

Abstract
Susceptibility to neural tube defects (NTDs), such as anencephaly and spina bifida is influenced by genetic and environmental factors including maternal nutrition. Maternal periconceptional supplementation with folic acid significantly reduces the risk of an NTD‐affected pregnancy, but does not prevent all NTDs, and “folic acid non‐responsive” NTDs continue to occur. Similarly, among mouse models of NTDs, some are responsive to folic acid but others are not.
Among nutritional factors, inositol deficiency causes cranial NTDs in mice while supplemental inositol prevents spinal and cranial NTDs in the curly tail (Grhl3 hypomorph) mouse, rodent models of hyperglycemia or induced diabetes, and in a folate‐deficiency induced NTD model. NTDs also occur in mice lacking expression of certain inositol kinases. Inositol‐containing phospholipids (phosphoinositides) and soluble inositol phosphates mediate a range of functions, including intracellular signaling, interaction with cytoskeletal proteins, and regulation of membrane identity in trafficking and cell division.
Myo‐inositol has been trialed in humans for a range of conditions and appears safe for use in human pregnancy. In pilot studies in Italy and the United Kingdom, women took inositol together with folic acid preconceptionally, after one or more previous NTD‐affected pregnancies. In nonrandomized cohorts and a randomized double‐blind study in the United Kingdom, no recurrent NTDs were observed among 52 pregnancies reported to date.
Larger‐scale fully powered trials are needed to determine whether supplementation with inositol and folic acid would more effectively prevent NTDs than folic acid alone. Birth Defects Research 109:68–80, 2017. © 2016 The Authors Birth Defects Research Published by Wiley Periodicals, Inc.
Keywords: neural tube defects, spina bifida, inositol, phosphoinositide, folic acid, clinical trial
Introduction
Maternal Nutrition during Pregnancy Influences Risk of NTDs
Failure in the process of neural tube closure during embryonic development results in severe birth defects termed neural tube defects, including anencephaly and spina bifida. Patterns of inheritance of NTDs indicate a major genetic contribution to risk of NTDs in the developing fetus, while it is also clear that non‐genetic, environmental, factors play a key role (Greene and Copp, 2014). These may include exogenous agents including anti‐epileptic drugs, such as valproic acid, the mycotoxin fumonisin or maternal exposures, such as high temperature resulting from fever (Gelineau‐van Waes et al., 2009; Wlodarczyk et al., 2012; Copp et al., 2013).
The area of most extensive study has been the potential contribution of maternal nutrition, given the observation that vitamin levels and risk of an affected pregnancy were both associated with socioeconomic status (Leck, 1974; Smithells et al., 1976; Au et al., 2010). Attention has particularly focused on vitamins relating to folate one‐carbon metabolism. Hence, maternal blood folate and vitamin B12 status are independent risk factors for NTDs, while elevated homocysteine, an inversely related biomarker of impaired folate status, is also associated with NTDs (Kirke et al., 1993; Blom et al., 2006).
The corollary to NTD risk conferred by sub‐optimal maternal vitamin status is that supplementation with specific nutritional factors may have a protective effect. Clinical trials have demonstrated a protective effect of periconceptional folic acid supplementation against NTD recurrence (after a first affected pregnancy) and occurrence (Smithells et al., 1980; MRC Vitamin Study Research Group, 1991; Czeizel and Dudás, 1992). The clear evidence for a preventive effect of folic acid led to recommendations that women planning pregnancy should take a folic acid supplement.
In addition, public health initiatives to implement mandatory food fortification have been associated with a reduction in NTD prevalence in many countries (Blencowe et al., 2010; Crider et al., 2011; Rosenthal et al., 2014). Nevertheless, the findings of clinical trials, monitoring of supplement use and case studies of recurrent NTDs all indicate that some NTDs are not preventable by folic acid (termed folic acid‐resistant or non‐responsive) (MRC Vitamin Study Research Group, 1991; Mosley et al., 2009; Bupp et al., 2015).
The recognition that some NTDs are not preventable by folic acid engendered interest in other nutritional factors that may contribute to prevention of NTDs. In this review, we focus on a potential role for another nutrient, inositol.
Inositol and Its Derivatives Participate in a Diverse Range of Cellular Functions
Inositol (1,2,3,4,5,6‐hexahydroxycyclohexane) is a naturally occurring simple six carbon sugar alcohol, sometimes referred to as a pseudo‐vitamin (vitamin Bh or B8), although it can be synthesized from glucose by means of conversion of glucose 6‐phosphate to inositol 1‐phosphate (Fig. 1). There are nine stereoisomers of inositol, of which myo‐, D‐chiro‐, scyllo‐, epi‐, muco‐, and neo‐inositol occur naturally, with the predominant form being myo‐inositol (Michell, 2008; Leung et al., 2011). Adults typically consume approximately 1 g of myo‐inositol per day which is present in a variety of foods including nuts, vegetables, and citrus fruits (Holub, 1987; Croze and Soulage, 2013). Plants, particularly legumes, oil seeds, and grains, are particularly rich in myo‐inositol hexakisphosphate (IP6; phytic acid); this is mostly hydrolyzed to free inositol before absorption from the gut (Michell, 2008; Schlemmer et al., 2009). However, owing to its ion‐chelating properties, an excess of phytate from dietary sources could theoretically hinder absorption of cations, such as Ca2+, from the gut (Wilson et al., 2015). Myo‐inositol in the nonphosphorylated form, typically available in vitamin supplements, does not have this property. Although less abundant, D‐chiro‐inositol is also obtained from dietary sources, principally in the methylated form 3‐O‐methyl‐D‐chiro‐inositol (pinitol) (Negishi et al., 2015).
Figure 1.
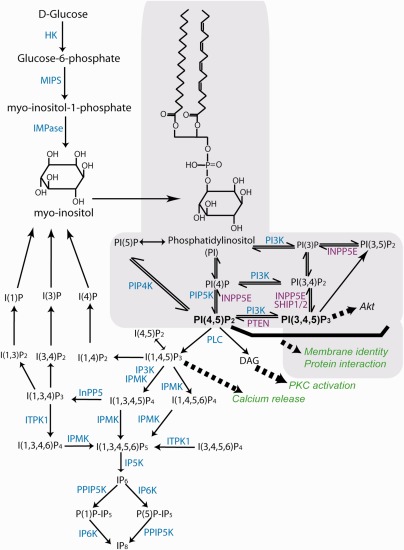
Outline of pathways for synthesis of inositol phosphates and phosphoinositides. Inositol can be obtained from dietary sources or synthesized from D‐glucose by the sequential action of hexokinase (HK), myo‐inositol‐1‐phosphate synthase (MIPS), and inositol monophosphatase (IMPas). The structure of myo‐inositol and phosphatidylinositol are shown (inositol lipids are shaded gray). These molecules form the backbone for synthesis of inositol phosphate (IP) and phosphoinositide (PI) molecules, mediated by action of multiple kinases and/or phosphatases. Enzymes that are discussed in the text are indicated (kinases in blue text, phosphatases in purple text). Notably, several phosphorylation/dephosphorylation steps can be mediated by several enzymes. For example, a number of kinases exist as multiple isoforms (e.g., PIP5K and PI3K [PI3‐kinase]), while several phosphatases can act to dephosphorylate PI(3,4,5)P3 (e.g., INPP5E, SHIP1, and SHIP2). In addition, some enzymes act at multiple steps (e.g. IPMK acts to phosphorylate inositol at a number of steps). Examples of downstream functional effects of key molecules are indicated in italics (green text). PLC, phospholipase C; DAG, diacylglycerol; PKC, protein kinase C; IPMK, inositol phosphate multikinase; ITPK1, inositol 1,3,4‐triphosphate 5/6 kinase.
Inositol is incorporated as the polar head‐group of membrane lipids based on phosphatidylinositol (PI), which is synthesized from myo‐inositol and cytidine diphosphate‐diacylglycerol (CDP‐DAG). Hence, phosphorylation of inositol at carbons 3, 4, and 5 generates a series of phosphoinositides which have multiple cellular functions in signaling and membrane dynamics (Di Pietro and De Camilli, 2006). When not bound to lipids, free inositol can carry a phosphate group at each of its six carbons (IP1 to IP6). IP1, IP2, and IP3, are generated by hydrolysis of PIP2 and subsequent recycling of inositol, while more highly phosphorylated forms (IP4, IP5, and IP6) and pyro‐phosphates (e.g., PP‐IP5 equivalent to IP7) are generated by the action of specific kinases (Livermore et al., 2016). Hence, intracellular inositol is present in a complex variety of lipid‐bound inositides as well as soluble inositol phosphates and pyrophosphates (Fig. 1).
Functions of phosphoinositides and inositol polyphosphates
The phosphoinositides PI(4,5)P2 and PI(3,4,5)P3 are key mediators of major signaling pathways that influence diverse cellular functions (Fig. 1). These pathways are reviewed extensively elsewhere (Di Paolo and De Camilli, 2006; Michell, 2008; Bridges and Saltiel, 2015). In brief, hydrolysis of PI(4,5)P2 by phospholipase C generates the second messengers inositol tri‐phosphate (IP3) and diacylglycerol (DAG). IP3 stimulates release of intracellular calcium while DAG stimulates several isoforms of protein kinase C (Nishizuka, 1992; Berridge, 1993;). Alternatively, phosphorylation of PIP2 at the 3‐position of inositol by PI3‐kinases generates PI(3,4,5)P3 which recruits downstream targets, including the serine‐threonine kinase Akt (Burgering and Coffer, 1995; Larue and Bellacosa, 2005; Hawkins and Stephens, 2015). The regulation of PI(3,4,5)P3 abundance by activation of PI3‐kinases and phosphatases such as PTEN regulates a range of downstream cellular properties including proliferation, motility and polarity (Tsujita and Itoh, 2015).
In parallel with their role in signaling pathways, phosphoinositides interact with numerous proteins and thereby provide an interface between membranes and the cytosol in a wide range of cellular contexts (Janmey and Lindberg, 2004; Posor et al., 2015). Regulation of the abundance of phosphorylated forms of PI involves the concerted action of a series of different kinases and phosphatases. For example, PI(4,5)P2 can be generated from PI(4)P by the action of PI4P 5‐kinases or to a much lesser extent from PI(5)P by the action of PI5P 4‐kinases (Fig. 1). There are several family members for each kinase, together with several corresponding phosphatases. Hence, there appears to be a complex regulation of the abundance of differing PIs.
Localized activity of specific kinases and phosphatases is thought to generate dynamic spatially restricted enrichment of particular PIs which can modulate membrane dynamics (Janmey and Lindberg, 2004; Saarikangas et al., 2010; Cauvin and Echard, 2015). For example, coordination of endocytosis and vesicle trafficking depends on specific PI pools. Clathrin‐mediated endocytosis requires PI(4,5)P2, with switching between PI types providing compartment identity as vesicles progress along the endocytic route (Croise et al., 2014; Posor et al., 2015). PIs, particularly PI(4,5)P2, can be coupled to the underlying F‐actin cytoskeleton by means of “linker” proteins such as class I myosins and Ezrin/Radixin/Moesin proteins, while PIP2 can also suppress proteins such as cofilin that mediate actin disassembly (Janmey and Lindberg, 2004; Tsujita and Itoh, 2015). Modulation of PI metabolism, for example through hydrolysis of PIP2, can thereby contribute to local alterations in membrane‐cytoskeleton tension during cell shape changes and rearrangement (Tsujita and Itoh, 2015). These properties of various PIs are implicated in membrane ruffling and micropinocytosis (Araki et al., 2007; Posor et al., 2015) as well as several events during cell division including spindle orientation, cell rounding and abcission (Field et al., 2005; Cauvin and Echard, 2015).
Among the proteins that interact with phosphoinositides and/or link PIs to F‐actin, a number are implicated in neural tube closure and/or NTDs. For example, several of the regulatory roles of PIs involve a close functional interaction with small GTPases, including Rac1, with which they associate at the membrane. Notably, disruption of cytoskeletal structure or turnover and impaired function of small GTPases are implicated in NTDs in several mouse models (Copp and Greene, 2010; Escuin et al., 2015; Rolo et al., 2016). Loss of function of another protein, MARCKS, which links PIP2 to the actin cytoskeleton also results in NTDs in mice (Stumpo et al., 1995; McLaughlin et al., 2002).
Inositol Is Required for Neural Tube Closure
Inositol deficiency causes cranial NTDs in mice
Interest in the influence of inositol levels on NTD predisposition was stimulated by nutritional studies in which rat embryos were cultured in defined conditions through the period of neural tube closure (Cockroft, 1979, 1988). Specific omission of several vitamins caused growth retardation or developmental abnormalities but NTDs occurred only in the absence of inositol. Similarly, deficiency of inositol in the culture medium causes cranial NTDs in mouse embryos, whereas deficiency of other vitamins, including folic acid, does not (Cockroft et al., 1992). Although inositol deficiency caused cranial NTDs among non‐mutant strains of mice, increased sensitivity was observed among cultured embryos of the curly tail (ct) strain, which are genetically predisposed to spinal and cranial NTDs (Cockroft et al., 1992). Under normal conditions curly tail embryos show only around 3 to 7% incidence of cranial NTDs (exencephaly). However, inositol‐deficiency caused much higher rates of cranial NTDs (∼70%) and an increased concentration of inositol was required to correct these defects than in other strains tested (Cockroft et al., 1992).
Inositol metabolism and neural tube defects in mouse models
Analysis of mice carrying gene‐trap alleles that result in loss or diminished expression of inositol kinases suggests that both phosphoinositides and inositol polyphosphates play key cellular functions essential for neural tube closure.
Cranial NTDs occur in embryos lacking PIP5KIγ, one of three isoforms of phosphatidylinositol‐4‐phosphate 5‐kinase that mediates synthesis of PI(4,5)P2 (Wang et al., 2007). Of interest, cranial NTDs also occur in mice lacking inositol polyphosphate 5‐phosphatase E (encoded by Inpp5E), which hydrolyses PI(4,5)P2 or PI(3,4,5)P3, and hence opposes the effect of PIP5KIγ on levels of PI(4)P (Jacoby et al., 2009). Given the functions of PI(4,5)P2 not only as substrate for phospholipase C‐mediated signaling by means of diacylglycerol and IP3 but also in membrane identity/trafficking, cytoskeletal organization and cell division, impaired production could potentially disturb neurulation through several different cellular mechanisms.
Recent studies suggest an additional possible mechanism relating to a requirement for regulation of phosphoinositides in regulating biogenesis and stability of primary cilia. PIP5KIγ localises to the basal body of cilia and appears to be essential for ciliogenesis, probably by depleting PI(4)P (Chavez et al., 2015; Xu et al., 2016). In contrast, Inpp5E opposes the action of PIP(5)KIγ during initial events of ciliogenesis (Xu et al., 2016), but appears to be required within established cilia to maintain their structural stability (Bielas et al., 2009; Plotnikova et al., 2015). Hence, phosphoinositide homeostasis appears to play a key role in cilia formation and function. In addition to cranial NTDs, mice lacking Inpp5E exhibit features of ciliopathies such as polycystic kidneys and polydactyly (Jacoby et al., 2009), while mutations in INPP5E are found in the ciliopathies Joubert Syndrome and MORM Syndrome (Bielas et al., 2009; Jacoby et al., 2009). Notably, cranial NTDs are observed in several other mouse mutants in association with abnormal cilia (Murdoch and Copp, 2010).
In addition to phosphoinositides, several functions have been ascribed to inositol phosphates beyond the function of IP3 in intracellular calcium signaling. In particular, generation of higher inositol polyphosphates appears to be required during the stage of neural tube closure. Mice that are deficient for Ipmk, encoding an inositol phosphate multikinase (Impk, also known as Ipk2), fail to develop beyond E9.5 (Frederick et al., 2005). Furthermore, embryos with reduced expression of Itpk1, encoding inositol 1,3,4,‐triphosphate 5/6‐kinase show growth retardation and cranial NTDs (frequency of ∼12%) (Wilson et al., 2009). These inositol kinases are crucial for production of the highly phosphorylated forms of inositol IP4, IP5, and IP6, the most abundant of which within cells is inositol hexakisphosphate (IP6) (Wilson et al., 2015). Impk may also act as a PI3‐kinase and can mediate transcriptional regulation through kinase‐independent functions (Kim et al., 2015).
Intriguingly, IP4 has been found to play a key role in modulating Rho GTPase activity and F‐actin during epithelial repair in Xenopus embryos (Soto et al., 2013). Given the requirement for Rho kinase‐dependent regulation of actomyosin turnover during mammalian neurulation (Escuin et al., 2015), it is tempting to speculate that inositol polyphosphates could directly influence the closure process.
Glycosylphosphatidylinositol‐anchored proteins and NTDs
In addition to PIs and IPs, inositol forms a structural component of glycophosphoinositide (GPI) anchors that link many different proteins to cell surfaces (Englund, 1993; Paulick and Bertozzi, 2008). Inositol glycans, derived from hydrolysis of GPI‐anchors, are also proposed to act as second messengers in certain signaling reactions responsive to insulin (Nascimento et al., 2006; Croze and Soulage, 2013).
Although GPI‐anchors may represent only a small proportion of cellular inositol it is intriguing that cranial NTDs arise in knockout mice for genes encoding several different GPI‐anchored proteins including Rgma (repulsive guidance molecule A; Niederkofler et al., 2004), Efna5 (ephrinA5) (Holmberg et al., 2000), and Folr1 (folate receptor 1, folate‐binding protein1; Piedrahita et al., 1999). Moreover, treatment of cultured embryos with phosphatidylinositol‐specific phospholipase C, which cleaves GPI anchors, disturbs spinal neural tube closure (O'Shea and Kaufman, 1980; Abdul‐Aziz et al., 2009).
Inositol Supplementation in Models of NTDs
Uptake of inositol by the embryo and generation of inositol phosphoinositides and inositol polyphosphates are required for cranial neurulation (Table 1). This raises the question of whether supplemental inositol may prevent NTDs in some models.
Table 1.
Experimental Models in Which Inositol Status or Metabolism Is Associated with Neural Tube Closure
| Inositol‐related deficits in NTD causation | NTD type | Comments/mechanism | Reference | |
|---|---|---|---|---|
| Inositol deficiency | Cultured mouse or rat embryos | Exencephaly | NTDs in non‐mutant and mutant strains. Higher incidence in ct strain | Cockroft, 1992 |
| PI4P5KIγ null | PIP kinase generates PI(4,5)P2 | Exencephaly |
Disordered actin Cilia defect? |
Wang, 2007 |
| Inpp5e null | PI(4,5)P2 and PI(3,4,5)P2 phosphatase | Exencephaly | Unstable cilia and ciliopathy phenotypes | Jacoby, 2009 |
| Itpk1 hypomorph | Generates IP(1,3,4,5)P4 and IP(1,3,4,6)P4 | Exencephaly &/or spina bifida | Deficit of higher IPs | Wilson, 2009 |
| NTD prevention by inositol in experimental models | ||||
| Curly tail mouse | Embryo culture; Oral; I.P. injection; sub‐cutaneous route | Spina bifida |
Corrects proliferation defect. Requires activity of PKC isoforms. Chiro‐inositol more effective than myo‐inositol |
Greene, 1997; Cogram, 2002; Cogram, 2004 |
| Hyperglycemia | High glucose in embryo culture | Exencephaly | Restores inositol levels. Arachidonic acid signalling? | Baker, 1990; Hashimoto, 1990; |
| Diabetes | Streptozotocin‐induced diabetes | Exencephaly | Restores inositol levels? | Khandelwal, 1998 |
| Folate‐deficient NTDs | Dietary folate deficiency in wild‐type strain (ct genetic background) | Exencephaly | Unknown | Burren, 2010 |
Prevention of spina bifida in the curly tail mouse
Although inositol deficiency was found to exacerbate cranial NTDs (see above), the main neural tube closure phenotype in the curly tail strain involves a defect of spinal neurulation affecting approximately 50% of embryos. Failure to complete closure of the posterior neuropore (PNP) at the end of spinal neurulation results in spina bifida in around 15% of embryos. A further 35 to 40% exhibit only a tail flexion defect, a phenotype which results from delayed completion of PNP closure (Van Straaten and Copp, 2001). Unlike in the cranial region, inositol deficiency did not further exacerbate the progression of spinal neurulation in cultured curly tail embryos or delay closure in the nonmutant CBA/Ca strain (Greene and Copp, 1997). However, increasing the myo‐inositol concentration normalized PNP closure in cultured embryos (Greene and Copp, 1997). This protective effect was confirmed by in vivo supplementation by means of maternal intra‐peritoneal injection, oral dosing or subcutaneous infusion (Greene and Copp, 1997; Cogram et al., 2002), with each strategy leading to a significant reduction in the frequency of spina bifida.
Interestingly, an alternative enantiomer of inositol, D‐chiro‐inositol showed a greater protective effect than myo‐inositol, with a larger reduction in incidence of spina bifida at the same dose, and maintained effectiveness even at a lower dose (Cogram et al., 2002). Whether myo‐ and chiro‐inositol act through the same mechanism is still unknown. Interconversion of the two enantiomers can be mediated by an endogenous epimerase (Pak et al., 1992; Sun et al., 2002), although other groups report limited interconversion in vivo (Lin et al., 2009). Notably, NTDs in the curly tail model are not preventable by folic acid and provide a model for “folic acid‐resistant” NTDs. Identification of a protective therapy such as inositol, therefore, raises the question of whether this approach may also have utility in preventing some human NTDs.
Supplemental inositol could potentially act to overcome the underlying causative defect responsible for NTDs and/or enhance the normal processes required for progression of neural tube closure. Completion of PNP closure does not occur prematurely in nonmutant embryos supplemented with inositol (Greene and Copp, 1997), which would argue that inositol may prevent NTDs by normalizing an underlying developmental abnormality. In the curly tail model, spina bifida results from a proliferation defect in the hindgut endoderm (Copp et al., 1988). This creates a growth imbalance between dorsal and ventral tissues, leading to increased ventral curvature of the caudal region of the embryo that mechanically opposes closure leading to spina bifida (Brook et al., 1991). This defect is thought to result from reduced expression of the transcription factor Grhl3. The curly tail strain is homozygous for a hypomorphic allele of Grhl3 and reinstatement of Grhl3 expression normalizes spinal neurulation (Ting et al., 2003; Gustavsson et al., 2007).
However, penetrance of defects is also strongly influenced by modifiers in the genetic background, such as a polymorphic variant of lamin B1 (de Castro et al., 2012). Inositol supplementation increases proliferation in the hindgut endoderm of curly tail embryos, which would explain the preventive effect on spinal NTDs (Cogram et al., 2004). Consistent with this hypothesis, spina bifida was also found to be preventable by combinations of thymidine and purine nucleotides, which also normalize proliferation in the hindgut (Leung et al., 2013).
In cultured embryos labelled inositol is taken up and incorporated into inositol phosphates and inositol‐containing phospholipids. This appears to be necessary for inositol's protective effect in curly tail embryos as inhibition of inositol phosphate recycling abolishes the protective effect (Greene and Copp, 1997). Increased production of PtdIns(4,5)P2 could potentially have a direct ameliorating effect on the underlying proliferation defect in curly tail embryos, owing to the its functions in cell division (Cauvin and Echard, 2015). In addition, among potential downstream effects following PtdIns(4,5)P2 hydrolysis, activation of protein kinase C appears to play a key role in prevention of spinal NTDs because the protective effect of inositol is (i) mimicked by short‐term treatment with a phorbol ester activator of PKC, and (ii) abrogated by broad spectrum and isoform‐specific PKC inhibitors (Greene and Copp, 1997; Cogram et al., 2004).
Inositol does not prevent NTDs in all models
The protective effect of inositol against spina bifida is not universal in mouse NTD models. For example, while prevention of NTDs in the curly tail model was confirmed in an independent laboratory, no response was observed among Grhl3 null mice (Ting et al., 2003). This is likely due to the greater severity of NTDs, which are fully penetrant in the complete knockout of Grhl3 compared with diminished expression in the curly tail hypomorph. In the context of cranial NTDs, it is also possible that inositol acts in the curly tail strain to normalize effects of genetic modifiers rather than the effects of Grhl3 mutation. For example, cranial NTDs in the curly tail strain appear to largely be a feature of the genetic background as, unlike spina bifida, they are not prevented by transgenic reinstatement of Grhl3 expression (Sudiwala et al., 2016). Spinal closure defects in the Bent tail mouse, carrying an X chromosome deletion that encompasses Zic3 (Franke et al., 2003; Klootwijk et al., 2004), and in Mekk4 knockouts (Chi et al., 2005) were also found to be unresponsive to inositol treatment.
Inositol and susceptibility to NTDs conferred by maternal diabetes
NTDs occur in cultured rat and mouse embryos exposed to hyperglycemia or streptozotocin‐induced diabetes (Sadler, 1980; Cockroft, 1984), both of which elicit depletion of tissue inositol levels (Hod et al., 1986; Sussman and Matschinsky, 1988; Hashimoto et al., 1990). Supplementation with inositol in cultured embryos corrects the hyperglycemia‐induced growth retardation and NTDs (Baker et al., 1990; Hashimoto et al., 1990; Hod et al., 1990). Similarly, oral administration of myo‐inositol led to a reduction in the frequency of diabetes‐induced abnormalities in rat embryos (Khandelwal et al., 1998) (Table 1).
Supplemental inositol restores tissue inositol levels, but could plausibly also act through pathways downstream of inositol phosphates or phosphoinositides. Arachidonic acid can be derived from diacylglycerol which is produced upon hydrolysis of PIP2. The protective effect of inositol against hyperglycemia in vitro is reversed by indomethacin, an inhibitor of arachidonic acid metabolism (Baker et al., 1990). Conversely, arachidonic acid and some derivative prostaglandins have a protective effect against NTDs in the hyperglycinemia and streptozotocin models (Goldman et al., 1985; Pinter et al., 1986). Whether inositol acts to prevent hyperglycemia and/or diabetes‐related NTDs through stimulating arachidonic acid signaling, upstream of prostaglandins and leukotrienes, or this pathway is required in parallel with inositol for cranial neurulation is not clear. Unlike, phorbol esters, arachidonic acid did not substitute for inositol in prevention of spinal NTDs in the curly tail model (Greene and Copp, 1997).
Prevention of NTDs resulting from folate deficiency?
While inositol reduces NTD frequency in the curly tail mouse, in which folic acid is not preventive, it is important to ask whether inositol can also prevent NTDs in models that are responsive to folic acid. Supplemental folic acid has been found to reduce the frequency of NTDs resulting from mutation of the Pax3 transcription factor in the Sp2H and Sp (alleles of Splotch mice) models (Fleming and Copp, 1998; Wlodarczyk et al., 2006), whereas myo‐inositol was not protective (Wlodarczyk et al., 2006). In dietary models, folate‐deficiency does not cause NTDs unless in the presence of a predisposing genetic mutation or background, such as Pax3 deficiency (Burren et al., 2008; Heid et al., 1992). Embryos with a genetic background similar to the curly tail strain but wild‐type for Grhl3 (+ct/+ct) do not develop cranial NTDs under normal dietary conditions but exhibit exencephaly when made folate‐deficient (Burren et al., 2010). As expected, NTDs in this model can be prevented by maternal administration of folic acid. Notably, however, myo‐inositol has a similar protective effect (Burren et al., 2010), raising the possibility that in some circumstances inositol could overcome NTD‐predisposing abnormalities imposed by sub‐optimal folate status. In this context, we cannot currently rule out a role for inositol, by means of its GPI‐anchor function, in promoting enhanced folate uptake by means of Folr1.
Inositol Status and Human NTDs
Few studies have systematically determined maternal inositol status in relation to NTD risk. Estimates of dietary myo‐inositol intake from a food frequency questionnaire did not suggest a strong association with risk of NTDs (Shaw et al., 2005). Moreover, determination of serum myo‐inositol concentration in mothers and their infants with spina bifida did not show significant variation from controls (63–102 subjects in each group), although a trend toward lower values was recognized (Groenen et al., 2003). However, among the lowest maternal serum myo‐inositol concentrations (lowest decile), there was a significant association with having a child with spina bifida (Groenen et al., 2003). If these findings are representative of maternal inositol status during pregnancy, one could speculate that insufficient inositol may contribute to development of NTDs in the offspring. Further studies on this relationship are warranted as evidence suggests that blood myo‐inositol levels may change during pregnancy (Groenen et al., 2006). Of note, in a more recent study in which samples were collected during pregnancy, maternal plasma IP6 was found be significantly diminished among NTD cases compared with controls (60 in each group) (Guan et al., 2014).
Considering potential genetic influences on inositol uptake or metabolism, analysis of variants in a myo‐inositol transporter, SLC5A11, did not suggest an association with spina bifida in a Dutch population (Groenen et al., 2004). However, investigation of maternal ITPK1 genotype, whose deficiency can cause NTDs in mice (Wilson et al., 2009), suggests a potential role in NTDs. A single nucleotide polymorphism (SNP) ‐based case–control analysis in a Chinese population, revealed a significant association of NTD with four tag SNPs in maternal ITPK1 that was principally attributable to association with spina bifida. Among the associated SNPs, rs3783903 also appeared to be predictive of lower ITPK1 expression in blood and plasma inositol levels (Guan et al., 2014).
Inositol Supplementation in Human Pregnancy
The findings of a preventive effect of a nutrient supplement, such as inositol, in rodent NTD models raises several questions, including: (1) are inositol supplements safe to use during pregnancy in humans, and (2) could inositol prevent some human NTDs?
In mouse studies, neither myo‐ nor chiro‐inositol had notable side‐effects in the supplemented dams or in the developing fetuses (Cogram et al., 2002). As described above, inositol supplementation has beneficial effects in several NTD models without apparent deleterious effects. However, in mice treated with valproic acid, an anti‐epileptic with known teratogenic properties, inositol was found to exacerbate the induced developmental abnormalities (Massa et al., 2006). Use of anti‐epileptics was an exclusion criterion in a subsequent clinical trial of inositol during pregnancy (Greene et al., 2016).
In adult humans, inositol has been tested for a variety of conditions (Table 2), including psoriasis, panic disorder, depression, and eating disorders (Benjamin et al., 1995; Levine et al., 1995; Chengappa et al., 2000; Gelber et al., 2001; Allan et al., 2004). Daily dosage ranging from 6 g to as high as 18 g did not result in major side effects, although mild flatulence or diarrhea was reported by a few patients. No side effects have been reported for doses less than 6 g per day. Overall, the outcomes of studies in adults do not suggest that supplemental inositol has deleterious effects (Croze and Soulage, 2013).
Table 2.
Clinical Use of myo‐Inositol
| Condition | Study size (total inositol and placebo groups) | Dose (daily, myo‐inositol unless stated) | Outcome | References |
|---|---|---|---|---|
| Adult conditions | ||||
| Psoriasis in patients taking lithium | 15 | 6 g for 1 week | Beneficial effect in psoriasis | Allan, 2004 |
| Bulimia nervosa and binge eating | 12 | 18 g for 6 weeks | Indication of therapeutic value | Gelber, 2001 |
| Add‐on treatment for bipolar disorder | 24 | 12 g 6 weeks | Indication of beneficial effect | Chengappa, 2000 |
| Panic disorder | 21 | 12 g for 4 weeks | Decline in frequency & severity | Benjamin, 1995 |
| Depression | 28 | 12 g for 4 weeks | Improved score in depression scale | Levine, 1995 |
| Metabolic syndrome | 80 | 2 g for 6 months | Improved blood pressure parameters | Giordano, 2011 |
| Treatment prior to or during pregnancy | ||||
| Polycystic ovary syndrome | 20 | 2 g for 12 weeks | Improved insulin sensitivity & menstrual cycle activity | Genazzani, 2008 |
| Polycystic ovary syndrome | 92 | 4 g for 14 weeks | Impoved ovarian function | Gerli, 2007 |
| Polycystic ovary syndrome | 42 | 4 g for 12–16 weeks | Improved insulin sensitivity & ovulation | Costantino, 2009 |
| Elevated fasting glucose | 75 | 4 g throughout pregnancy | Reduced incidence of gestational diabetes | Matarrelli, 2013 |
| Risk of gestational diabetes | 220 | 2 g from 1st trimester | Reduced incidence of gestational diabetes | D'Anna, 2013 |
| Gestational diabetes | 69 | 4 g for 8 weeks | Decline in fasting glucose | Corrado, 2011 |
| NTDs ‐ recurrence | 12 non‐randomised (15 pregnancies) | 0.5 or 1 g daily to 60 days pregnancy | No recurrent NTDs (0/17 babies) |
Cavalli and Copp, 2002
Cavalli, 2011 |
| NTDs ‐ recurrence |
47 randomised 22 non‐randomised |
1 g daily to 12 weeks pregnancy (+ folic acid) | No NTDs among pregnancies in inositol groups (14 randomised and 21 non‐randomised). 3 NTDs in non‐supplemented groups | Greene, 2016 |
Examples of conditions in which myo‐inositol has been tested for potential beneficial effects.
An important additional consideration in any potential novel periconceptional preventive therapy for NTDs is the requirement for safety in the fetus and pregnant mother. The inositol concentration is higher in reproductive organs than in blood, and concentration in the ovaries is under hormonal control (Lewin et al., 1982). Several clinical trials in women with polycystic ovary syndrome have suggested a beneficial effect of inositol (myo‐ or chiro‐) supplements in improving ovarian function, hormone status and menstrual activity, with no reported side effects at typical doses of 2 to 4 g daily (Table 2; Nestler et al., 1999; Gerli et al., 2007; Genazzani et al., 2008; Costantino et al., 2009; reviewed by Dinicola et al., 2014).
Furthermore, similar doses of inositol have been trialed in randomized studies during pregnancy for treatment and/or prevention of gestational diabetes (Corrado et al., 2011; D'Anna et al., 2013; Matarrelli et al., 2013), as well as in postmenopausal women with metabolic syndrome (Giordano et al., 2011). These studies were based on the hypothesis that inositol would be beneficial, as myo‐ and D‐chiro‐inositol and their methylated derivatives (sequoyital and D‐pinitol, respectively) were reported to have insulin‐sensitizing properties in experimental models of hyperglycemia and diabetes (Croze and Soulage, 2013). In women considered at risk owing to a family history of type 2 diabetes (D'Anna et al., 2013) or elevated fasting glucose (Matarrelli et al., 2013), there was a lower incidence of gestational diabetes in the myo‐inositol group compared with placebo. Supplementation (2 g inositol, twice daily) throughout pregnancy from the first trimester onward was not associated with side effects or increased risk of adverse pregnancy outcomes (reviewed by Croze and Soulage, 2013; Noventa et al., 2016).
Prevention of NTDs by Inositol?
Taking into account the prevalence of NTDs, the sample size required for investigation of a potential novel therapy in a “first occurrence” trial is likely to be prohibitively large. As a result, inositol supplementation has so far been studied solely in women with one or more previous NTD‐affected pregnancies (Table 2). Such women are known to be at considerably higher risk of a subsequent NTD‐affected pregnancy than women without a history of NTDs: the recurrence risk after a single affected pregnancy is typically quoted as 3.1%, rising to 11.8% after two affected pregnancies (Seller, 1981). A cohort of 12 women in Italy took myo‐inositol and folic acid pre‐ and postconceptionally after at least one previous affected pregnancy. Folic acid use was reported in the majority of their previous pregnancies which suggests that most of these NTDs were “folic acid‐resistant”. Fifteen subsequent pregnancies were followed up and 17 babies were born without NTDs (Cavalli and Copp, 2002; Cavalli et al., 2011). These findings encourage the view that there may be a contribution of inositol toward prevention of NTD recurrence. However, the number of pregnancies was too small to draw firm conclusions and the nonrandomized study design was a potential source of bias.
Where ethically and logistically possible, a randomized controlled trial is generally considered the optimal approach to determine the efficacy of a novel therapy. Given the known protective effect of folic acid, it would be unethical to conduct a trial without providing it to all participants. Hence, a potential trial design would test whether inositol + folic acid is more effective than placebo + folic acid. The MRC Vitamin Study recruited around 1800 women who had a previous NTD‐affected pregnancy and the outcome of 1200 subsequent pregnancies was ascertained (approximately 600 in the combined folic acid supplemented groups and 600 in combined nonfolic acid groups). The recurrence rate in the folic acid groups was 1% compared with 3.5% in the other groups. Detection of a further significant reduction in recurrence rate, due to inositol supplementation, should require a similar or larger sample size.
Another consideration is that any trial for prevention of NTDs now takes place in the context of use of folic acid supplement use and/or food fortification on a population level, including among women without a previous history of NTDs. Hence, it could be hypothesized that those NTDs which occur now may be more enriched for folic acid–non‐responsive NTDs than at the time of the folic acid trials in the 1980s, when use of folic acid supplements was not widespread. Similar lack of response in a subsequent pregnancy would thus lead to a higher rate of recurrence than in the MRC Vitamin Study. However, in the United Kingdom, it is estimated that only around 30 to 35% of women take periconceptional folic acid (Bestwick et al., 2014) and food fortification is not in place. Therefore, the likely recurrence rate in a folic acid supplemented study group is difficult to predict.
A pilot double‐blind randomized clinical trial of inositol was performed in the United Kingdom, with recruitment during 2009 to 2013 to make further progress toward determining whether use of inositol supplements with folic acid would have a greater protective effect than folic acid alone. The PONTI (Prevention Of Neural Tube defects by Inositol) study recruited women with one or more previous affected NTD pregnancies, who were planning a further pregnancy (Greene et al., 2016). The study design involved randomization of equal numbers of pregnancies to inositol (1 g daily) + folic acid (5 mg daily) or placebo + folic acid groups and, among 99 eligible women who contacted the study center, 47 agreed to be randomized. The majority of randomized women (46 of 47) had a history of one previous affected pregnancy (31 spina bifida and 15 anencephaly; one woman had two previous pregnancies with anencephaly). Among 14 established pregnancies in the inositol + folic acid group, all led to the birth of unaffected babies. Among 19 established pregnancies in the placebo + folic acid group, 18 led to normal outcomes, while one fetus was diagnosed with anencephaly on ultrasound (Greene et al., 2016).
One aim of the study was to evaluate the feasibility of recruiting women at “high risk” of an NTD pregnancy into a randomized study. In fact, many women (around half of those who contacted the study team) were unwilling to enter the randomized study; almost all cited the possibility of inclusion in the placebo group as their reason for declining randomization. This was despite provision of folic acid to all participants and the unproven nature of inositol's preventive effect. A cohort of 22 women who met the eligibility criteria for the PONTI study but decided against randomization, agreed to be followed up in subsequent pregnancies. Among this nonrandomized group 19 women, 6 of whom had experienced two previous affected pregnancies, used peri‐conceptional supplementation with folic acid (at least 5 mg daily) and inositol (typically 1 g daily). They reported 21 pregnancies without NTDs. A further three nonrandomized women, who subsequently took only 5 mg folic acid reported one normal pregnancy outcome and two pregnancies that were terminated following a diagnosis of anencephaly.
Conclusions and Future Prospects
Inositol, inositol phosphoinsitides, and inositol polyphosphates are a group of molecules that fulfil a diverse range of cellular functions. Supply and metabolism of inositol is required for cranial neural tube closure, while supplemental inositol can prevent spinal and cranial NTDs in various experimental models. Notably, these include the curly tail strain, whose multigenic causation perhaps models the etiology of human NTDs more representatively than single gene knockouts. In addition, inositol has been implicated in ameliorating NTDs in diabetes‐induced and folate‐deficiency models, each of which are implicated in human NTDs. Exogenous inositol could potentially act to overcome the underlying causative defect responsible for NTDs and/or enhance the normal processes required for progression of neural tube closure. Indeed, in the curly tail model, inositol has been found to counteract the genetically determined imbalance of cell proliferation in the region of caudal neurulation. Given the multiple functions of inositol in differing cellular signaling pathways and processes it is also plausible that there are differing metabolic and cellular mechanisms underlying the effects of inositol in the cranial and spinal regions and in the various models where it has been shown to act. Further studies will address these questions.
Clinical use of inositol in adults and during pregnancy does not indicate safety concerns at the doses tested to date and beneficial effects on development of gestational diabetes have been reported. In the context of NTD prevention, the findings of recent non‐randomized and randomized studies are encouraging but these studies are currently of insufficient size to demonstrate a significant protective effect of inositol. Moreover, studies to date have been recurrence trials so the effect of inositol on first occurrence of NTDs has not yet been formally investigated. We propose that a fully powered randomized controlled study is justified to test whether inositol + folic acid has a greater protective effect against NTDs than folic acid alone. Such a study will likely require a multi‐site, multi‐country design. Nevertheless, if inositol does prove to be protective it is a simple, inexpensive compound that could potentially be used on a population‐wide level, to further enhance primary prevention of NTDs.
Supported by grants from the Medical Research Council (G0401315, G0601546, G0802163), Sparks (09ICH01), and Wellcome Trust (087525). The authors are supported by the National Institute for Health Research (NIHR) Biomedical Research Centre at Great Ormond Street Hospital and University College London.
References
- Abdul‐Aziz NM, Turmaine M, Greene ND, Copp AJ. 2009. EphrinA‐EphA receptor interactions in mouse spinal neurulation: implications for neural fold fusion. Int J Dev Biol 53:559–568. [DOI] [PubMed] [Google Scholar]
- Allan SJ, Kavanagh GM, Herd RM, Savin JA. 2004. The effect of inositol supplements on the psoriasis of patients taking lithium: a randomized, placebo‐controlled trial. Br J Dermatol 150:966–969. [DOI] [PubMed] [Google Scholar]
- Araki N, Egami Y, Watanabe Y, Hatae T. 2007. Phosphoinositide metabolism during membrane ruffling and macropinosome formation in EGF‐stimulated A431 cells. Exp Cell Res 313:1496–1507. [DOI] [PubMed] [Google Scholar]
- Au KS, Ashley‐Koch A, Northrup H. 2010. Epidemiologic and genetic aspects of spina bifida and other neural tube defects. Dev Disabil Res Rev 16:6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker L, Piddington R, Goldman A, et al. 1990. Myo‐inositol and prostaglandins reverse the glucose inhibition of neural tube fusion in cultured mouse embryos. Diabetologia 33:593–596. [DOI] [PubMed] [Google Scholar]
- Benjamin J, Levine J, Fux M, et al. 1995. Double‐blind, placebo‐controlled, crossover trial of inositol treatment for panic disorder. Am J Psychiatry 152:1084–1086. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. 1993. Inositol trisphosphate and calcium signalling. Nature 361:315–325. [DOI] [PubMed] [Google Scholar]
- Bestwick JP, Huttly WJ, Morris JK, Wald NJ. 2014. Prevention of neural tube defects: a cross‐sectional study of the uptake of folic acid supplementation in nearly half a million women. PLoS One 9:e89354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas SL, Silhavy JL, Brancati F, et al. 2009. Mutations in INPP5E, encoding inositol polyphosphate‐5‐phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 41:1032–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencowe H, Cousens S, Modell B, Lawn J. 2010. Folic acid to reduce neonatal mortality from neural tube disorders. Int J Epidemiol 39(Suppl 1):i110–i121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom HJ, Shaw GM, Den Heijer M, Finnell RH. 2006. Neural tube defects and folate: case far from closed. Nat Rev Neurosci 7:724–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges D, Saltiel AR. 2015. Phosphoinositides: key modulators of energy metabolism. Biochim Biophys Acta 1851:857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook FA, Shum ASW, Van Straaten HWM, Copp AJ. 1991. Curvature of the caudal region is responsible for failure of neural tube closure in the curly tail (ct) mouse embryo. Development 113:671–678. [DOI] [PubMed] [Google Scholar]
- Bupp CP, Sarasua SM, Dean JH, Stevenson RE. 2015. When folic acid fails: insights from 20 years of neural tube defect surveillance in South Carolina. Am J Med Genet A 167A:2244–2250. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. 1995. Protein kinase B (c‐Akt) in phosphatidylinositol‐3‐OH kinase signal transduction. Nature 376:599–602. [DOI] [PubMed] [Google Scholar]
- Burren KA, Savery D, Massa V, et al. 2008. Gene‐environment interactions in the causation of neural tube defects: folate deficiency increases susceptibility conferred by loss of Pax3 function. Hum Mol Genet 17:3675–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burren KA, Scott JM, Copp AJ, Greene ND. 2010. The genetic background of the curly tail strain confers susceptibility to folate‐deficiency‐induced exencephaly. Birth Defects Res A Clin Mol Teratol 88:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauvin C, Echard A. 2015. Phosphoinositides: lipids with informative heads and mastermind functions in cell division. Biochim Biophys Acta 1851:832–843. [DOI] [PubMed] [Google Scholar]
- Cavalli P, Copp AJ. 2002. Inositol and folate‐resistant neural tube defects. J Med Genet 39:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli P, Tonni G, Grosso E, Poggiani C. 2011. Effects of inositol supplementation in a cohort of mothers at risk of producing an NTD pregnancy. Birth Defects Res A Clin Mol Teratol 91:962–965. [DOI] [PubMed] [Google Scholar]
- Chavez M, Ena S, Van Sande J, et al. 2015. Modulation of ciliary phosphoinositide content regulates trafficking and sonic hedgehog signaling output. Dev Cell 34:338–350. [DOI] [PubMed] [Google Scholar]
- Chengappa KN, Levine J, Gershon S, et al. 2000. Inositol as an add‐on treatment for bipolar depression. Bipolar Disord 2:47–55. [DOI] [PubMed] [Google Scholar]
- Chi H, Sarkisian MR, Rakic P, Flavell RA. 2005. Loss of mitogen‐activated protein kinase kinase kinase 4 (MEKK4) results in enhanced apoptosis and defective neural tube development. Proc Natl Acad Sci U S A 102:3846–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockroft DL. 1979. Nutrient requirements of rat embryos undergoing organogenesis in vitro. J Reprod Fertil 57:505–510. [DOI] [PubMed] [Google Scholar]
- Cockroft DL. 1984. Abnormalities induced in cultured rat embryos by hyperglycaemia. Br J Exp Pathol 65:625–636. [PMC free article] [PubMed] [Google Scholar]
- Cockroft DL. 1988. Changes with gestational age in the nutritional requirements of postimplantation rat embryos in culture. Teratology 38:281–290. [DOI] [PubMed] [Google Scholar]
- Cockroft DL, Brook FA, Copp AJ. 1992. Inositol deficiency increases the susceptibility to neural tube defects of genetically predisposed (curly tail) mouse embryos in vitro. Teratology 45:223–232. [DOI] [PubMed] [Google Scholar]
- Cogram P, Hynes A, Dunlevy LPE, et al. 2004. Specific isoforms of protein kinase C are essential for prevention of folate‐resistant neural tube defects by inositol. Hum Mol Genet 13:7–14. [DOI] [PubMed] [Google Scholar]
- Cogram P, Tesh S, Tesh J, et al. 2002. D‐chiro‐inositol is more effective than myo‐inositol in preventing folate‐resistant mouse neural tube defects. Hum Reprod 17:2451–2458. [DOI] [PubMed] [Google Scholar]
- Copp AJ, Crolla JA, Brook FA. 1988. Prevention of spinal neural tube defects in the mouse embryo by growth retardation during neurulation. Development 104:297–303. [DOI] [PubMed] [Google Scholar]
- Copp AJ, Greene NDE. 2010. Genetics and development of neural tube defects. J Pathol 220:217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Stanier P, Greene ND. 2013. Neural tube defects: recent advances, unsolved questions, and controversies. Lancet Neurol 12:799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado F, D'Anna R, Di Vieste G, et al. 2011. The effect of myoinositol supplementation on insulin resistance in patients with gestational diabetes. Diabet Med 28:972–975. [DOI] [PubMed] [Google Scholar]
- Costantino D, Minozzi G, Minozzi E, Guaraldi C. 2009. Metabolic and hormonal effects of myo‐inositol in women with polycystic ovary syndrome: a double‐blind trial. Eur Rev Med Pharmacol Sci 13:105–110. [PubMed] [Google Scholar]
- Crider KS, Bailey LB, Berry RJ. 2011. Folic acid food fortification‐its history, effect, concerns, and future directions. Nutrients 3:370–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croise P, Estay‐Ahumada C, Gasman S, Ory S. 2014. Rho GTPases, phosphoinositides, and actin: a tripartite framework for efficient vesicular trafficking. Small GTPases 5:e29469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croze ML, Soulage CO. 2013. Potential role and therapeutic interests of myo‐inositol in metabolic diseases. Biochimie 95:1811–1827. [DOI] [PubMed] [Google Scholar]
- Czeizel AE, Dudás I. 1992. Prevention of the first occurrence of neural‐tube defects by periconceptional vitamin supplementation. N Engl J Med 327:1832–1835. [DOI] [PubMed] [Google Scholar]
- D'Anna R, Scilipoti A, Giordano D, et al. 2013. myo‐Inositol supplementation and onset of gestational diabetes mellitus in pregnant women with a family history of type 2 diabetes: a prospective, randomized, placebo‐controlled study. Diabetes Care 36:854–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro SC, Malhas A, Leung KY, et al. 2012. Lamin b1 polymorphism influences morphology of the nuclear envelope, cell cycle progression, and risk of neural tube defects in mice. PLoS Genet 8:e1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pietro G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657. [DOI] [PubMed] [Google Scholar]
- Dinicola S, Chiu TT, Unfer V, et al. 2014. The rationale of the myo‐inositol and D‐chiro‐inositol combined treatment for polycystic ovary syndrome. J Clin Pharmacol 54:1079–1092. [DOI] [PubMed] [Google Scholar]
- Englund PT. 1993. The structure and biosynthesis of glycosyl phosphatidylinositol protein anchors. Annu Rev Biochem 62:121–138. [DOI] [PubMed] [Google Scholar]
- Escuin S, Vernay B, Savery D, et al. 2015. Rho‐kinase‐dependent actin turnover and actomyosin disassembly are necessary for mouse spinal neural tube closure. J Cell Sci 128:2468–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field SJ, Madson N, Kerr ML, et al. 2005. PtdIns(4,5)P2 functions at the cleavage furrow during cytokinesis. Curr Biol 15:1407–1412. [DOI] [PubMed] [Google Scholar]
- Fleming A, Copp AJ. 1998. Embryonic folate metabolism and mouse neural tube defects. Science 280:2107–2109. [DOI] [PubMed] [Google Scholar]
- Franke B, Klootwijk R, Lemmers B, et al. 2003. Phenotype of the neural tube defect mouse model bent tail is not sensitive to maternal folinic acid, myo‐inositol, or zinc supplementation. Birth Defects Res Part A Clin Mol Teratol 67:979–984. [DOI] [PubMed] [Google Scholar]
- Frederick JP, Mattiske D, Wofford JA, et al. 2005. An essential role for an inositol polyphosphate multikinase, Ipk2, in mouse embryogenesis and second messenger production. Proc Natl Acad Sci U S A 102:8454–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelber D, Levine J, Belmaker RH. 2001. Effect of inositol on bulimia nervosa and binge eating. Int J Eat Disord 29:345–348. [DOI] [PubMed] [Google Scholar]
- Gelineau‐van Waes J, Voss KA, Stevens VL, et al. 2009. Maternal fumonisin exposure as a risk factor for neural tube defects. Adv Food Nutr Res 56:145–181. [DOI] [PubMed] [Google Scholar]
- Genazzani AD, Lanzoni C, Ricchieri F, Jasonni VM. 2008. Myo‐inositol administration positively affects hyperinsulinemia and hormonal parameters in overweight patients with polycystic ovary syndrome. Gynecol Endocrinol 24:139–144. [DOI] [PubMed] [Google Scholar]
- Gerli S, Papaleo E, Ferrari A, Di Renzo GC. 2007. Randomized, double blind placebo‐controlled trial: effects of myo‐inositol on ovarian function and metabolic factors in women with PCOS. Eur Rev Med Pharmacol Sci 11:347–354. [PubMed] [Google Scholar]
- Giordano D, Corrado F, Santamaria A, et al. 2011. Effects of myo‐inositol supplementation in postmenopausal women with metabolic syndrome: a perspective, randomized, placebo‐controlled study. Menopause 18:102–104. [DOI] [PubMed] [Google Scholar]
- Goldman AS, Baker L, Piddington R, et al. 1985. Hyperglycemia‐induced teratogenesis is mediated by a functional deficiency of arachidonic acid. Proc Natl Acad Sci U S A 82:8227–8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene ND, Copp AJ. 2014. Neural tube defects. Annu Rev Neurosci 37:221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene ND, Leung KY, Gay V, et al. 2016. Inositol for the prevention of neural tube defects: a pilot randomised controlled trial. Br J Nutr 115:974–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene NDE, Copp AJ. 1997. Inositol prevents folate‐resistant neural tube defects in the mouse. Nat Med 3:60–66. [DOI] [PubMed] [Google Scholar]
- Groenen PM, Klootwijk R, Schijvenaars MM, et al. 2004. Spina bifida and genetic factors related to myo‐inositol, glucose, and zinc. Mol Genet Metab 82:154–161. [DOI] [PubMed] [Google Scholar]
- Groenen PM, Peer PG, Wevers RA, et al. 2003. Maternal myo‐inositol, glucose, and zinc status is associated with the risk of offspring with spina bifida. Am J Obstet Gynecol 189:1713–1719. [DOI] [PubMed] [Google Scholar]
- Groenen PM, Roes EM, Peer PG, et al. 2006. Myo‐inositol, glucose and zinc concentrations determined in the preconceptional period, during and after pregnancy. Eur J Obstet Gynecol Reprod Biol 127:50–55. [DOI] [PubMed] [Google Scholar]
- Guan Z, Wang J, Guo J, et al. 2014. The maternal ITPK1 gene polymorphism is associated with neural tube defects in a high‐risk Chinese population. PLoS One 9:e86145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson P, Greene ND, Lad D, et al. 2007. Increased expression of Grainyhead‐like‐3 rescues spina bifida in a folate‐resistant mouse model. Hum Mol Genet 16:2640–2646. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Akazawa S, Akazawa M, et al. 1990. Effects of hyperglycaemia on sorbitol and myo‐inositol contents of cultured embryos: treatment with aldose reductase inhibitor and myo‐inositol supplementation. Diabetologia 33:597–602. [DOI] [PubMed] [Google Scholar]
- Hawkins PT, Stephens LR. 2015. PI3K signalling in inflammation. Biochim Biophys Acta 1851:882–897. [DOI] [PubMed] [Google Scholar]
- Heid MK, Bills ND, Hinrichs SH, Clifford AJ. 1992. Folate deficiency alone does not produce neural tube defects in mice. J Nutr 122:888–894. [DOI] [PubMed] [Google Scholar]
- Hod M, Star S, Passonneau J, et al. 1990. Glucose‐induced dysmorphogenesis in the cultured rat conceptus: prevention by supplementation with myo‐inositol. Isr J Med Sci 26:541–544. [PubMed] [Google Scholar]
- Hod M, Star S, Passonneau JV, et al. 1986. Effect of hyperglycemia on sorbitol and myo‐inositol content of cultured rat conceptus: failure of aldose reductase inhibitors to modify myo‐inositol depletion and dysmorphogenesis. Biochem Biophys Res Commun 140:974–980. [DOI] [PubMed] [Google Scholar]
- Holmberg J, Clarke DL, Frisén J. 2000. Regulation of repulsion versus adhesion by different splice forms of an Eph receptor. Nature 408:203–206. [DOI] [PubMed] [Google Scholar]
- Holub BJ. 1987. The cellular forms and functions of the inositol phospholipids and their metabolic derivatives. Nutr Rev 45:65–71. [DOI] [PubMed] [Google Scholar]
- Jacoby M, Cox JJ, Gayral S, et al. 2009. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet 41:1027–1031. [DOI] [PubMed] [Google Scholar]
- Janmey PA, Lindberg U. 2004. Cytoskeletal regulation: rich in lipids. Nat Rev Mol Cell Biol 5:658–666. [DOI] [PubMed] [Google Scholar]
- Khandelwal M, Reece EA, Wu YK, Borenstein M. 1998. Dietary myo‐inositol therapy in hyperglycemia‐induced embryopathy. Teratology 57:79–84. [DOI] [PubMed] [Google Scholar]
- Kim E, Beon J, Lee S, et al. 2015. IPMK: a versatil regulator of nuclear signalling events. Adv Biol Regul doi:10.1016/j.bior.2015.11.005. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Kirke PN, Molloy AM, Daly LE, et al. 1993. Maternal plasma folate and vitamin B12 are independent risk factors for neural tube defects. Q J Med 86:703–708. [PubMed] [Google Scholar]
- Klootwijk R, Schijvenaars MMVA, Mariman ECM, Franke B. 2004. Further characterization of the genetic defect of the Bent tail mouse, a mouse model for human neural tube defects. Birth Defects Res Part A Clin Mol Teratol 70:880–884. [DOI] [PubMed] [Google Scholar]
- Larue L, Bellacosa A. 2005. Epithelial‐mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 24:7443–7454. [DOI] [PubMed] [Google Scholar]
- Leck I. 1974. Causation of neural tube defects: clues from epidemiology. Br Med Bull 30:158–163. [DOI] [PubMed] [Google Scholar]
- Leung KY, De Castro SC, Savery D, et al. 2013. Nucleotide precursors prevent folic acid‐resistant neural tube defects in the mouse. Brain 136:2836–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KY, Mills K, Burren KA, et al. 2011. Quantitative analysis of myo‐inositol in urine, blood and nutritional supplements by high‐performance liquid chromatography tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 879:2759–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine J, Barak Y, Gonzalves M, et al. 1995. Double‐blind, controlled trial of inositol treatment of depression. Am J Psychiatry 152:792–794. [DOI] [PubMed] [Google Scholar]
- Lewin LM, Yannai Y, Melmed S, Weiss M. 1982. myo‐inositol in the reproductive tract of the female rat. Int J Biochem 14:147–150. [DOI] [PubMed] [Google Scholar]
- Lin X, Ma L, Gopalan C, Ostlund RE. 2009. d‐ chiro‐Inositol is absorbed but not synthesised in rodents. Br J Nutr 102:1426–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore TM, Azevedo C, Kolozsvari B, et al. 2016. Phosphate, inositol and polyphosphates. Biochem Soc Trans 44:253–259. [DOI] [PubMed] [Google Scholar]
- Massa V, Wlodarczyk B, Giavini E, Finnell RH. 2006. Myo‐inositol enhances teratogenicity of valproic acid in the mouse. Birth Defects Res A Clin Mol Teratol 76:200–204. [DOI] [PubMed] [Google Scholar]
- Matarrelli B, Vitacolonna E, D'Angelo M, et al. 2013. Effect of dietary myo‐inositol supplementation in pregnancy on the incidence of maternal gestational diabetes mellitus and fetal outcomes: a randomized controlled trial. J Matern Fetal Neonatal Med 26:967–972. [DOI] [PubMed] [Google Scholar]
- McLaughlin S, Wang J, Gambhir A, Murray D. 2002. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct 31:151–175. [DOI] [PubMed] [Google Scholar]
- Michell RH. 2008. Inositol derivatives: evolution and functions. Nat Rev Mol Cell Biol 9:151–161. [DOI] [PubMed] [Google Scholar]
- Mosley BS, Cleves MA, Siega‐Riz AM, et al. 2009. Neural tube defects and maternal folate intake among pregnancies conceived after folic acid fortification in the United States. Am J Epidemiol 169:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MRC Vitamin Study Research Group . 1991. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet 338:131–137. [PubMed] [Google Scholar]
- Murdoch JN, Copp AJ. 2010. The relationship between Hedgehog signalling, cilia and neural tube defects. Birth Defects Res A Clin Mol Teratol 88:633–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento NR, Lessa LM, Kerntopf MR, et al. 2006. Inositols prevent and reverse endothelial dysfunction in diabetic rat and rabbit vasculature metabolically and by scavenging superoxide. Proc Natl Acad Sci U S A 103:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi O, Mun'im A, Negishi Y. 2015. Content of methylated inositols in familiar edible plants. J Agric Food Chem 63:2683–2688. [DOI] [PubMed] [Google Scholar]
- Nestler JE, Jakubowicz DJ, Reamer P, et al. 1999. Ovulatory and metabolic effects of D‐chiro‐inositol in the polycystic ovary syndrome. N Engl J Med 340:1314–1320. [DOI] [PubMed] [Google Scholar]
- Niederkofler V, Salie R, Sigrist M, Arber S. 2004. Repulsive guidance molecule (RGM) gene function is required for neural tube closure but not retinal topography in the mouse visual system. J Neurosci 24:808–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y. 1992. Intracellular signalling by hydrolysis of phospholipids and activation of protein kinase C. Science 158:607–613. [DOI] [PubMed] [Google Scholar]
- Noventa M, Vitagliano A, Quaranta M, et al. 2016. Preventive and therapeutic role of dietary inositol supplementation in periconceptional period and during pregnancy: a summary of evidences and future applications. Reprod Sci 23:278–288. [DOI] [PubMed] [Google Scholar]
- O'Shea KS, Kaufman MH. 1980. Phospholipase C induced neural tube defects in the mouse embryo. Experientia 36:1217–1219. [DOI] [PubMed] [Google Scholar]
- Pak Y, Huang LC, Lilley KJ, Larner J. 1992. In vivo conversion of [3H]myoinositol to [3H]chiroinositol in rat tissues. J Biol Chem 267:16904–16910. [PubMed] [Google Scholar]
- Paulick MG, Bertozzi CR. 2008. The glycosylphosphatidylinositol anchor: a complex membrane‐anchoring structure for proteins. Biochemistry 47:6991–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedrahita JA, Oetama B, Bennett GD, et al. 1999. Mice lacking the folic acid‐binding protein Folbp1 are defective in early embryonic development. Nat Genet 23:228–232. [DOI] [PubMed] [Google Scholar]
- Pinter E, Reece EA, Leranth CZ, et al. 1986. Arachidonic acid prevents hyperglycemia‐associated yolk sac damage and embryopathy. Am J Obstet Gynecol 155:691–702. [DOI] [PubMed] [Google Scholar]
- Plotnikova OV, Seo S, Cottle DL, et al. 2015. INPP5E interacts with AURKA, linking phosphoinositide signaling to primary cilium stability. J Cell Sci 128:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posor Y, Eichhorn‐Grunig M, Haucke V. 2015. Phosphoinositides in endocytosis. Biochim Biophys Acta 1851:794–804. [DOI] [PubMed] [Google Scholar]
- Rolo A, Savery D, Armer HEJ, et al. 2016. Regulation of cell protrusions by small GTPases during fusion of the neural folds. eLife 5:pii: e13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal J, Casas J, Taren D, et al. 2014. Neural tube defects in Latin America and the impact of fortification: a literature review. Public Health Nutr 17:537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarikangas J, Zhao H, Lappalainen P. 2010. Regulation of the actin cytoskeleton‐plasma membrane interplay by phosphoinositides. Physiol Rev 90:259–289. [DOI] [PubMed] [Google Scholar]
- Sadler TW. 1980. Effects of maternal diabetes on early embryogenesis: II. Hyperglycemia‐induced exencephaly. Teratology 21:349–356. [DOI] [PubMed] [Google Scholar]
- Schlemmer U, Frolich W, Prieto RM, Grases F. 2009. Phytate in foods and significance for humans: food sources, intake, processing, bioavailability, protective role and analysis. Mol Nutr Food Res 53(Suppl 2):S330–S375. [DOI] [PubMed] [Google Scholar]
- Seller MJ. 1981. Recurrence risks for neural tube defects in a genetic counselling clinic population. J Med Genet 18:245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw GM, Carmichael SL, Yang W, Schaffer DM. 2005. Periconceptional dietary intake of myo‐inositol and neural tube defects in offspring. Birth Defects Res A Clin Mol Teratol 73:184–187. [DOI] [PubMed] [Google Scholar]
- Smithells RW, Sheppard S, Schorah CJ. 1976. Vitamin deficiencies and neural tube defects. Arch Dis Child 51:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithells RW, Sheppard S, Schorah CJ, et al. 1980. Possible prevention of neural‐tube defects by periconceptional vitamin supplementation. Lancet 1:339–340. [DOI] [PubMed] [Google Scholar]
- Soto X, Li J, Lea R, et al. 2013. Inositol kinase and its product accelerate wound healing by modulating calcium levels, Rho GTPases, and F‐actin assembly. Proc Natl Acad Sci U S A 110:11029–11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpo DJ, Bock CB, Tuttle JS, Blackshear PJ. 1995. MARCKS deficiency in mice leads to abnormal brain development and perinatal death. Proc Natl Acad Sci U S A 92:944–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudiwala S, De Castro SC, Leung KY, et al. 2016. Formate supplementation enhances folate‐dependent nucleotide biosynthesis and prevents spina bifida in a mouse model of folic acid‐resistant neural tube defects. Biochimie doi:10.1016/j.biochi.2016.02.010. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun TH, Heimark DB, Nguygen T, et al. 2002. Both myo‐inositol to chiro‐inositol epimerase activities and chiro‐inositol to myo‐inositol ratios are decreased in tissues of GK type 2 diabetic rats compared to Wistar controls. Biochem Biophys Res Commun 293:1092–1098. [DOI] [PubMed] [Google Scholar]
- Sussman I, Matschinsky FM. 1988. Diabetes affects sorbitol and myo‐inositol levels of neuroectodermal tissue during embryogenesis in rat. Diabetes 37:974–981. [DOI] [PubMed] [Google Scholar]
- Ting SB, Wilanowski T, Auden A, et al. 2003. Inositol‐ and folate‐resistant neural tube defects in mice lacking the epithelial‐specific factor Grhl‐3. Nat Med 9:1513–1519. [DOI] [PubMed] [Google Scholar]
- Tsujita K, Itoh T. 2015. Phosphoinositides in the regulation of actin cortex and cell migration. Biochim Biophys Acta 1851:824–831. [DOI] [PubMed] [Google Scholar]
- Van Straaten HWM, Copp AJ. 2001. Curly tail: a 50‐year history of the mouse spina bifida model. Anat Embryol (Berl) 203:225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lian L, Golden JA, et al. 2007. PIP5KI gamma is required for cardiovascular and neuronal development. Proc Natl Acad Sci U S A 104:11748–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MP, Hugge C, Bielinska M, et al. 2009. Neural tube defects in mice with reduced levels of inositol 1,3,4‐trisphosphate 5/6‐kinase. Proc Natl Acad Sci U S A 106:9831–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MS, Bulley SJ, Pisani F, et al. 2015. A novel method for the purification of inositol phosphates from biological samples reveals that no phytate is present in human plasma or urine. Open Biol 5:150014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk BJ, Palacios AM, George TM, Finnell RH. 2012. Antiepileptic drugs and pregnancy outcomes. Am J Med Genet A 158A:2071–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk BJ, Tang LS, Triplett A, et al. 2006. Spontaneous neural tube defects in splotch mice supplemented with selected micronutrients. Toxicol Appl Pharmacol 213:55–63. [DOI] [PubMed] [Google Scholar]
- Xu Q, Zhang Y, Wei Q, et al. 2016. Phosphatidylinositol phosphate kinase PIPKIgamma and phosphatase INPP5E coordinate initiation of ciliogenesis. Nat Commun 7:10777. [DOI] [PMC free article] [PubMed] [Google Scholar]