

Abstract
Sudden cardiac death (SCD) is defined as natural death due to cardiac causes, heralded by abrupt loss of consciousness within 1 h of the onset of acute symptoms; pre-existing heart disease may have been known to be present but the time and mode of death are unexpected. Prediction and prevention of SCD is an area of active investigation, but considerable challenges persist that limit the efficacy and cost-effectiveness of available methodologies. It was well-recognized that optimization of SCD risk stratification would require integration of multi-disciplinary efforts at the bench and bedside, with studies in the general population. This integration has yet to be effectively accomplished. There is also increasing awareness that more investigation needs to be directed toward the identification of early predictors of SCD. Significant advancements have recently occurred for risk prediction in the inherited channelopathies and other inherited conditions that predispose to SCD, but there is much to be accomplished in this regard for the more common complex phenotypes, such as SCD among patients with coronary artery disease. A multimodality imaging approach is actually the most important tool to provide comprehensive information on different pathophysiological mechanisms related to SCD.
Keywords: Cardiovascular imaging, channelopathies, ICD, sudden cardiac death, ventricular arrhythmias
SUDDEN CARDIAC DEATH
The term Sudden Cardiac Death (SCD) has been used for several centuries and throughout this time different authors have debated how to define it most appropriately. SCD is defined as follows: ‘Natural death due to cardiac causes, heralded by abrupt loss of consciousness within 1 h of the onset of acute symptoms; pre-existing heart disease may have been known to be present, but the time and mode of death are unexpected’.[1]
The key concepts in the definition of sudden death are the non-traumatic nature of the event and the fact that sudden death should be unexpected and instantaneous.
Prediction and prevention of SCD is an area of active investigation, but considerable challenges persist that limit the efficacy and cost-effectiveness of available methodologies.[2] It was well recognized that optimization of SCD risk stratification would require integration of multi-disciplinary efforts at the bench and bedside, with studies in the general population. This integration has yet to be effectively accomplished. There is also increasing awareness that more investigation needs to be directed towards the identification of early predictors of SCD.[3]
Significant advancements have recently occurred for risk prediction in the inherited channelopathies and other inherited conditions that predispose to SCD, such as hypertrophic cardiomyopathy.[4] However, there is much to be accomplished in this regard for the more common complex phenotypes, such as SCD among patients with coronary artery disease.
Epidemiology
SCD is the leading cause of death in industrialized countries and is responsible for 60-70% of all deaths of cardiovascular origin.
Overall incidence of SCD is between 0.36 and 1.28 per 1000 inhabitants per year.[5]
The most frequently documented arrhythmia is ventricular fibrillation (75-80%) while bradyarrhythmias contribute in minor proportion. The single most important cause of death in the adult population of the industrialized world is SCD due to coronary disease. In 5-10% of cases, SCD occurs in the absence of coronary artery disease and heart failure[1] [Figure 1].
Figure 1.
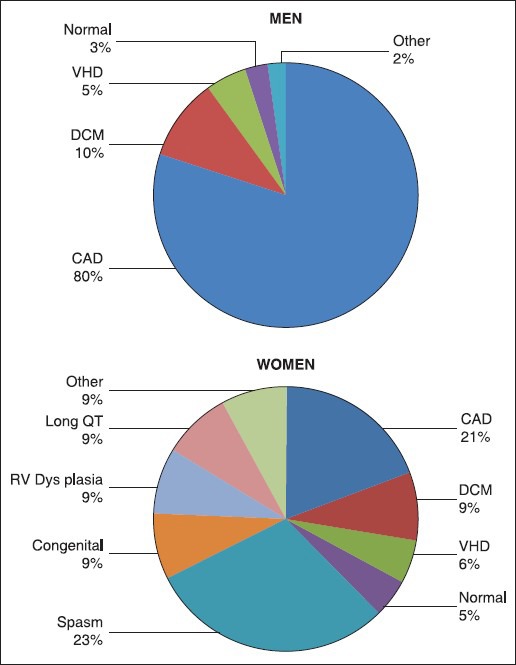
Structural heart disease in cardiac arrest survivors
Approximately 65% of cardiac arrests occur in the presence of witnesses.[6]
Incidence varies with age, sex, and the presence or absence of cardiovascular disease.
The peak incidence of the disease is between 45 and 75 years with prevalence in males of younger age, which is attenuated until the male: female ratio becomes about 2:1 in the decade 65-74 years.
The incidence of sudden cardiac death also shows a circadian rhythm with prevalence between 6 a.m. and noon.
Patophysiology
The pathophysiology of SCD is complex and is believed to require the interaction between a transient event and an underlying substrate. This process induces electric instability and lethal ventricular arrhythmias followed by hemodynamic collapse. Population studies in many industrialized countries have demonstrated that the risk factors for SCD are predominantly the same as those for atherosclerotic coronary disease, namely increasing age, male gender, family history of coronary artery disease, increased LDL cholesterol, hypertension, smoking, and diabetes mellitus. SCD may occur also as a consequence of an inherited genetic abnormality affecting the key proteins of the heart. Evidence supporting the existence of a genetic “susceptibility factor” predisposing to SCD has emerged from large-scale epidemiological studies that have demonstrated a familial association of SCD.
The practical implications of current knowledge about the genetic basis of SCD are to encourage the assessment of family history in survivors of SCD. Once familial clustering of cardiac arrests or SCD has been established, the presence of a monogenic disorder (Brugada syndrome, long QT syndrome, hypertrophic cardiomyopathy, etc.) should be carefully evaluated, particularly if these events occurred at a young age.
Coronary artery disease (CAD) is still the most common substrate underlying SCDs in the Western world, being responsible for 75% of SCD. Cardiomyopathies (dilated, hypertrophic, and arrhythmogenic right ventricular cardiomyopathy) and primary electric disorders related to channelopathies account for most of the remainder. In 5% of SCDs or cardiac arrests, a significant cardiac abnormality is not found after extensive evaluation or at autopsy.
CAD predisposes to SCD in three general settings:
Acute myocardial infarction,
Ischemia without infarction, and
Structural alterations such as scar formation or ventricular dilatation secondary to prior infarction or chronic ischemia.
Presumably, the mechanism of SCD in cases without acute myocardial infarction (MI) is an electric event due to a ventricular arrhythmia triggered by ischemia or other arrhythmogenic stimuli in the setting of a chronically diseased heart.[7] Ventricular fibrillation degenerates to asystole over the course of several minutes; as a result, the majority of SCD patients demonstrate asystole or pulseless electric activity when first examined by rescue teams.
The overwhelming majority of SCD occurs in the general population and approximately 55% of men and at least 68% of women have no clinically recognized heart disease prior SCD.
Furthermore, several electrocardiographic risk markers have emerged from cohorts and community-based studies more likely reflect the general population. These include a prolonged QT interval[8] and the T peak-T end interval.[9]
There is a considerable interest in using markers that reflect arrhythmia substrates more directly, and therefore, enrich the prediction of SCD events. Invasive electrophysiological testing using programmed cardiac stimulation adds considerable specificity to identification of patient populations with ischemic heart disease who are at risk for SCD and who, therefore, may benefit from ICD therapy. In contrast to invasive electrophysiological testing, non-invasive tests for predicting SCD are clearly more attractive in a clinical strategy for widespread screening.
SINGLE CONDITIONS
Scd in myocardial infarction
The most important cause of SCD is represented by ischemic heart disease of which it represents the most acute manifestation.[10] In patients with chronic post-infarction ischemic heart disease, the incidence of SCD, one year after the event, is 3-5%. In this population, the risk factors are: Previous MI, left ventricular dysfunction (FE ≤35%), inducible sustained ventricular tachycardia. In such patients, the presence of an arrhythmogenic substrate, acute ischemia, severe contractile deficit, electrolyte disturbances, of modulating factors (changes in the autonomic nervous system, drugs) are variously represented in determining SCD.[11]
Ischemia, from a pathogenic point of view, is involved in the onset of ventricular arrhythmias during myocardial infarction in a different manner depending on the distance in time of onset of symptoms. In the early hours, acute ischemia causes multiple ionic and metabolic alterations that lead to electrophysiological heterogeneity at the cellular level, at the base of re-entry circuits. In the 24-72 hours following the alterations of the action potential ischemia causes, the abnormal pulse generation in the islands of tissue survived and in the border area: It is a mechanism of enhanced automation and/or triggered activity.[12]
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy is a relatively common cardiac disorder in which sudden unexpected death is the more devastating component, occurring throughout life, but particularly in young asymptomatic patients. This disease has a prevalence of about two cases per 1,000 young adults with an incidence of SCD, which is 2-4% per year in adults and 4-6% per years in children and adolescents.[13] In these individuals, the main risk factors for SCD are represented by family history of SCD, history of SCD or sustained ventricular tachycardia, recurrent syncope, multiple episodes of non-sustained ventricular tachycardia (NSVT), and especially a massive hypertrophy of the left ventricle. Regarding the latter factor, it has been seen, in fact, that the entity ventricular hypertrophy is directly correlated with the risk of SCD and is an independent predictor of prognosis in the short and long term.[14]
Dilated cardiomiopathy
It represents the substrate of about 10% of SCD in the adult population and mortality ranges from 10% to 50% annually, depending on the severity of the pathology. In a series of 14 old studies involving 1432 patients, the mortality rate after four years of follow-up was 42%, with 28% of deaths classified as “sudden”.[15]
The presence of NSVT in these patients identifies those at high risk, and the presence of return pulses along an accessory beam is an important cause of ventricular tachycardia; the terminal event can also be a condition of asystole or electromechanical dissociation, especially in patients in whom there is already a significant impairment of ventricular function.
Arrhythmogenic right ventricular cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy is one the major causes of SCD in the pre-CAD age group. Although predictive markers of SCD have not yet been defined in large prospective studies, SCD occurs more frequently in patients with extensive right ventricular changes and in those with left ventricular involvement.[16]
The prevalence of the disease is not well-defined and varies from 1:1000 to 1:10000 per year with an annual incidence of SCD of 2%. Exercise is often a precipitating factor in the pathogenesis of terminal arrhythmic event, which is typically a ventricular fibrillation (while atrioventricular disorders are rare). The left ventricle involvement, however, seems to be the main predictor of SCD, while, in risk stratification, is still doubtful the importance of familiarity, early onset (<20 years), electrocardiographic abnormalities of repolarization, of previous syncope.
Athletic heart
Sudden and unexpected death in young, trained athletes is predominantly due to underlying and usually unsuspected congenital cardiovascular disease. The most important of these appear to be hypertrophic cardiomyopathy, anomalous origin of coronary artery, and arrhythmogenic right ventricular cardiomyopathy.[17]
Scd and channelopathies
In the last 15 years, the results of mutation screening in sudden unexplained death syndrome or sudden infant death syndrome, the so-called molecular autopsy, have been reported in several studies. The identification of the genes underlying the inherited arrhythmogenic syndromes has greatly contributed to the understanding of the substrate for arrhythmias development but, more importantly, it has provided major practical information that are helpful when managing affected individuals. The identification of a mutation allows us to establish the diagnosis independently from the electrocardiographic features and the arrhythmic manifestations. When screening family members of a genotyped proband, an unexpectedly large number of carriers of the genetic defect are identified among relatives that were considered non-affected based on clinical evaluation.[1]
Based on current evidence, it seems that genetic information alone or in combination with other clinical variables could represent a novel and useful parameter for risk stratification with immediate contribution to clinical practice.
Although genotyping is already worth being pursued in all patients with inherited genetic disorders, it is expected that its contribution to medical practice will increase when some of the gaps in understanding of the links between DNA defects and clinical phenotype are filled.
(Gene connection for the heart: A project of the study group on molecular basis of arrythmias. Fondazione Salvatore Maugeri http://pc4.fsm.it:81/cardmoc/).
In analogy with what happens in the genetics of inherited cardiomyopathies, in arrhythmogenic disorders, the “same” phenotype is associated with multiple genetic defects; this phenomenon is called “genetic heterogeneity”.
Channelopathies associated with prolonged repolarization
Long QT syndrome (LQTS)
LQTS is defined as an arrythmogenic disorder in the structurally normal heart presenting with QT prolongation that is often associated with peculiar ST-T wave morphology, syncope, and sudden death.[18]
The number of genes known to cause LQTS has steadily increased over the last 15 years. However, even after extended screening, mutations in the first three genes identified in the early 1990s (KCNQ1, KCNH2, and SCN5A) still account for the vast majority (80-90%) of patients.
LQTS diagnosis should be considered in all patients presenting with QTc >440 milliseconds in male patients and >460 milliseconds in female patients. Except for the Timothy syndrome and rare SCN5A mutations, the presence of structural abnormalities and of acquired causes of QT prolongation should be excluded. Given the presence of incomplete penetrance and QT variability, exercise stress testing, Holter monitoring, and pharmacological challenge may be useful as diagnostic tests.[19]
Once diagnosis is established, beta-blockers therapy is recommended. LQT1 patients respond very well to beta-blockers.[20] Compared with LQT1 patients, LQT2 and LQT3 patients have a higher recurrence rate of arrhythmic events while on therapy.[21]
The implantation of an implantable cardioverter-defibrillator (ICD) may be considered on evidence of beta-blocker failure and in selected high-risk individuals.
Channelopathies associated with abbreviated repolarization and conduction defects
LQTS is caused by a loss-of-function mutation in a channel that conducts a repolarizing current and a gain-of-function mutation in a channel that carries a depolarizing current. When the opposite effects occur, mutations cause completely different diseases such as Brugada syndrome (BrS), short-QT syndrome, early repolarization syndrome, sinus node dysfunction, and progressive conduction defects.
Brugada syndrome (brs)
BrS is characterized by ST segment elevation with “coved” morphology in the right precordial leads and complete or incomplete right bundle-branch block.[22] This ECG pattern is intermittent and may be unmasked by pharmacological challenge with sodium channel blockers such as procainamide, flecainide, ajmaline, or pilsicainide. The onset of ventricular arrhythmias causes the occurrence of syncope and may lead to sudden death, usually at rest. Syndrome manifestations occur more frequently in young males (with male:female ratio of 8:1) aged between 30 and 40 years.[23]
The electrophysiological mechanisms of BrS are still not completely known. It has been suggested that the arrhythmogenic substrate is due to the altered balance of inward and outward currents that is too much in favor of the latter in the early action potential phases (mainly during phase 1, loss of action potential dome), particularly in the epicardial layers.
This “selective” action potential shortening increases transmural dispersion and favors re-entry.[24]
Management of BrS is based on the use of ICD in selected high-risk individuals. No drug has been definitely proven effective in reducing the cardiac arrest burden. It is, therefore, clear that risk assessment is a key issue in this condition to tailor the use of ICD therapy.
Short-QT syndrome
Another newly discovered disease that increases the risk of SCD in young people is the short QT syndrome caused by a genetic defect in a channel for potassium, produced by the KCNJ2 gene and clinically characterized by arrhythmias and repeated unexplained fainting.[25]
Short-QT syndrome is described as a disorder characterized by abbreviated QT interval, ventricular and atrial arrhythmias, and SCD. There have been 70 short-QT syndrome cases reported worldwide, with the mean QTc value in the entire population of 310 milliseconds (The upper limit set by the majority of groups is between 340 and 350 milliseconds). Symptomatic (sudden death or cardiac arrest) individuals, accounting for 25% of the total, tend to present with shorter QTc (average, 300 milliseconds).
Channelopathies associated with abnormal calcium handling
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is characterized by polymorphic ventricular tachycardia in the absence of structural cardiac abnormalities, clinically described by recurrent syncopal episodes that occur in people who often have a family history of such episodes.
CPVT is characterized by a high incidence of cardiac events among untreated patients (79% in patients up to 40 years of age) and 30% incidence of SCD.[26]
Investigations directed to disclose the molecular basis of CPVT led to the identification of mutations of two genes, the ryanodine receptor RyR2 and the cardiac calsequestrin CASQ2, which are associated with the autosomal dominant and recessive forms of CPVT, respectively.
When a CPVT diagnosis is established, beta-blockers should be administered. Although this approach affords protection in the majority of patients, 30% experience at least one arrhythmic event while on therapy.[27] In these cases an ICD may be indicated.
Clinical manifestation and therapy
The onset of cardiac arrest, which is the mechanism that precedes SCD, can be associated with the typical symptoms of acute cardiac events, i.e., chest pain, dyspnea or orthopnea, fatigue, sudden onset of sustained tachycardia, and palpitations. When symptoms appear, the complete loss of consciousness follows, and in the absence of early intervention, within a few minutes, death occurs. However, in many cases, the onset of SCD is completely unexpected, without warning signs.
Dramatic survival rates that we find in the literature vary from 2% to 44%[28] depending on appropriateness and rapidity of the therapeutic intervention, with a mortality rate of 100% if the patient is not resuscitated.
In the event of a cardiac arrest, execution of a correct procedure for cardiopulmonary resuscitation within a few minutes after the event is the only chance of survival for the patient.
The obvious difficulties and poor results reached by the “treatment” of cardiac arrest, however, emphasize the importance of prevention that is divided into primary and secondary according to the presence or absence of a history of potentially fatal tachyarrhythmias and/or cardiorespiratory arrest.
However, SCD prevention is largely an issue of primary prevention because only a small proportion, less than 5%, of people who experience SCD have a history of episodes of ventricular tachycardia or fibrillation.
It is, therefore, crucial to correct risk factors for cardiovascular mortality and identify populations at increased risk for SCD, which may be favored by specific therapeutic aids. Although therapies exist for treatment of life-threatening ventricular tachyarrhythmias and prevention of myocardial infarction/coronary artery plaque rupture, significant challenges exist in identifying the individual patient within population subgroups who is at substantial personal risk of these events, and in whom the most intensive therapies could and should be applied.
Regarding drug therapy, it is now clear that many of the anti-arrhythmic drugs can cause anomalies that can become life-threatening arrhythmias.[29]
Randomized clinical trials support the use of implantable defibrillators for mortality reduction in specific populations at high risk for SCD.[1]
The implementation of primary prevention of SCD should also consider the costs of conducting a screening of candidates, which is much more accurate, in order to identify those at highest risk that are able to obtain the greatest benefit from the installation of the defibrillator.
Considering the above limitations in the application and dissemination of strategies for primary and secondary prevention, the utilization and implementation of therapeutic strategies that can positively influence the mortality of patients with cardiopulmonary arrest outside the hospital seem fundamental.
ROLE OF IMAGING TECHNIQUES IN SCD SUBSET
The accurate identification of patients at risk for SCD is a mission of research in cardiology and cardiac imaging. Multiple invasive and noninvasive tests have been evaluated but currently no optimal strategy for risk stratification exists. Table 1 Advantages of Non Invasive Imaging Modalities
Table 1.
Advantages of non-invasive imaging modalities
| No radiation burden |
| No iodinated contrast |
| Quantification of left ventricular ejection fraction |
| Quantification of left ventricular volume indexes |
| Evaluation of right ventricular pathology |
| Differentiation of types of cardiomyopathy (ischemic, non-ischemic, infiltrative) |
| Visualization and quantification of scar |
| Area of no-reflow in ischemic cardiomyopathy |
| Interstitial fibrosis |
| Evaluation for anomalous coronary artery |
Regardless of the underlying structural heart disease, the use of ICD for secondary prevention of SCD is supported by extensive evidence demonstrating the superiority of ICD over antiarrhythmic drug therapy in reducing SCD and all-cause mortality of patients resuscitated from cardiac arrest.[30] However, recommendations for ICD implantation for primary prevention in patients with other structural heart diseases, such as hypertrophic cardiomyopathy or right ventricular arrhythmogenic dysplasia, rely on prospective registries or retrospectively analyzed series and predictive markers of SCD, including imaging ones, have not been definitively established.
Left ventricular ejection fraction (EF) by echocardiography is the most widely used measure of left ventricular systolic function. It is promptly accessible, easy, and fast to measure. Echocardiography is a Class I indication (Level of Evidence B) for patients with ventricular arrythmias suspected of having structural heart disease as well for the subset of patients at high risk for the development of serious ventricular arrhythmias or SCD, such as those with cardiomyopathies, MI survivors, or relatives of patients with inherited disorders associated with SCD.[1]
Reduced EF is the principal risk factor for overall mortality and SCD in the heart failure population. The relationship between EF and death due to heart failure and ventricular arrhythmias in patients with MI and non-ischemic dilated cardiomyopathy is well-established.[31,32,33,34]
The prognostic value of impaired left ventricular function for overall mortality and SCD persists despite progress in treatments for acute MI.[35]
However, we should consider that the relationship between SCD and EF could be spurious in some way, considering the null effect of ICD implantation in the earliest phases (i.e., >40 days) after MI or ischemic events, thus suggesting that low LVEF may be as much a marker for death due to progressive pump failure as it is for death due to SCD.[36,37]
More strikingly, the vast majority of SCD is dramatically observed in otherwise healthy subjects or trained athletes, with supposed normal or super-normal EF, thus revealing the limited role of this parameter in risk assessment and claiming for new imaging markers of risk.[38]
As a correlate, it should also be considered that many patients with indications to ICD based on EF criteria, according to current guidelines,[1] experience the side effects of this therapy (inappropriate shocks; device related complications).[39] These observations underscore the limited accuracy of EF to identify patients at high risk for SCD.
A multimodality imaging approach is actually the most important tool to provide comprehensive information on different pathophysiological mechanisms related to SCD [Table 2].[40]
Table 2.
Imaging techniques and their role in SCD risk stratification (*also forensic application)
| Imaging Technique | Parameter | Utility |
|---|---|---|
| Echocardiography | Ejection Fraction (EF) | Ischemic Heart Disease; Dilated Cardiomyopathy |
| Wall Thickness | Hypertrophic Cardiomyopathy | |
| Right Ventricle | Right Ventricular Dysplasia | |
| Ischemia/Viability(Stress) | Ischemic Heart Disease | |
| Speckle Tracking Imaging | Regional deformation; tissue characterization | |
| CMR* | Ischemia/Viability | Ischemic Heart Disease; Cardiomyopathies |
| Tissue Characterization | Right Ventricular Dysplasia | |
| Scar | Ischemic Heart Disease; Cardiomyopathies | |
| Tagged MRI | Regional deformation | |
| MRI tractography | Fiber distribution | |
| SPECT | Ischemia/Viability | Ischemic Heart Disease |
| Scar | Ischemia; Cardiomyopathies | |
| Sympathetic Innervation | Heart Failure | |
| PET | Hybernating Myocardium | Heart failure; ischemia |
| Sympathetic Innervation | Heart Failure Cardiomyopathies | |
| Computed Tomography* | Tissue Characterization | Ischemic Heart Disease; Cardiomyopathies |
Based on Bertini M, Schalij MJ, Bax JJ, Delgado V. Emerging role of multimodality imaging to evaluate patients at risk for sudden cardiac death. Circ Cardiovasc Imaging 2012;5:525-35
Beside EF, echocardiography is able to offer a more deep insight into cardiac structural alterations, as well-functional implications of conditions at risk for SCD. Its intrinsic low costs, makes echocardiography the baseline ideal tool for screening of relatives of patients with aborted SCD or inherited arrythmogenic cardiomyopathies. For example, evaluation of wall motion anomalies, wall thickness, or right ventricle is able to reveal in a simple, fast, and cost-saving manner the presence of ischemia, pathological hypertrophy (suspect for hypertrophic cardiomyopathy; high risk for SCD when septal hypertrophy is > 30 mm) or hypothesize right ventricular dysplasia, demanding further investigations [Figure 2].
Figure 2.
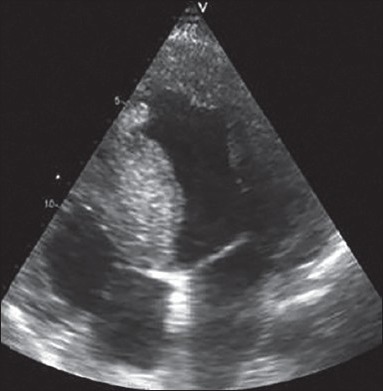
Assessment of arrhythmogenic substrate in hypertrophic cardiomyopathy: On 2D echocardiography, septum thickness >30 mm is a risk factor for ventricular arrhythmias
Recent advances in tissue deformation imaging (speckle tracking) reveal the great potential for echocardiography to associate mechanical properties to genetic alterations (channelopathies), linking electrical to anatomical substrate. Speckle tracking imaging also allows a better evaluation of ischemic myocardium (peri-infarcted areas) with subsequent implication for risk stratification. The presence of impaired segmental longitudinal strain in the peri-infarct zone was independently associated with an increased risk of appropriate ICD therapy for VF/TV.[41]
Subtle electric derangements may cause mechanical dysfunction. Two dimensional speckle tracking echocardiography has unraveled inhomogeneous myocardial contraction within the left ventricle in patients with long-QT syndrome. In 101 genotyped long-QT mutation carriers, including 48 symptomatic patients with documented ventricular arrhythmias, longitudinal, and circumferential LV myocardial strain was measured with two-dimensional speckle tracking echocardiography. Subendocardial (longitudinal) and midmyocardial (circumferential) mechanical dispersions were significantly larger in symptomatic carriers as compared with asymptomatic carriers.[42]
Increasing evidence suggests that the presence and extent of myocardial tissue heterogeneity with regions of scar and interstitial fibrosis provide a substrate for ventricular arrhythmias that is believed to be the major cause of SCD, both in ischemic and non-ischemic cardiomyopathy.[43]
Cardiac magnetic resonance (CMR) is a non-invasive imaging modality that visualizes and quantifies myocardial scar with late gadolinium enhancement (LGE), with growing evidence delineating its additive value in identifying patients at higher risk for SCD with proven histopathological correlation[44,45] [Figure 3].
Figure 3.
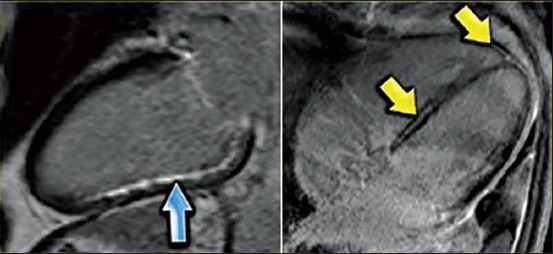
Role of CMR in the differential diagnosis: LEFT: Long axis late enhancement image in a patient with an inferior wall infarction with subendocardial enhancement in the territory of the right coronary artery RIGHT: 4-chamber late enhancement image in a patient with idiopathic dilated cardiomyopathy with midmyocardial enhancement
A minor but important role in tissue characterization is recognized also for contrast-enhanced multi-detector row computed tomography.[46]
CMR has no ionizing radiation burden or iodinate contrast use, is able to quantify more accurately than standard echocardiography chamber volumes and EF, allows a direct visualization of coronary arteries excluding possible anomalies, and is actually a good standard in evaluation of right ventricle (dysplasia).
Because of its high spatial resolution, CMR differentiates the various scar patterns and detects areas with interstitial fibrosis (necrosis) or edema (myocarditis; acute ischemia).[47,48]
There is growing evidence delineating the strength and additive value of CMR in identifying patients at risk for SCD.
The extent of myocardial scar is an independent predictor of ventricular arrhythmias.[49,50] Delayed contrast-enhanced CMR has the highest spatial resolution for assessment of scar tissue and has further increased the understanding of the pathophysiology of SCD in patients with ischemic heart disease. Initial reports demonstrate the superiority of myocardial scar burden on contrast-enhanced CMR over EF for prediction of ventricular arrhythmias.[51] Also, in hypertrophic and dilated cardiomyopathy, CMR revealed the importance of scar burden in determining SCD.
In hypertrophic cardiomyopathy, variable patterns of contrast-enhancement can be observed: Transmural and non-transmural contrast-enhancement in focal, multifocal, or confluent patterns.
Tagged CMR is able to accurately evaluate tissue deformation, offering a deep insight into myocardial mechanics.
Nuclear imaging as well as stress echocardiography and, more recently, myocardial perfusion CMR provide further risk stratification of patients with suspected ischemic heart disease by assessing ischemia and viability that may influence the arrhythmogenic substrate. Exercise or pharmacological stress testing are Class I recommendation in patients with arrythmias and intermediate probability of having ischemic heart disease.[1]
Even if two-dimensional and contrast echocardiography remain the imaging techniques of choice for arrhythmogenic right ventricular cardiomyopathy diagnosis, contrast-enhanced MRI has emerged as a valuable tool to characterize the right ventricular myocardium. Regional akinesia, dyskinesia, or dyssinchronus right ventricular contraction along with right ventricular dilatation or impaired systolic function assessed with MRI are major diagnostic criteria. Contrast-enhanced MRI has demonstrated to be a valid alternative to endomyocardial biopsy. This fibrous or fibrofatty replacement of the right ventricular myocardium appears as bright, hyperenhanced areas on contrast-enhanced MRI.
Radionuclide techniques are also able to assess the extent of scar formation.[52,53] With thallium-201 (201Tl) or technetium-99m (99mTc) sestamibi/tetrofosmin Single Photon Emission Computed Tomography (SPECT) imaging, myocardial scar is visualized as fixed perfusion defects, and it is associated with risk of adverse events, including arrythmic events and SCD.
SPECT has shown to be an effective method of risk stratification in patients with known CAD but relatively preserved EF (>35%). The presence of ischemia is a strong prognostic indicator also in hypertrophic cardiomyopathy.[54]
Association between myocardial viability assessed with Positron Emission Tomography (PET) and risk of ventricular arrhythmias has also been explored. 18-fluorine (18F) fluorodeoxyglucose PET allows evaluation of hibernating myocardium and flow-metabolism mismatch pattern (myocardial hypoperfusion with enhanced glucose myocardial metabolism), a pattern at risk for arrythmic events.[55]
Other non-invasive imaging modalities such as stress myocardial perfusion CMR and stress echocardiography have also provided important prognostic information.[56,57] Particularly, integrated myocardial ischemia and scar assessment with vasodilator stress CMR perfusion and contrast-enhanced CMR has demonstrated to be a powerful risk stratification tool.
Cardiac sympathetic innervation imaging has emerged as an important risk stratification tool for patients with ischemic heart failure and non-ischemic cardiomyopathies. Using radiolabeled norepinephrine analogs, the anatomic integrity and function of the sympathetic nerve terminals of the heart can be evaluated with SPECT and PET imaging.[58]
Carbon-11 (11C) hydroxyephedrine (HED) is the most widely used PET tracer to quantify the density of sympathetic nerve terminals and its cardiac uptake correlates with norepinephrine tissue concentration.[59] Reduced retention of 11C-HED on PET images indicates the presence of regional sympathetic denervation, at risk for arrythmic events. The SPECT tracer 123-iodine (123I)-metaiodobenzylguanidine (MIBG) mimics the uptake, storage and release of norepinephrine in the sympathetic nerve endings. However, 123I-MIBG is not metabolized and does not interact with postsynaptic receptors allowing visualization of the sympathetic postganglionic presynaptic fibers using both planar and SPECT imaging.
Imaging techniques also play a very important role in forensic post-mortem practice.[60] In the last 20 years, computed tomography and MRI have been increasingly used in routine practice and research. Ideally, these techniques, confirming autoptic findings, should furnish a faster and non-invasive diagnostic insight to pathologist.
Post-mortem CT angiography (PMCTA) enables the investigation of the vascular lumen.[61] At present, coronary arteries are investigated using PMCTA and/or Post-mortem Magnetic Resonance angiography (PMMRA).[62]
Post-mortem Magnetic Resonance enables the detection of myocardial infarction and the estimation of infarct age based on signal behavior as well myocardial structure in cardiomyopathies.[63]
CONCLUDING REMARKS: FUTURE DEVELOPMENT
Thanks to the work of several groups, the community has witnessed a remarkable growth of the awareness of inherited arrhythmogenic disease. The role of these conditions as key determinants of juvenile sudden death is now largely recognized. Currently, there is a large gap between the clinically useful information that we may derive from genetic testing of the more prevalent variants and the scant insight provided by the screening of rare genetic forms that account for 1% to 5% of all cases.
The goal for the future is to fulfill the transition from a population-based management to personalized therapeutic strategies.
There are actually no models for SCD risk prediction among the general population despite multiple studies reporting on individual risk factors. Risk stratification algorithms based on findings from epidemiological studies that evaluate traditional cardiovascular risk factors, lifestyle, and dietary habits, imaging and biological markers, and genetic variants in combination may aid in the identification of susceptible subgroups within the population. It will also be critical to determine whether novel markers associate with SCD to a greater extent than with other manifestations of heart disease.
The heritability of SCD remains poorly understood with the current data. Because of the rapid development of next-generation sequencing technologies, large-scale sequencing projects are becoming possible that will allow the examination of rare genetic variation as a component of SCD risk. SCD is a complex disorder that has been a research and clinical focus for several decades. As our understanding of this condition continues to improve with epidemiological studies, experimental investigations, and clinical trials, strategies to reduce the incidence and lethality of SCD across the population remain important priorities. Low-risk interventions and therapies that are directed toward cardiovascular disease in general and SCD specifically will likely help reduce the burden of SCD in the population.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114:e385–484. doi: 10.1161/CIRCULATIONAHA.106.178233. [DOI] [PubMed] [Google Scholar]
- 2.Chugh SS, Reinier K, Teodorescu C, Evanado A, Kehr E, Al Samara M, et al. Epidemiology of sudden cardiac death: Clinical and research implications. Prog Cardiovasc Dis. 2008;51:213–28. doi: 10.1016/j.pcad.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chugh SS. Early identification of risk factors for sudden cardiac death. Nat Rev Cardiol. 2010;7:318–26. doi: 10.1038/nrcardio.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maron BJ, Spirito P, Shen WK, Haas TS, Formisano F, Link MS, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405–12. doi: 10.1001/jama.298.4.405. [DOI] [PubMed] [Google Scholar]
- 5.Thel MC, O’Connor CM. Cardiopulmonary resuscitation: Historical perspective to recent investigations. Am Heart J. 1999;137:39–48. doi: 10.1016/s0002-8703(99)70458-8. [DOI] [PubMed] [Google Scholar]
- 6.de Vreede-Swagemakers JJ, Gorgels AP, Dubois-Arbouw WI, van Ree JW, Daemen MJ, Houben LG, et al. Out-of-hospital cardiac arrest in the 1990's: A population-based study in the Maastricht area on incidence, characteristics and survival. J Am Coll Cardiol. 1997;30:1500–5. doi: 10.1016/s0735-1097(97)00355-0. [DOI] [PubMed] [Google Scholar]
- 7.Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473–82. doi: 10.1056/NEJMra000650. [DOI] [PubMed] [Google Scholar]
- 8.Chugh SS, Reinier K, Singh T, Uy-Evanado A, Socoteanu C, Peters D, et al. Determinants of prolonged QT interval and their contribution to sudden death risk in coronary artery disease: The Oregon Sudden Unexpected Death Study. Circulation. 2009;119:663–70. doi: 10.1161/CIRCULATIONAHA.108.797035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panikkath R, Reinier K, Uy-Evanado A, Teodorescu C, Hattenhauer J, Mariani R, et al. Prolonged Tpeak-to-tend interval on the resting ECG is associated with increased risk of sudden cardiac death. Circ Arrhythm Electrophysiol. 2011;4:441–7. doi: 10.1161/CIRCEP.110.960658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myerburg RJ, Kessler KM, Castellanos A. Sudden cardiac death: Epidemiology, transient risk, and intervention assessment. Ann Intern Med. 1993;119:1187–97. doi: 10.7326/0003-4819-119-12-199312150-00006. [DOI] [PubMed] [Google Scholar]
- 11.Roberts WC, Kragel AH, Gertz SD, Roberts CS. Coronary arteries in unstable angina pectoris, acute myocardial infarction, and sudden coronary death. Am J. 1994;127:1588–93. doi: 10.1016/0002-8703(94)90390-5. [DOI] [PubMed] [Google Scholar]
- 12.Ehlert FA, Goldberger JJ. Cellular and pathophysiological mechanisms of ventricular arrhythmias in acute ischemia and infarction. Pacing Clin Electrophysiol. 1997;20:966–75. doi: 10.1111/j.1540-8159.1997.tb05501.x. [DOI] [PubMed] [Google Scholar]
- 13.Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–55. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- 14.Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342:1778–85. doi: 10.1056/NEJM200006153422403. [DOI] [PubMed] [Google Scholar]
- 15.Tamburro P, Wilber D. Sudden death in idiopathic dilated cardiomyopathy. Am Heart J. 1992;124:1035–45. doi: 10.1016/0002-8703(92)90989-9. [DOI] [PubMed] [Google Scholar]
- 16.Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J Am Coll Cardiol. 1997;30:1512–20. doi: 10.1016/s0735-1097(97)00332-x. [DOI] [PubMed] [Google Scholar]
- 17.Maron BJ, Pelliccia A. The heart of trained athletes: Cardiac remodeling and the risks of sports, including sudden death. Circulation. 2006;114:1633–44. doi: 10.1161/CIRCULATIONAHA.106.613562. [DOI] [PubMed] [Google Scholar]
- 18.Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. 2008;51:2291–300. doi: 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 19.Hekkala AM, Viitasalo M, Vaananen H, Swan H, Toivonen L. Abnormal repolarization dynamics revealed in exercise test in long QT syndrome mutation carriers with normal resting QT interval. Europace. 2010;12:1296–301. doi: 10.1093/europace/euq184. [DOI] [PubMed] [Google Scholar]
- 20.Vincent GM, Schwartz PJ, Denjoy I, Swan H, Bithell C, Spazzolini C, et al. High efficacy of beta-blockers in long-QT syndrome type 1: Contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215–21. doi: 10.1161/CIRCULATIONAHA.108.772533. [DOI] [PubMed] [Google Scholar]
- 21.Priori SG, Napolitano C, Schwartz PJ, Grillo M, Bloise R, Ronchetti E, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341–4. doi: 10.1001/jama.292.11.1341. [DOI] [PubMed] [Google Scholar]
- 22.Brugada J, Brugada R, Antzelevitch C, Towbin J, Nademanee K, Brugada P. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation. 2002;105:73–8. doi: 10.1161/hc0102.101354. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki H, Torigoe K, Numata O, Yazaki S. Infant case with a malignant form of Brugada syndrome. J Cardiovasc Electrophysiol. 2000;11:1277–80. doi: 10.1046/j.1540-8167.2000.01277.x. [DOI] [PubMed] [Google Scholar]
- 24.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: Report of the second consensus conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–70. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 25.Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96:800–7. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 26.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 27.Sy RW, Gollob MH, Klein GJ, Yee R, Skanes AC, Gula LJ, et al. Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2011;8:864–71. doi: 10.1016/j.hrthm.2011.01.048. [DOI] [PubMed] [Google Scholar]
- 28.Myerburg RJ, Interian A, Jr, Mitrani RM, Kessler KM, Castellanos A. Frequency of sudden cardiac death and profiles of risk. Am J Cardiol. 1997;80:10–9F. doi: 10.1016/s0002-9149(97)00477-3. [DOI] [PubMed] [Google Scholar]
- 29.Cleland JG, Ghosh J, Freemantle N, Kaye GC, Nasir M, Clark AL, et al. Clinical trials update and cumulative meta-analyses from the American College of Cardiology: WATCH, SCD-HeFT, DINAMIT, CASINO, INSPIRE, STRATUS-US, RIO-Lipids and cardiac resynchronisation therapy in heart failure. Eur J Heart Fail. 2004;6:501–8. doi: 10.1016/j.ejheart.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 30.Ezekowitz JA, Armstrong PW, McAlister FA. Implantable cardioverter defibrillators in primary and secondary prevention: A systematic review of randomized, controlled trials. Ann Intern Med. 2003;138:445–52. doi: 10.7326/0003-4819-138-6-200303180-00007. [DOI] [PubMed] [Google Scholar]
- 31.Bigger JT, Jr, Fleiss JL, Kleiger R, Miller JP, Rolnitzky LM. The relationships among ventricular arrhythmias, left ventricular dysfunction, and mortality in the 2 years after myocardial infarction. Circulation. 1984;69:250–8. doi: 10.1161/01.cir.69.2.250. [DOI] [PubMed] [Google Scholar]
- 32.Volpi A, De Vita C, Franzosi MG, Geraci E, Maggioni AP, Mauri F, et al. Determinants of 6-month mortality in survivors of myocardial infarction after thrombolysis. Results of the GISSI-2 data base. The Ad hoc Working Group of the Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto Miocardico (GISSI)-2 Data Base. Circulation. 1993;88:416–29. doi: 10.1161/01.cir.88.2.416. [DOI] [PubMed] [Google Scholar]
- 33.Risk stratification and survival after myocardial infarction. N Engl J Med. 1983;309:331–6. doi: 10.1056/NEJM198308113090602. [DOI] [PubMed] [Google Scholar]
- 34.Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994;331:1564–75. doi: 10.1056/NEJM199412083312307. [DOI] [PubMed] [Google Scholar]
- 35.Huikuri HV, Tapanainen JM, Lindgren K, Raatikainen P, Makikallio TH, Juhani Airaksinen KE, et al. Prediction of sudden cardiac death after myocardial infarction in the beta-blocking era. J Am Coll Cardiol. 2003;42:652–8. doi: 10.1016/s0735-1097(03)00783-6. [DOI] [PubMed] [Google Scholar]
- 36.Hohnloser SH, Kuck KH, Dorian P, Roberts RS, Hampton JR, Hatala R, et al. Prophylactic use of an implantable cardioverter-defibrillator after acute myocardial infarction. N Engl J Med. 2004;351:2481–8. doi: 10.1056/NEJMoa041489. [DOI] [PubMed] [Google Scholar]
- 37.Bigger JT., Jr Prophylactic use of implanted cardiac defibrillators in patients at high risk for ventricular arrhythmias after coronary-artery bypass graft surgery. Coronary Artery Bypass Graft (CABG) Patch Trial Investigators. N Engl J Med. 1997;337:1569–75. doi: 10.1056/NEJM199711273372201. [DOI] [PubMed] [Google Scholar]
- 38.Myerburg RJ, Mitrani R, Interian A, Jr, Castellanos A. Interpretation of outcomes of antiarrhythmic clinical trials: Design features and population impact. Circulation. 1998;97:1514–21. doi: 10.1161/01.cir.97.15.1514. [DOI] [PubMed] [Google Scholar]
- 39.Moss AJ, Greenberg H, Case RB, Zareba W, Hall WJ, Brown MW, et al. Long-term clinical course of patients after termination of ventricular tachyarrhythmia by an implanted defibrillator. Circulation. 2004;110:3760–5. doi: 10.1161/01.CIR.0000150390.04704.B7. [DOI] [PubMed] [Google Scholar]
- 40.Bertini M, Schalij MJ, Bax JJ, Delgado V. Emerging role of multimodality imaging to evaluate patients at risk for sudden cardiac death. Circ Cardiovasc Imaging. 2012;5:525–35. doi: 10.1161/CIRCIMAGING.110.961532. [DOI] [PubMed] [Google Scholar]
- 41.Ng AC, Bertini M, Borleffs CJ, Delgado V, Boersma E, Piers SR, et al. Predictors of death and occurrence of appropriate implantable defibrillator therapies in patients with ischemic cardiomyopathy. Am J Cardiol. 2010;106:1566–73. doi: 10.1016/j.amjcard.2010.07.029. [DOI] [PubMed] [Google Scholar]
- 42.Haugaa KH, Amlie JP, Berge KE, Leren TP, Smiseth OA, Edvardsen T. Transmural differences in myocardial contraction in long-QT syndrome: Mechanical consequences of ion channel dysfunction. Circulation. 2010;122:1355–63. doi: 10.1161/CIRCULATIONAHA.110.960377. [DOI] [PubMed] [Google Scholar]
- 43.Wu TJ, Ong JJ, Hwang C, Lee JJ, Fishbein MC, Czer L, et al. Characteristics of wave fronts during ventricular fibrillation in human hearts with dilated cardiomyopathy: Role of increased fibrosis in the generation of reentry. J Am Coll Cardiol. 1998;32:187–96. doi: 10.1016/s0735-1097(98)00184-3. [DOI] [PubMed] [Google Scholar]
- 44.Moon JC, Reed E, Sheppard MN, Elkington AG, Ho SY, Burke M, et al. The histologic basis of late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;43:2260–4. doi: 10.1016/j.jacc.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 45.Wagner A, Mahrholdt H, Holly TA, Elliott MD, Regenfus M, Parker M, et al. Contrast-enhanced MRI and routine single photon emission computed tomography (SPECT) perfusion imaging for detection of subendocardial myocardial infarcts: An imaging study. Lancet. 2003;361:374–9. doi: 10.1016/S0140-6736(03)12389-6. [DOI] [PubMed] [Google Scholar]
- 46.Schuleri KH, Centola M, George RT, Amado LC, Evers KS, Kitagawa K, et al. Characterization of peri-infarct zone heterogeneity by contrast-enhanced multidetector computed tomography: A comparison with magnetic resonance imaging. J Am Coll Cardiol. 2009;53:1699–707. doi: 10.1016/j.jacc.2009.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flett AS, Hayward MP, Ashworth MT, Hansen MS, Taylor AM, Elliott PM, et al. Equilibrium contrast cardiovascular magnetic resonance for the measurement of diffuse myocardial fibrosis: Preliminary validation in humans. Circulation. 2010;122:138–44. doi: 10.1161/CIRCULATIONAHA.109.930636. [DOI] [PubMed] [Google Scholar]
- 48.Iles L, Pfluger H, Phrommintikul A, Cherayath J, Aksit P, Gupta SN, et al. Evaluation of diffuse myocardial fibrosis in heart failure with cardiac magnetic resonance contrast-enhanced T1 mapping. J Am Coll Cardiol. 2008;52:1574–80. doi: 10.1016/j.jacc.2008.06.049. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt A, Azevedo CF, Cheng A, Gupta SN, Bluemke DA, Foo TK, et al. Infarct tissue heterogeneity by magnetic resonance imaging identifies enhanced cardiac arrhythmia susceptibility in patients with left ventricular dysfunction. Circulation. 2007;115:2006–14. doi: 10.1161/CIRCULATIONAHA.106.653568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klem I, Weinsaft JW, Bahnson TD, Hegland D, Kim HW, Hayes B, et al. Assessment of myocardial scarring improves risk stratification in patients evaluated for cardiac defibrillator implantation. J Am Coll Cardiol. 2012;60:408–20. doi: 10.1016/j.jacc.2012.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bello D, Fieno DS, Kim RJ, Pereles FS, Passman R, Song G, et al. Infarct morphology identifies patients with substrate for sustained ventricular tachycardia. J Am Coll Cardiol. 2005;45:1104–8. doi: 10.1016/j.jacc.2004.12.057. [DOI] [PubMed] [Google Scholar]
- 52.van der Burg AE, Bax JJ, Boersma E, Pauwels EK, van der Wall EE, Schalij MJ. Impact of viability, ischemia, scar tissue, and revascularization on outcome after aborted sudden death. Circulation. 2003;108:1954–9. doi: 10.1161/01.CIR.0000091410.19963.9A. [DOI] [PubMed] [Google Scholar]
- 53.Machecourt J, Longere P, Fagret D, Vanzetto G, Wolf JE, Polidori C, et al. Prognostic value of thallium-201 single-photon emission computed tomographic myocardial perfusion imaging according to extent of myocardial defect. Study in 1,926 patients with follow-up at 33 months. J Am Coll Cardiol. 1994;23:1096–106. doi: 10.1016/0735-1097(94)90597-5. [DOI] [PubMed] [Google Scholar]
- 54.Sorajja P, Chareonthaitawee P, Ommen SR, Miller TD, Hodge DO, Gibbons RJ. Prognostic utility of single-photon emission computed tomography in adult patients with hypertrophic cardiomyopathy. Am Heart J. 2006;151:426–35. doi: 10.1016/j.ahj.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 55.Dorbala S, Hachamovitch R, Curillova Z, Thomas D, Vangala D, Kwong RY, et al. Incremental prognostic value of gated Rb-82 positron emission tomography myocardial perfusion imaging over clinical variables and rest LVEF. JACC Cardiovasc Imaging. 2009;2:846–54. doi: 10.1016/j.jcmg.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elhendy A, Chapman S, Porter TR, Windle J. Association of myocardial ischemia with mortality and implantable cardioverter-defibrillator therapy in patients with coronary artery disease at risk of arrhythmic death. J Am Coll Cardiol. 2005;46:1721–6. doi: 10.1016/j.jacc.2005.04.065. [DOI] [PubMed] [Google Scholar]
- 57.Steel K, Broderick R, Gandla V, Larose E, Resnic F, Jerosch-Herold M, et al. Complementary prognostic values of stress myocardial perfusion and late gadolinium enhancement imaging by cardiac magnetic resonance in patients with known or suspected coronary artery disease. Circulation. 2009;120:1390–400. doi: 10.1161/CIRCULATIONAHA.108.812503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flotats A, Carrio I. Cardiac neurotransmission SPECT imaging. J Nucl Cardiol. 2004;11:587–602. doi: 10.1016/j.nuclcard.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 59.Bengel FM, Schwaiger M. Assessment of cardiac sympathetic neuronal function using PET imaging. J Nucl Cardiol. 2004;11:603–16. doi: 10.1016/j.nuclcard.2004.06.133. [DOI] [PubMed] [Google Scholar]
- 60.Michaud K, Grabherr S, Jackowski C, Bollmann MD, Doenz F, Mangin P. Postmortem imaging of sudden cardiac death. Int J Legal Med. 2014;128:127–37. doi: 10.1007/s00414-013-0819-6. [DOI] [PubMed] [Google Scholar]
- 61.Michaud K, Grabherr S, Doenz F, Mangin P. Evaluation of postmortem MDCT and MDCT-angiography for the investigation of sudden cardiac death related to atherosclerotic coronary artery disease. Int J Cardiovasc Imaging. 2012;28:1807–22. doi: 10.1007/s10554-012-0012-x. [DOI] [PubMed] [Google Scholar]
- 62.Ruder TD, Bauer-Kreutz R, Ampanozi G, Rosskopf AB, Pilgrim TM, Weber OM, et al. Assessment of coronary artery disease by post-mortem cardiac MR. Eur J Radiol. 2012;81:2208–14. doi: 10.1016/j.ejrad.2011.06.042. [DOI] [PubMed] [Google Scholar]
- 63.Jackowski C, Christe A, Sonnenschein M, Aghayev E, Thali MJ. Postmortem unenhanced magnetic resonance imaging of myocardial infarction in correlation to histological infarction age characterization. Eur Heart J. 2006;27:2459–67. doi: 10.1093/eurheartj/ehl255. [DOI] [PubMed] [Google Scholar]