weinshenker BG, Wolinsky JS. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58:840–846. doi: 10.1002/ana.20703. http://dx.doi.org/10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- 12.Dalton CM, Brex PA, Miszkiel KA, Fernando K, MacManus DG, Plant GT, Thompson AJ, Miller DH. New T2 lesions enable an earlier diagnosis of multiple sclerosis in clinically isolated syndromes. Ann Neurol. 2003;53:673–676. doi: 10.1002/ana.10580. http://dx.doi.org/10.1002/ana.10580. [DOI] [PubMed] [Google Scholar]
- 13.Barkhof F, Filippi M, Miller DH, Scheltens P, Campi A, Polman CH, Comi G, Adèr HJ, Losseff N, Valk J. Comparison of MR imaging criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain. 1997;120:2059–2069. doi: 10.1093/brain/120.11.2059. http://dx.doi.org/10.1093/brain/120.11.2059. [DOI] [PubMed] [Google Scholar]
- 14.Tintoré M, Rovira A, Martínez MJ, Rio J, Díaz-Villoslada P, Brieva L, Borrás C, Grivé E, Capellades J, Montalban X. Isolated demyelinating syndromes: comparison of different MR imaging criteria to predict conversion to clinically definite multiple sclerosis. Am J Neuroradiol. 2000;21:702–706. [PMC free article] [PubMed] [Google Scholar]
- 15.Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Montalban X, O’Connor P, Sandberg-Wollheim M, Thompson AJ, Waubant E, Weinshenker B, Wolinsky JS. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. http://dx.doi.org/10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swanton JK, Rovira A, Tintore’ M, Altmann DR, Barkhof F, Filippi M, Huerga E, Miszkiel KA, Plant GT, Polman C, Rovaris M, Thompson AJ, Montalban X, Miller DH. MRI criteria for multiple sclerosis in patients presenting with clinically isolated syndromes: a multicentre retrospective study. Lancet Neurol. 2007;6:677–686. doi: 10.1016/S1474-4422(07)70176-X. http://dx.doi.org/10.1016/S1474-4422(07)70176-X. [DOI] [PubMed] [Google Scholar]
- 17.Swanton JK, Fernando K, Dalton CM, Miszkiel KA, Thompson AJ, Plant GT, Miller DH. Modification of MRI criteria for multiple sclerosis in patients with clinically isolated syndromes. J Neurol Neurosurg Psychiatry. 2006;77:830–833. doi: 10.1136/jnnp.2005.073247. http://dx.doi.org/10.1136/jnnp.2005.073247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodin DS, Bates D. Treatment of early multiple sclerosis: the value of treatment initiation after a first clinical episode. Mult Scler. 2009;15:1175–1182. doi: 10.1177/1352458509107007. http://dx.doi.org/10.1177/1352458509107007. [DOI] [PubMed] [Google Scholar]
- 19.Comi G, De Stefano N, Freedman MS, Barkhof F, Polman CH, Uitdehaag BM, Casset-Semanaz F, Hennessy B, Moraga MS, Rocak S, Stubinski B, Kappos L. Comparison of two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): a phase 3 randomised controlled trial. Lancet Neurol. 2012;11:33–41. doi: 10.1016/S1474-4422(11)70262-9. http://dx.doi.org/10.1016/S1474-4422(11)70262-9. [DOI] [PubMed] [Google Scholar]
- 20.Kappos L, Polman CH, Freedman MS, Edan G, Hartung HP, Miller DH, Montalban X, Barkhof F, Bauer L, Jakobs P, Pohl C, Sandbrink R. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology. 2006;67:1242–1249. doi: 10.1212/01.wnl.0000237641.33768.8d. http://dx.doi.org/10.1212/01.wnl.0000237641.33768.8d. [DOI] [PubMed] [Google Scholar]
- 21.Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ, Simonian NA, Slasor PJ, Sandrock AW. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med. 2000;343:898–904. doi: 10.1056/NEJM200009283431301. http://dx.doi.org/10.1056/NEJM200009283431301. [DOI] [PubMed] [Google Scholar]
- 22.O’Connor P, Kinkel RP, Kremenchutzky M. Efficacy of intramuscular interferon beta-1a in patients with clinically isolated syndrome: analysis of subgroups based on new risk criteria. Mult Scler. 2009;15:728–34. doi: 10.1177/1352458509103173. http://dx.doi.org/10.1177/1352458509103173. [DOI] [PubMed] [Google Scholar]
- 23.Comi G, Martinelli V, Rodegher M, Moiola L, Bajenaru O, Carra A, Elovaara I, Fazekas F, Hartung HP, Hillert J, King J, Komoly S, Lubetzki C, Montalban X, Myhr KM, Ravnborg M, Rieckmann P, Wynn D, Young C, Filippi M. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374:1503–1511. doi: 10.1016/S0140-6736(09)61259-9. http://dx.doi.org/10.1016/S0140-6736(09)61259-9. [DOI] [PubMed] [Google Scholar]
- 24.Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernández O, Hartung H, Seeldrayers P, Sørensen PS, Rovaris M, Martinelli V, Hommes OR. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet. 2001;357:1576–1582. doi: 10.1016/s0140-6736(00)04725-5. http://dx.doi.org/10.1016/S0140-6736(00)04725-5. [DOI] [PubMed] [Google Scholar]
- 25.Galetta SL. The controlled high risk Avonex multiple sclerosis trial (CHAMPS Study) J Neuroophthalmol. 2001;21:292–295. doi: 10.1097/00041327-200112000-00013. http://dx.doi.org/10.1097/00041327-200112000-00013. [DOI] [PubMed] [Google Scholar]
- 26.Siva A. Immunotherapy for clinically isolated syndrome? Not necessarily. Nat Clin Pract Neurol. 2008;4:236–237. doi: 10.1038/ncpneuro0774. http://dx.doi.org/10.1038/ncpneuro0774. [DOI] [PubMed] [Google Scholar]
- 27.Patrucco L, Rojas JI, Miguez JS, Cristiano E. Application of the McDonald 2010 criteria for the diagnosis of multiple sclerosis in an Argentinean cohort of patients with clinically isolated syndromes. Mult Scler. 2013;19:1297–1301. doi: 10.1177/1352458513475492. http://dx.doi.org/10.1177/1352458513475492. [DOI] [PubMed] [Google Scholar]
- 28.Holdeman NR, Nguyen T, Tang RA. Demyelinating optic neuritis presenting as a clinically isolated syndrome. Optometry. 2012;83:9–18. doi: 10.1016/j.optm.2011.10.010. http://dx.doi.org/10.1016/j.optm.2011.10.010. [DOI] [PubMed] [Google Scholar]