

Abstract
Although inappropriate activation of the Wnt/β-catenin pathway has been implicated in the development of hepatocellular carcinoma (HCC), the role of this signaling in liver carcinogenesis remains unclear. To investigate this issue, we constructed a mutant mouse strain, Apclox/lox, in which exon 14 of the tumor-suppressor gene adenomatous polyposis coli (Apc) is flanked by loxP sequences. i.v. injection of adenovirus encoding Cre recombinase (AdCre) at high multiplicity [109 plaque-forming units (pfu) per mouse] inactivated the Apc gene in the liver and resulted in marked hepatomegaly, hepatocyte hyperplasia, and rapid mortality. β-Catenin signaling activation was demonstrated by nuclear and cytoplasmic accumulation of β-catenin in the hepatocytes and by the induction of β-catenin target genes (glutamine synthetase, glutamate transporter 1, ornithine aminotransferase, and leukocyte cell-derived chemotaxin 2) in the liver. To test a long-term oncogenic effect, we inoculated mice with lower doses of AdCre (0.5 × 109 pfu per mouse), compatible with both survival and persistence of β-catenin-activated cells. In these conditions, 67% of mice developed HCC. β-Catenin signaling was strongly activated in these Apc-inactivated HCCs. The HCCs were well, moderately, or poorly differentiated. Indeed, their histological and molecular features mimicked human HCC. Thus, deletion of Apc in the liver provides a valuable model of human HCC, and, in this model, activation of the Wnt/β-catenin pathway by invalidation of Apc is required for liver tumorigenesis.
Keywords: adenomatous polyposis coli, hepatocarcinogenesis, mouse model
Hepatocellular carcinoma (HCC) is the most frequent primary malignancy of the liver and accounts for >5% of all cancers worldwide. The worldwide incidence is estimated to be between 250,000 and 1.2 million new cases each year, and the disease causes 500,000 to 1 million deaths annually (1). It occurs in a previously diseased liver, and the causes of the underlying liver disease differ according to the geographical zone (aflatoxin B1 and hepatitis B virus in Africa and southern Asia and hepatitis C virus infection and alcohol in Japan, Egypt, North America, and Europe).
Although the genetic events responsible for either HCC initiation or HCC progression are not clear, they involve at least three carcinogenesis pathways: the p53, RB, and Wnt/β-catenin signaling pathways (2). In this last pathway, β-catenin plays a pivotal role. In nonproliferative epithelial cells, β-catenin is continuously phosphorylated by GSK3β in a complex with adenomatous polyposis coli (Apc)-axin/conductin and is quickly degraded through the ubiquitin/proteasome pathway. Aberrant activation of β-catenin signaling is observed in 30–40% of human HCC, mimicking Wnt stimulation, because of an abnormal stabilization of β-catenin. Consequently, β-catenin translocates into the nucleus to transcribe target genes in cooperation with lymphoid enhancer factor/T cell factor transcription factors (3). This phenomenon is related to mutations in β-catenin genes in 12–26% of human HCC and to mutations in AXIN1 or AXIN2 in 8–13% of human HCC (3, 4). Interestingly, no somatic mutation of the APC gene and no loss of heterozygosity of the 5q21 APC locus have been described in HCC, even though this event is implicated in >80% of colorectal cancers (5, 6). However, there are data implicating a loss of function of APC in liver carcinogenesis: infrequent hepatoblastomas have been described (7, 8) and two cases of HCC have been reported (9, 10) in familial adenomatous polyposis patients carrying a germ-line heterozygous mutation of the APC gene.
Improving our understanding of β-catenin-activated hepatocarcinogenesis is important. Indeed, β-catenin signaling defines a particular pathway of hepatocarcinogenesis that develops in a chromosomally stable environment, whereas tumors without β-catenin mutations develop in a context of multiple chromosomal alterations (11, 12). We previously created a transgenic model expressing a stabilized form of β-catenin (13, 14). Marked hepatomegaly was apparent shortly after the hepatic expression of the mutant. However, the premature death of the mice prevented analysis of hepatocarcinogenesis. Harada et al. (15), using an adenovirus-mediated delivery of a stabilized mutant of β-catenin, found that a controlled induction of this mutant in the liver, allowing survival of the animals, did not lead to liver tumorigenesis.
We investigated whether (i) Apc is functional in the liver and (ii) activating β-catenin signaling is an oncogenic event in the liver. Therefore, we established a mouse strain containing a double-mutant Apc allele, the exon 14 of which was flanked with loxP sequences (the Apclox/lox mouse). When Cre recombinase is expressed, the β-catenin degradation sites and the axin-binding sites of Apc protein are deleted. This strategy was used successfully to obtain the germinal heterozygous invalidation of Apc and efficiently inactivate Apc: These Apc+/Δex14 mice developed numerous Apc–/– intestinal polyps in a β-catenin-signaling-mediated manner (16). Using an adenovirus-mediated system to deliver Cre recombinase to the liver of Apclox/lox mice, we report here the oncogenic effect of a liver-restricted biallelic invalidation of Apc.
Materials and Methods
Obtaining Apc+/lox Mice. Embryonic stem cells and mice carrying one Apc trilox allele harboring both the floxed Apc exon 14 and the floxed hypoxanthine phosphoribosyltransferase (hprt) selection cassette are described in ref. 16. These mice were crossed with Cre-deletor mice to generate Apc+/lox mice that were deleted of the selection cassette and carried one Apc+ allele and one Apclox allele (Fig. 1 A and B) (17). Apc+/lox males were backcrossed six times with C57BL/6N females. Tail or liver DNA was used for genotyping by PCR with primers ApcInt13s and ApcInt14a (Fig. 1 A).
Fig. 1.

Activation of β-catenin signaling, hepatomegaly, and altered survival in Apc-inactivated mice infected with AdCre at high multiplicity. (A) Various alleles of Apc in floxed mice. Restriction sites: E, EcoRI; B, BamHI; X, XbaI; EV, EcoRV. Exons 11–15 are represented by black bars; primers and the lengths of the PCR fragments generated are indicated by arrows. (B) Cre delivery strategy. (C) Survival curve for Apclox/lox and Apc+/lox mice after injection of various doses of AdCre. (D) Liver as a percentage of body weight and proliferation quantified by Ki67 scores in Apclox/lox compared with Apc+/lox Cre-infected livers. *, P < 0.001; **, P = 0.0028. (E) PCR of DNA extracted from livers of Apc floxed mice showing the accumulation of the ApcΔex14 band in AdCre-treated livers compared with noninjected (NI) mouse livers. (F) β-Catenin immunostaining of membranes in Apc+/lox injected mice (Apc+/–). (G) Cytosolic and nuclear accumulation of β-catenin staining in injected, Apclox/lox mice (Apc–/–). (Scale bars in F and G:20 μm.)
Care of Mice and i.v. Injection of Adenovirus Encoding Cre Recombinase (AdCre). All animal procedures reported were carried out in accordance with French government regulations. Mice were housed in conventional conditions. The Ad5-CMV-Cre was prepared by Genethon and supplied at 1011 plaque-forming units (pfu)/ml. Diluted aliquots (150 μl) were injected into the retroorbital vein of 8-week-old mice after isoflurane anesthesia was administered.
Liver Samples. Mice were killed by cervical dislocation: Liver tumor and adjacent nontumoral liver samples were either snapfrozen in liquid nitrogen and stored at –70°C until molecular analyses or fixed in 4% formol for 12 h and then embedded in paraffin. First, tumors were identified macroscopically on 5-mm-thick liver slices and were analyzed by a pathologist (B. Terris, Hôpital Cochin, Paris) on hematoxylin/eosin (H&E)-stained and reticulin-stained liver sections. Then, microscopic tumor foci were identified and counted on H&E-stained and glutamine synthetase (GS)-stained liver sections. For each liver, four transections, at least 200 μm apart, were analyzed. Micronodular hepatocellular foci were scored “+” for 1 or 2 lesions detected in these conditions or “++” for 3–10 lesions (Table 1).
Table 1. Hepatocarcinogenesis in Apc–/– mice.
| No. of GS+ cells in nontumoral tissue†
|
Micronodular preneoplastic foci
|
No. of tumors
|
||||
|---|---|---|---|---|---|---|
| Mouse ID no* | WD | MD | PD | HCC size, mm | ||
| 1 | 6 | ++ | 4 | — | — | 4.2, 2, 1.5, 2 |
| 2 | 3 | + | — | 3 | — | 20, 6, 8 |
| 3 | 1.8 | + | — | — | 1 | 0.5 |
| 4 | 1.3 | 0 | — | — | 1 | 3 |
| 5, 6 | 0.6, 0.9 | 0 | — | — | — | — |
0, no lesions; +, 1 or 2 lesions; ++, 3-10 lesions. —, absent.
Mice were killed 8 months (1) or 9 months (2-6) after AdCre infection.
GS-expressing cells in nonperivenous areas per 1,000 hepatocytes.
Immunohistochemistry and Quantification. Paraffin-embedded liver sections were treated as described in ref. 18. Antibody references are listed in the Supporting Text, which is published as supporting information on the PNAS web site. For double-labeling, β-catenin was immunostained with NovaRed (Vector Laboratories) as the visualization substrate, and the targets [GS, glutamate transporter 1 (GLT1), ornithine aminotransferase (OAT), and leukocyte cell-derived chemotaxin 2 (LECT2)] were stained with Vector SG (Vector Laboratories). The percentages of hepatocytes stained for Ki67 or GS were quantified by using metamorph software, with 5,000–35,000 hepatocytes counted at 100-fold magnification. For GS, perivascular stained cells were excluded (physiological staining).
RNA Extraction and Analyses. Extraction of total RNA and Northern blot analysis were performed as described in ref. 18. Real-time quantitative RT-PCR analysis used reverse transcription by standard protocols (Invitrogen) and was performed in duplicate on a LightCycler apparatus (Roche Diagnostics) and expressed relative to 18S rRNA (18). The sequences of the PCR primers are available upon request.
Results
Liver-Targeted Apc Ablation Activates β-Catenin Signaling and Quickly Induces Major Phenotypic Changes. C57BL/6N Apc+/lox mice were intercrossed to generate Apclox/lox progeny, born at a Mendelian ratio and surviving to an age of up to 2 years without any physiological or histological abnormality. To selectively inactivate Apc in hepatocytes, Apclox/lox mice and Apc+/lox mice (as controls) were injected i.v. with a single dose of 109 pfu of AdCre. We checked that such injections infected mainly and massively the liver (data not shown), in accordance with previous studies (15, 19). As expected, PCR analysis revealed an almost complete conversion of the Apclox allele to the knockout ApcΔex14 allele in the infected livers of both Apc+/lox and Apclox/lox mice, leading to Apc+/Δex14 and ApcΔex14/Δex14 genotypes, respectively (Fig. 1 B and E). Loss of Apc in the liver led to a substantial mortality: 50% of the Apclox/lox mice died within 2 weeks of the injection, and 95% died within 2 months (Fig. 1C). No mortality was observed in the controls.
On day 7 (D7) after administration of 109 pfu of AdCre, Apclox/lox mice presented a significant hepatomegaly, the livers being 60% bigger than those of the controls (Fig. 1D). This hepatomegaly was associated with a significant increase in hepatocyte proliferation, as shown by the Ki67 staining score (Fig. 1D). Apc contributes to tumorigenesis by controlling cellular levels of β-catenin, so we studied β-catenin immunoreactivity in the livers of Apc-inactivated mice. Control mice exhibited a normal staining of β-catenin restricted to the membrane. In contrast, up to 95% of hepatocytes from the AdCre-injected Apclox/lox mice showed more extensive and mislocalized β-catenin staining in the cytoplasm and nucleus, characteristic of activated β-catenin signaling (Fig. 1 F and G). Accordingly, the loss of function of Apc in the liver led to a phenotype similar to that obtained after the overexpression of a stabilized mutant of β-catenin (13, 14). We named the AdCre-infected Apclox/lox and Apc+/lox animals Apc–/– and Apc+/–, respectively.
Dose-Dependent Activation of β-Catenin Signaling. AdCre infections at high doses resulted in the death of Apc–/– mice, preventing the search for hepatocarcinogenesis. We therefore administered lower doses of the adenovirus. After injection with 0.5 × 109 pfu of AdCre, 15% of the Apc–/– mice died within 2 months, whereas no mortality was observed with 0.25 × 109 pfu of AdCre (Fig. 1C). Loss of Apc function was evaluated both by the cytosolic-nuclear staining of β-catenin and by immunolabeling for GS, the product of one of its target genes in the liver (20). In control livers, after infection with 109 pfu of AdCre, GS was detected in a small ring of hepatocytes surrounding the centrolobular vein, consistent with its normal staining pattern in the liver (Fig. 2A). In Apc–/– livers at 109 pfu of AdCre, most of the hepatocytes showed β-catenin staining in the nucleus, the cytoplasm, or both, and most also presented GS immunoreactivity (Fig. 2 A). As the dose of adenovirus decreased, both β-catenin and GS staining became mosaic. Thus, this approach revealed a dose dependence of the number of hepatocytes with activated β-catenin signaling.
Fig. 2.

Dose-dependent Apc inactivation and cell-autonomous induction of liver-specific β-catenin target genes. (A) β-Catenin and GS immunostaining of Apc+/– and Apc–/– mice injected with various doses of AdCre. (B and C) Coimmunostaining of β-catenin and its liver-specific targets (GS, GLT1, and LECT2, in blue) in perivenous (B) and nonperivenous (C) areas. β-Catenin stains red-brown at the membrane of every hepatocyte and accumulates in the cytosol and nuclei of Apc–/– hepatocytes. CV, centrolobular vein; PS, portal space. (Scale bars: A, 100 μm; B and C, 20 μm.) (D) Percentage of hepatocytes that are GS+ in nonperivenous areas of Apc–/– mice, according to dose of AdCre. Filled ovals represent mice with a hepatomegaly (liver > 8.5% of body weight), and open ovals represent mice without hepatomegaly.
The genes for the glutamate transporter GLT1 and the chemokine-like protein LECT2 are also β-catenin targets in the liver (18, 20). To confirm that they are good markers of β-catenin-activated hepatocytes, Apc–/– livers infected with 0.5 × 109 pfu of AdCre were coimmunostained for these proteins and β-catenin. In normal livers, these target genes are expressed only in the hepatocytes surrounding the centrolobular vein area. Indeed, we observed such perivenous staining: cytosolic for GS and LECT2 and membranous for the transporter protein GLT1 (Fig. 2B). However, elsewhere in the lobule, GS-positive (GS+) cells were detected; they all exhibited a dark-blue cytoplasmic staining for GS and a red-brown β-catenin staining in the nucleus (Fig. 2C). Similarly, every nuclear β-catenin-stained cell presented membranous GLT1 staining and cytoplasmic LECT2 staining (Fig. 2C). These observations imply that the control of expression of GS, GLT1, and LECT2 genes by β-catenin signaling is cell-autonomous.
This result allowed β-catenin-activated cells to be followed by counting GS-stained hepatocytes in Apc–/– livers, outside the perivenous area. In Apc–/– mice infected with 109 pfu of AdCre, 70–95% of hepatocytes were GS+, whereas only 30–75% at 0.5 × 109 pfu and 1–30% at 0.25 × 109 pfu of AdCre were positive (Fig. 2D). All of the Apc–/– livers in which >70% of hepatocytes were GS+ displayed significant hepatomegaly (the liver being at least 8.5% of the body weight) (Fig. 2D). No significant hepatomegaly and no hepatocyte proliferation were detected in Apc–/– mice in which <70% of hepatocytes were GS+ (Fig. 2D and data not shown).
These findings imply that the development of hepatomegaly, linked to hepatocyte proliferation, correlates with the number of hepatocytes with β-catenin signaling and requires the loss of function of Apc in at least 70% of the hepatocytes.
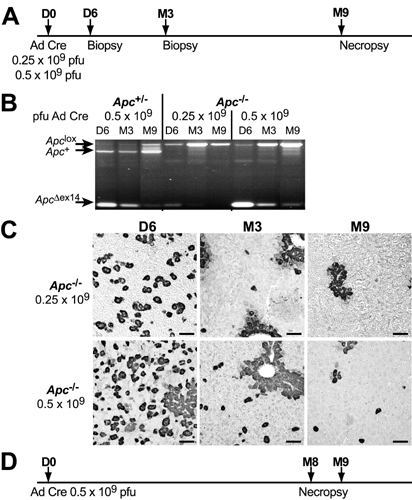
Tumorigenesis in Apc–/– Hepatocytes. Monitoring the persistence of the Apc–/– hepatocytes would be useful when analyzing the oncogenic role of Apc loss in the liver. Indeed, many adenovirus-infected cells are eliminated by the immune response (21). Two liver-biopsy specimens were collected from Apc floxed mice 6 days (D6) and 3 months (M3) after infection with 0.25 or 0.5 × 109 pfu of AdCre, and mice were killed 9 months later (see Fig. 6, which is published as supporting information on the PNAS web site). PCR analysis of DNA from both Apc+/– and Apc–/– livers confirmed a progressive loss of the ApcΔex14 band between D6 and M3, and this loss was most substantial after administration of 0.25 × 109 pfu of AdCre (Fig. 6). Only the livers injected at 0.5 × 109 pfu of AdCre (for both Apc+/– and Apc–/– animals) still contained hepatocytes with the ApcΔex14 allele at 9 months: a maximum of 6 hepatocytes per 1,000 cells (Table 1). Thus, we used this dose of AdCre for the subsequent studies.
Apc floxed male mice infected with 0.5 × 109 pfu of AdCre were monitored for 8 or 9 months (Fig. 6). Ten Apc+/– mice (controls) displayed no hepatic abnormality (data not shown). Ten Apc–/– mice were analyzed; four did not contain any GS+ hepatocytes and developed no liver tumor. Of the six mice that still possessed GS+ hepatocytes, four (67%) developed micronodules and/or a total of nine tumors (Table 1). These HCCs were histologically typed as trabecular and well differentiated (WD) (Table 1), moderately differentiated (MD) (Fig. 3 D–F), or poorly differentiated (PD) (Fig. 3 G–I). The ApcΔex14 allele was amplified from the DNA of every tumor, showing Apc loss (Fig. 3C). β-Catenin staining was nuclear and cytoplasmic (Fig. 3 E and H) and was associated with GS immunostaining (Fig. 3 F and I–K). Nuclear β-catenin immunostaining also was observed in micronodular hepatocellular foci (data not shown), which were easily detectable because of their GS-positivity (Fig. 3K and Table 1).
Fig. 3.

Hepatocarcinogenesis from Apc–/– hepatocytes. (A and B) HCC1 and HCC2 developed 8 and 9 months, respectively, after AdCre infection. (C) PCR analysis from tumoral DNA shows the loss of the Apclox allele and the gain of the ApcΔex14 allele. (D–F) MD HCCs. (G–I) PD HCCs. Hematoxylin/eosin staining shows atypical hepatocytes organized in a trabecular (D) or pseudoglandular (G) pattern; β-catenin mislocalizes in the nucleus (E and H), and GS is overexpressed (F and I). Nontumoral (NT) and tumoral (T) tissues are delineated by a dashed line. (J and K) GS immunostaining in macronodular HCCs (J) and in a micronodular preneoplastic lesion (K), indicated by arrows. (Scale bars: A and B, 1 cm; D–I, 50 μm; J and K, 1 mm.)
The β-catenin activation in isolated GS+ cells, hepatocellular foci, and HCC suggests that the tumors arose from a single Apc–/– GS+ cell. In our model, Apc loss leading to β-catenin signaling is thus sufficient to induce the development of hepatocellular foci that develop into HCC.
Molecular Analysis of Apc–/– Tumors. We tested the expression of several β-catenin target genes to compare the transcriptional program late in β-catenin-activated liver tumors and early after β-catenin signaling due to Apc inactivation. Consistent with immunocytochemical findings (Fig. 2C), the GS, GLT1, and LECT2 genes were clearly overexpressed in Apc–/– livers on D7, as was another liver β-catenin target, the ornithine aminotransferase (OAT) gene (20) (Fig. 4A). These genes were also up-regulated in Apc–/– liver tumors and in WD HCCs extracted from PK/c-Myc livers, which contain β-catenin-stabilizing mutations (3, 4) (Fig. 4A). Interestingly, the level of expression of these genes decreased as the differentiation status of the tumors decreased: GS and GLT1 mRNA levels were lower in the MD carcinoma, and LECT2 and OAT mRNAs were almost undetectable (Fig. 4A, HCC2). This observation was confirmed by the absence of staining for LECT2 in PD Apc–/– HCCs (data not shown). These results mimic those of human HCC, in which GS and GLT1 are good markers of β-catenin-activated tumors but LECT2 and OAT are not systematically up-regulated (18, 20).
Fig. 4.

Comparative expressions of β-catenin-dependent target genes in Apc–/– hyperplastic livers and in β-catenin-activated HCC. (A) Northern blot analysis of livers from adult (3–5 months old) Apc floxed mice, not injected (0) or on D7 after injection of 109 pfu of AdCre (109). Apc–/– and PK/c-Myc tumoral (T) or nontumoral (NT) livers are also shown. HCC1 and HCC2 are the WD and MD HCCs shown in Fig. 3 A and B, respectively. Note for c-Myc the presence of two bands in PK/c-Myc livers: The lower band is the c-Myc transgene (tg) mRNA, and the upper band is the endogenous c-Myc mRNA. OAT, ornithine aminotransferase; 28S and 18S are the rRNAs. (B) Real-time quantitative RT-PCR analysis of cyclin D1 (CycD1) and c-Myc mRNAs extracted from D7 infected livers, HCC1 and HCC2. For analyses at D7, three 1-month-old and four 5-month-old animals were analyzed in each group. No significant differences were observed, except those marked by an asterisk, where P =0.016.
We also analyzed the two canonical β-catenin target genes, encoding cyclin D1 and c-Myc (22, 23). Although these two genes are frequently up-regulated in HCC, a direct link between activation of the β-catenin pathway and the genes' overexpression in the liver is controversial (24–27). Cyclin D1 mRNA was induced in adult Apc–/– livers on D7 and in both Apc–/– and PK/c-Myc liver tumors (Fig. 4). We had previously described no increase in cyclin D1 mRNA in juvenile livers of transgenic mice expressing a stabilized β-catenin mutant (14). We analyzed the expression of cyclin D1 by real-time quantitative RT-PCR in juvenile Apc–/– livers and confirmed that it is not induced in response to a β-catenin signal in juvenile livers (Fig. 4B). No significant induction of c-Myc was detected in hyperplastic Apc–/– livers or in WD Apc–/– tumors (HCC1), although it was up-regulated in MD Apc–/– tumors (HCC2) and PK/c-Myc tumors (Fig. 4). Thus, although cyclin D1 is induced by β-catenin signaling in adult livers, c-Myc is not. The increased expression of c-Myc in these liver tumors may be a result of another signaling pathway (or pathways) activated during the tumoral process. These observations suggest that the genetic program induced by β-catenin is heterogeneous and depends on cell- or tissue-specific factors, the postnatal development status of the liver, and the differentiation status of the tumor.
Genetic Events Elicited for Apc–/– HCC Development. Apc–/– tumors appeared after a latency period of at least 8 months and were relatively infrequent. We thus searched for additional genetic alterations in Apc–/– HCC. Even if in most chemically induced HCCs, mutations of β-catenin are exclusive of those of H-Ras (28, 29), a recent study using mouse technology proposed that the forced activation of H-Ras could be the genetic event that cooperates with activated β-catenin to cause hepatocarcinogenesis (30). Therefore, we analyzed two micronodules and the nine HCCs identified for known hot spot mutations on codons 12–13 and codon 61 of the H-Ras gene (31). No H-Ras mutation was observed in any of these samples (data not shown).
Moreover, p53 inactivation is a frequent event selected for human hepatocarcinogenesis (2). We analyzed two micronodules and nine Apc–/– HCCs to detect a nuclear immunostaining of p53, which is known to reveal the inactivation of the p53 pathway in cancers (32). Only one HCC stained positively for p53 in the nuclei of transformed cells. Interestingly, it was a PD HCC (Fig. 5). This observation suggests that inactivating the p53 pathway can be involved in hepatocarcinogenesis from an Apc–/– hepatocyte.
Fig. 5.

p53 immunostaining of Apc–/– HCC. (A) p53 accumulation in a hyperplastic liver of large T antigen-expressing ATIII-SV40 transgenic mice (37), used as a positive control for nuclear staining. (B) No p53 staining was seen in an Apc–/– MD HCC. (C) Intense p53 staining was seen in the nuclei of tumoral cells from an Apc–/– PD HCC. (Scale bars: 50 μm; 20 μmin Insets.)
Discussion
Here, we demonstrate that aberrant β-catenin signaling is a genetic event able to elicit the development of HCC. Consequently, we have constructed a relevant mouse model of human HCC.
The histopathology and the molecular characteristics of the HCC developing in the Apc–/– HCC model shared many similarities to human HCC. This conditional model has at least three advantages over conventional transgenic models used previously, allowing faithful reproduction of human HCC. (i) Like all sporadic human tumors, the Apc–/– tumors result from a somatic initiating event that occurs in a single cell and grow in a genetically wild-type environment, unlike the other mouse HCC models, in which the initiating event is targeted to all of the hepatocytes. (ii) The genetic event results in activation of the β-catenin pathway, one of the main carcinogenesis pathways implicated in human HCC. (iii) Inactivation of the p53 pathway is an event that cooperates with β-catenin signaling to elicit the development of Apc–/– tumors. Interestingly, this event was found in one Apc–/– HCC with poor differentiation, as is the case in human HCC (33, 34).
Harada et al. (15, 30) had observed no tumoral development 6 months after activation of β-catenin signaling in hepatocytes. The phenotypes of β-catenin-activated and Apc-inactivated mouse livers are strikingly similar in short-term studies. According to these similarities, the long latency period and the infrequency of Apc–/– tumorigenesis can be the reasons for the apparent contradiction between Harada's studies and ours. However, this discrepancy also could be linked to the method of activation of β-catenin in the two studies, i.e., a gain of function of β-catenin in Harada's work and a loss of function of Apc in our study. One might argue that the loss of Apc may favor hepatocarcinogenesis more than the gain of β-catenin function does. Particularly, potential tumor-suppressor roles for Apc protein, other than degrading β-catenin, have been proposed and should be tested in a liver context (35).
Because forced biallelic inactivation of Apc in the liver can induce liver tumorigenesis in our model, it is surprising that the loss of APC is such an infrequent event in human HCC (5, 6). Our results show that Apc is able to degrade β-catenin in the liver; thus, the limiting factor presumably is that APC inactivation does not occur in human liver. Loss of heterozygosity (LOH) is the major mechanism of APC inactivation in the intestine; thus, it is plausible that LOH of the APC locus is tissue-dependent, as is the case for the BRCA1 tumor-suppressor gene (36). Alternatively, it is possible that APC LOH occurs in hepatocytes but that, for an unknown reason, this event is not positively selected in the liver for tumor development.
In conclusion, we have developed a relevant mouse model of HCC involving activation by β-catenin. The genetic events that cooperate with β-catenin activation to result in tumorigenesis should be investigated further. Such work will facilitate the discovery of new cancer genes and pathways involved in this multistep tumorigenesis that should be relevant to human HCC. This mouse model may prove a valuable tool to test novel therapeutic and chemopreventive approaches.
Supplementary Material
Acknowledgments
We thank Dr. Béatrice Romagnolo for helpful discussions and critical feedback; Prof. Benoit Terris for expert histopathological studies; Dr. M. Watanabe (Hokkaido University School of Medicine, Sapporo, Japan), Dr. S. Yamagoe (National Institute of Infectious Diseases, Tokyo), and Dr. C. Caron de Fromontel (Institut National de la Santé et de la Recherche Médicale U590, Lyon, France) for antibodies; Servane Le Plenier for technical assistance; Véronique Fauveau for biopsy sampling; and Vincent Zuliani (Genethon, Evry, France) and the vector core of Genethon, supported by the Association Francaise Contre les Myopathies, for providing the AdCre vector. This work was supported by the Institut National de la Santé et de la Recherche Médicale, the Comité de Paris de la Ligue Nationale Contre le Cancer, the Association pour la Recherche Contre le Cancer, the Ministère Délégué á la Recherche, Action Concertée Incitative Biologie du Développement et Physiologie Intégrative, and the GIS Maladies Rares.
Author contributions: C.P. designed research; S.C., T.D., M.N.-K., C.G., and G.H. performed research; M.N.-K., G.H., and M.G. contributed new reagents/analytic tools; S.C., T.D., A.K., and C.P. analyzed data; and S.C. and C.P. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AdCre, adenovirus encoding Cre recombinase; Apc, adenomatous polyposis coli; D7, day 7; GLT1, glutamate transporter 1; GS, glutamine synthetase; GS+, immunostained for GS; HCC, hepatocellular carcinoma; LECT2, leukocyte cell-derived chemotaxin 2; MD, moderately differentiated; PD, poorly differentiated; pfu, plaque-forming units; WD, well differentiated.
References
- 1.Befeler, A. S. & Di Bisceglie, A. M. (2002) Gastroenterology 122, 1609–1619. [DOI] [PubMed] [Google Scholar]
- 2.Buendia, M. A. (2000) Semin. Cancer Biol. 10, 185–200. [DOI] [PubMed] [Google Scholar]
- 3.Giles, R. H., van Es, J. H. & Clevers, H. (2003) Biochim. Biophys. Acta 1653, 1–24. [DOI] [PubMed] [Google Scholar]
- 4.de La Coste, A., Romagnolo, B., Billuart, P., Renard, C. A., Buendia, M. A., Soubrane, O., Fabre, M., Chelly, J., Beldjord, C., Kahn, A. & Perret, C. (1998) Proc. Natl. Acad. Sci. USA 95, 8847–8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, T. C., Hsieh, L. L., Ng, K. F., Jeng, L. B. & Chen, M. F. (1998) Cancer Lett. 134, 23–28. [DOI] [PubMed] [Google Scholar]
- 6.Legoix, P., Bluteau, O., Bayer, J., Perret, C., Balabaud, C., Belghiti, J., Franco, D., Thomas, G., Laurent-Puig, P. & Zucman-Rossi, J. (1999) Oncogene 18, 4044–4046. [DOI] [PubMed] [Google Scholar]
- 7.Hughes, L. J. & Michels, V. V. (1992) Am. J. Med. Genet. 43, 1023–1025. [DOI] [PubMed] [Google Scholar]
- 8.Kurahashi, H., Takami, K., Oue, T., Kusafuka, T., Okada, A., Tawa, A., Okada, S. & Nishisho, I. (1995) Cancer Res. 55, 5007–5011. [PubMed] [Google Scholar]
- 9.Su, L. K., Abdalla, E. K., Law, C. H., Kohlmann, W., Rashid, A. & Vauthey, J. N. (2001) Cancer 92, 332–339. [DOI] [PubMed] [Google Scholar]
- 10.Gruner, B. A., DeNapoli, T. S., Andrews, W., Tomlinson, G., Bowman, L. & Weitman, S. D. (1998) J. Pediatr. Hematol. Oncol. 20, 274–278. [DOI] [PubMed] [Google Scholar]
- 11.Laurent-Puig, P., Legoix, P., Bluteau, O., Belghiti, J., Franco, D., Binot, F., Monges, G., Thomas, G., Bioulac-Sage, P. & Zucman-Rossi, J. (2001) Gastroenterology 120, 1763–1773. [DOI] [PubMed] [Google Scholar]
- 12.Calvisi, D. F., Factor, V. M., Ladu, S., Conner, E. A. & Thorgeirsson, S. S. (2004) Gastroenterology 126, 1374–1386. [DOI] [PubMed] [Google Scholar]
- 13.Romagnolo, B., Berrebi, D., Saadi-Keddoucci, S., Porteu, A., Pichard, A. L., Peuchmaur, M., Vandewalle, A., Kahn, A. & Perret, C. (1999) Cancer Res. 59, 3875–3879. [PubMed] [Google Scholar]
- 14.Cadoret, A., Ovejero, C., Saadi-Kheddouci, S., Souil, E., Fabre, M., Romagnolo, B., Kahn, A. & Perret, C. (2001) Cancer Res. 61, 3245–3249. [PubMed] [Google Scholar]
- 15.Harada, N., Miyoshi, H., Murai, N., Oshima, H., Tamai, Y., Oshima, M. & Taketo, M. M. (2002) Cancer Res. 62, 1971–1977. [PubMed] [Google Scholar]
- 16.Colnot, S., Niwa-Kawakita, M., Hamard, G., Godard, C., Le Plenier, S., Houbron, C., Romagnolo, B., Berrebi, D., Giovannini, M. & Perret, C. (2004) Lab. Invest. 84, 1619–1630. [DOI] [PubMed] [Google Scholar]
- 17.Leneuve, P., Colnot, S., Hamard, G., Francis, F., Niwa-Kawakita, M., Giovannini, M. & Holzenberger, M. (2003) Nucleic Acids Res. 31, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ovejero, C., Cavard, C., Perianin, A., Hakvoort, T., Vermeulen, J., Godard, C., Fabre, M., Chafey, P., Suzuki, K., Romagnolo, B., et al. (2004) Hepatology 40, 167–176. [DOI] [PubMed] [Google Scholar]
- 19.Wang, Y., Krushel, L. A. & Edelman, G. M. (1996) Proc. Natl. Acad. Sci. USA 93, 3932–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cadoret, A., Ovejero, C., Terris, B., Souil, E., Levy, L., Lamers, W. H., Kitajewski, J., Kahn, A. & Perret, C. (2002) Oncogene 21, 8293–8301. [DOI] [PubMed] [Google Scholar]
- 21.Yang, Y., Xiang, Z., Ertl, H. C. & Wilson, J. M. (1995) Proc. Natl. Acad. Sci. USA 92, 7257–7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He, T. C., Sparks, A. B., Rago, C., Hermeking, H., Zawel, L., da Costa, L. T., Morin, P. J., Vogelstein, B. & Kinzler, K. W. (1998) Science 281, 1509–1512. [DOI] [PubMed] [Google Scholar]
- 23.Shtutman, M., Zhurinsky, J., Simcha, I., Albanese, C., D'Amico, M., Pestell, R. & Ben-Ze'ev, A. (1999) Proc. Natl. Acad. Sci. USA 96, 5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anna, C. H., Iida, M., Sills, R. C. & Devereux, T. R. (2003) Toxicol. Appl. Pharmacol. 190, 135–145. [DOI] [PubMed] [Google Scholar]
- 25.Cui, J., Zhou, X., Liu, Y., Tang, Z. & Romeih, M. (2003) J. Gastroenterol. Hepatol. 18, 280–287. [DOI] [PubMed] [Google Scholar]
- 26.Gotoh, J., Obata, M., Yoshie, M., Kasai, S. & Ogawa, K. (2003) Carcinogenesis 24, 435–442. [DOI] [PubMed] [Google Scholar]
- 27.Prange, W., Breuhahn, K., Fischer, F., Zilkens, C., Pietsch, T., Petmecky, K., Eilers, R., Dienes, H. P. & Schirmacher, P. (2003) J. Pathol. 201, 250–259. [DOI] [PubMed] [Google Scholar]
- 28.Devereux, T. R., Anna, C. H., Foley, J. F., White, C. M., Sills, R. C. & Barrett, J. C. (1999) Oncogene 18, 4726–4733. [DOI] [PubMed] [Google Scholar]
- 29.Aydinlik, H., Nguyen, T. D., Moennikes, O., Buchmann, A. & Schwarz, M. (2001) Oncogene 20, 7812–7816. [DOI] [PubMed] [Google Scholar]
- 30.Harada, N., Oshima, H., Katoh, M., Tamai, Y., Oshima, M. & Taketo, M. M. (2004) Cancer Res. 64, 48–54. [DOI] [PubMed] [Google Scholar]
- 31.Buchmann, A., Bauer-Hofmann, R., Mahr, J., Drinkwater, N. R., Luz, A. & Schwarz, M. (1991) Proc. Natl. Acad. Sci. USA 88, 911–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soussi, T. (2000) Ann. N.Y. Acad. Sci. 910, 121–137, and discussion (2000) 910, 137–139. [DOI] [PubMed] [Google Scholar]
- 33.Nakano, A., Watanabe, N., Nishizaki, Y., Takashimizu, S. & Matsuzaki, S. (2003) Hepatol. Res. 25, 158–165. [DOI] [PubMed] [Google Scholar]
- 34.Nagao, T., Kondo, F., Sato, T., Nagato, Y. & Kondo, Y. (1995) Hum. Pathol. 26, 326–333. [DOI] [PubMed] [Google Scholar]
- 35.Fodde, R. (2003) Nat. Cell Biol. 5, 190–192. [DOI] [PubMed] [Google Scholar]
- 36.Monteiro, A. N. (2003) Trends Genet. 19, 312–315. [DOI] [PubMed] [Google Scholar]
- 37.Dubois, N., Bennoun, M., Allemand, I., Molina, T., Grimber, G., Daudet-Monsac, M., Abelanet, R. & Briand, P. (1991) J. Hepatol. 13, 227–239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}