

Abstract
Successful high-throughput characterization of intact proteins from complex biological samples by mass spectrometry requires instrumentation capable of high mass resolving power, mass accuracy, sensitivity, and spectral acquisition rate. These limitations often necessitate the performance of hundreds of LC–MS/MS experiments to obtain reasonable coverage of the targeted proteome, which is still typically limited to molecular weights below 30 kDa. The National High Magnetic Field Laboratory (NHMFL) recently installed a 21 T FT-ICR mass spectrometer, which is part of the NHMFL FT-ICR User Facility and available to all qualified users. Here we demonstrate top-down LC-21 T FT-ICR MS/MS of intact proteins derived from human colorectal cancer cell lysate. We identified a combined total of 684 unique protein entries observed as 3238 unique proteoforms at a 1% false discovery rate, based on rapid, data-dependent acquisition of collision-induced and electron-transfer dissociation tandem mass spectra from just 40 LC–MS/MS experiments. Our identifications included 372 proteoforms with molecular weights over 30 kDa detected at isotopic resolution, which substantially extends the accessible mass range for high-throughput top-down LC–MS/MS.
Keywords: FT-ICR, 21 tesla, top-down proteomics, FTMS
Graphical abstract
1. INTRODUCTION
Regulation of nearly every cellular process is directly linked to protein primary structure. Comprehensive knowledge of primary structure cannot be derived from the genome because translation of mRNA into protein does not dictate the composition of the final protein complement. Alternative splicing, single nucleotide polymorphisms, endogenous protein cleavages, and the vast number of ways in which any single protein can be post-translationally modified expand the number of theoretically possible proteoforms to well over a billion.1,2 Each proteoform has the innate potential to impact and affect different biological outcomes including phenotypes of health and disease. Mass spectrometry (MS) is an invaluable source of protein structural information, including amino acid sequence and identification and site-localization of post-translational modifications (PTMs).
The ultimate proteomics platform must be capable of unequivocal differentiation of closely related proteoforms, which requires intact analysis (i.e., top-down). However, top-down proteomics typically suffers from slow spectral acquisition rate, low signal-to-noise ratio (S/N), inefficient protein fragmentation, and increased spectral complexity.3 Tandem mass spectra typically contain many overlapping fragments that require high mass resolving power (RP) and spectral averaging to improve S/N. These obstacles have proven difficult for the burgeoning field of top-down proteomics, particularly for proteins larger than 30 kDa.
Fourier-transform ion cyclotron resonance (FT-ICR) MS offers the highest achievable mass RP and mass accuracy of any mass analyzer,4 which is especially important for sequence characterization of intact proteins and site-localization of PTMs. We recently described the design and initial performance of the first 21 tesla (T) FT-ICR mass spectrometer.5 At 21 T, increased ion cyclotron frequency provides higher mass RP at faster scan rates. The instrument design includes an external quadrupole ion trap (multipole storage device, MSD) located between the Velos ion trap and the ICR cell,6 which is used to store multiple accumulations of analyte precursor or fragment ions prior to high-resolution mass analysis in the ICR cell.7 The use of multiple ion accumulations improves spectral S/N more rapidly than spectral averaging and facilitates acquisition of multiple high-quality tandem mass spectra (MS/MS) on a time scale that is more compatible with chromatography.8 The front-end of the instrument is equipped with an electric discharge-based chemical ionization source that is used to create reagents for gas-phase ion/ion reactions such as electron-transfer dissociation (ETD).9,10 Electron-transfer and collision-induced dissociation (CID) techniques combine to produce hundreds of fragment ions, facilitating sequence determination and site-specific characterization of PTMs.
Here we demonstrate top-down proteomic analysis of human colorectal cancer cell lysate by liquid chromatography (LC)–MS/MS on a 21 T FT-ICR mass spectrometer. We identify hundreds of proteins expressed as thousands of unique proteoforms at a 1% false discovery rate (FDR), based on rapid, data-dependent acquisition of CID and ETD fragment ion spectra. Finally, we report significant progress toward the extension of high-throughput top-down proteomics to proteins larger than 30 kDa while maintaining isotopic resolution throughout the analysis.
2. EXPERIMENTAL PROCEDURES
2.1. Cell Culture
DLD-1 parental (KRas wt/G13D) human colorectal cancer cells (HD PAR-086, Horizon Discovery Limited, Cambridge, U.K.) were grown at 37 °C and 5% CO2 in RPMI 1640 (Corning-Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO) and 0.5% penicillin/streptomycin (Corning-Mediatech). Cells were trypsinized in 0.5% trypsin-EDTA (Life Technologies, Carlsbad, CA), resuspended in serum-free RPMI 1640, counted by hemocytometer, and washed twice in 10 mL of ice-cold 1× Dulbecco’s PBS (Life Technologies) prior to centrifugation at 200g for 10 min at 4 °C to remove residual media and serum proteins. Cell pellets, each comprising 2 × 107 cells, were stored dry at −80 °C prior to lysis and protein quantitation.
2.2. Sample Preparation
Cell pellets were thawed on ice and resuspended in 1 mL of 20 mM Tris, pH 7.5, containing 100 mM sodium chloride, 1% (w/v) N-lauroylsarcosine, and 1× final concentration of HALT protease/phosphatase inhibitor cocktail (EDTA-free) (Thermo Fisher Scientific, San Jose, CA). Lysates were incubated on ice for 20 min. Magnesium chloride was added to a final concentration of 1 mM, followed by 750 units of benzonase nuclease (Sigma-Aldrich). Lysates were incubated at 37 °C for 20 min, chilled on ice, and centrifuged at 16 800g for 15 min at 4 °C to pellet cellular debris. Total lysate protein concentration was determined by microplate BCA assay (Thermo Fisher Scientific). Following protein quantitation, 400 μg of protein from each lysate was precipitated in acetone and incubated at −80 °C overnight. Pellets were reconstituted in 100 μL of 1% (w/v) SDS containing 50 μM DTT and 1× Tris-acetate sample buffer (Expedeon, San Diego, CA). Samples were incubated at 95 °C for 5 min and centrifuged at 16 800g for 10 min at room temperature to pellet any remaining debris. Supernatants were loaded into a 10% acrylamide monomer (%T) gel-eluted liquid-fraction entrapment electrophoresis (GELFrEE) cartridge and resolved into 12 fractions according to the manufacturer’s protocol (GELFrEE 8100 Fractionation System, Expedeon). Aliquots (10 μL) from each GELFrEE fraction were resolved by SDS-PAGE and visualized by silver nitrate stain11 to evaluate total protein content (example shown in Figure S-1). Eluted fractions were stored at −80 °C. Directly prior to LC–MS/MS analysis, fractions were precipitated with a mixture of methanol, chloroform, and water to remove SDS.12 After the final MeOH wash, pellets were immediately reconstituted in 50 μL of ice-cold HPLC Solvent A (0.3% formic acid and 5% acetonitrile (v/v) in water; all MS grade) with gentle pipetting.
2.3. Liquid Chromatography
Reconstituted protein fractions were optionally diluted up to 5-fold in ice-cold HPLC solvent A (based on silver nitrate stain intensity) and analyzed by reverse-phase LC–MS/MS. For each injection, 5 μL was loaded onto an in-house-fabricated 360 μm o.d. × 150 μm i.d. fused-silica microcapillary trap column packed 2 to 3 cm with PLRP-S resin (5 μm particle, 1000 Å pore, Agilent Technologies, Palo Alto, CA) or with Poroshell 300-SB C8 resin (5 μm particle, 300 Å pore, Agilent Technologies). The nano-HPLC system (ACQUITY M-Class, Waters, Milford, MA) was operated at 2.5 μL/min for loading onto the trap column and washed with 95% A for 10 min. Separation was achieved on an in-house-fabricated 360 μm o.d. × 75 μm i.d. fused-silica microcapillary analytical column packed 15 cm with PLRP-S or C8 resin (same as corresponding trap columns). For Sample Set 1, all fractions were eluted at 0.3 μL/min using a gradient of 5–20% B in 5 min, 20–40% B in 20 min, 40–60% B in 40 min, 60–75% B in 15 min, and 75–95% B in 5 min (85 min total length). For Sample Set 2, fractions 5–8 were eluted with a gradient of 5–20% B in 5 min, 20–35% B in 20 min, 35–60% B in 75 min, 60–75% B in 15 min, and 75–95% B in 5 min (120 min total length). The gradients utilized solvent A, 0.3% formic acid and 5% acetonitrile in water, and solvent B, 47.5% acetonitrile, 47.5% 2-propanol, 4.7% water, and 0.3% formic acid (% all expressed as v/v). Following separation, samples were directly ionized by microelectrospray ionization using a 15 μm fused-silica PicoTip (New Objective, Woburn, MA) emitter, which was packed with 2–5 mm PLRP-S resin to minimize the formation of bubbles and promote stable ESI.
2.4. Mass Spectrometry
The instrument was operated in data-dependent mode using Xcalibur software (Thermo Fisher Scientific). Precursor (MS1) and product (MS2) ion spectra were collected in the ICR mass analyzer at 21 T. For Sample Set 1 (DLD-1 A), instrument parameters were set as follows: For MS1 spectra – RP = 150 000 at m/z 400 (~381 ms transient duration); 1E6 automatic gain control (AGC) target; 3 transients (microscans, μS) summed per spectrum. The RP was chosen as the minimum required for isotopic resolution of proteins up to ~60 kDa to maximize spectral acquisition rate and number of detected proteins and proteoforms. For MS2 spectra – RP = 150 000 at m/z 400; 7.5E5 AGC target; 4 fragment ion fills of the MSD; CID activation used 41% normalized collision, 10 ms activation period energy and 0.4q; 15 m/z isolation window; dynamic exclusion with a repeat count of 1, repeat duration of 240 s and an exclusion duration of 240 s. For Sample Set 2 (DLD-1 B and C), replicate injections alternated between CID and ETD fragmentation. Instrument parameters were as described above with the following exceptions: For fractions 1–4, ETD MS2 spectra – 2E5 precursor AGC target; 4E5 ETD reagent (fluoranthene) AGC target; 10 fragment ion fills of the MSD; 20 ms reaction period. For fractions 5–8, MS1 spectra – RP = 300 000 at m/z 400 (~762 ms transient duration); 1E6 AGC target; 3 ion fills of the MSD; 2 μS. For fractions 5–8, MS2 spectra −12 ms ETD reaction period; 12 ETD fragment ion fills of the MSD; CID activation used 0.25q. Total data acquisition times were as follows: DLD-1 A fractions 1–8, 880 min (110 min/run, 8 LC–MS/MS runs); DLD-1 B and C, fractions 1–4, 1760 min (110 min/run, 16 LC–MS/MS runs); DLD-1 B and C, fractions 5–8, 2320 min (145 min/run, 16 LC–MS/MS runs). In sum, the data took 82.7 h to acquire.
2.5. Data Analysis
Raw data were submitted to the National Resource for Translational and Developmental Proteomics (Northwestern University, Evanston, IL) for processing on the TDPortal high-performance computing environment at Northwestern University (available for academic collaborators here: http://nrtdp.northwestern.edu/tdportal-request/). Instrument data were then processed using an in-house-developed spectral averaging strategy that involved averaging 30 s windows of reduced profile MS1 data prior to converting data to centroid and use of Thermo Fisher’s Xtract deconvolution algorithm to decharge and deisotope the spectra. For all analyses, a standardized three-pronged search strategy was employed against a database of ~1 × 107 candidate proteoforms created from the 2016_04 release of the Swiss-Prot human proteome. The search strategy used a tree made of three modes as defined for ProSight PTM 2.013: first a narrow absolute mass search (with an MS1 tolerance of 2.2 Da and 10 ppm tolerance for MS2), then a biomarker search (akin to a no-enzyme type search in peptide data analysis; MS1 and MS2 tolerance of 10 ppm), and finally a wide absolute mass search (MS1 tolerance of 200 Da and 10 ppm tolerance for MS2, with Δm mode activated to find unexpected modifications). Data derived from each biological replicate were analyzed separately. Searches for all raw files were completed in 46.9 h.
Estimations of FDR instantaneous q-values at the protein entry and proteoform level were performed using an in-house-developed target-decoy system to be detailed in a separate manuscript. In brief, local FDR calculations were performed by the method previously introduced in ref 14, which involved searching a scrambled database with the MS1 and MS2 data. All resulting hits were taken as incorrect; the distribution of incorrect hits was then used to estimate the null distribution of the scoring metrics. The probability of a forward hit receiving the observed score, or better, due to chance alone was calculated from this null distribution and then combined with conservative corrections for multiple tests15 to accurately estimate FDR with minimum influence from unknown dependencies. Global FDR calculations were calculated by pooling the local FDR results from multiple search strategies and employing the “pick best” approach.16 Note that an FDR is determined at multiple levels (e.g., protein and proteoform), so by default we chose to report only those proteoforms that map to protein entries scoring at an FDR of 1% or better, such that each reported proteoform asserted to be present links to a protein entry. Post-search results were analyzed using the TDViewer version 0.9.0.10 (http://topdownviewer.northwestern.edu). Lists of identified proteins and proteoforms at 1% FDR were exported into Microsoft Excel and either collated into Venn diagrams with Venny 2.1 (http://bioinfogp.cnb.csic.es/tools/venny/) or compared within histograms generated by Microsoft Excel 2013. Specific examples highlighted below were manually validated. The .tdReport files, containing lists of identified protein entries and proteoforms, their respective observed sequence coverage, q-values, C-scores, and other statistics, as well as all .raw files and the .txt file used in the creation of the search database are available for download from the MassIVE repository with identifier/username MSV000079978 (ftp://massive.ucsd.edu/MSV000079978/).
3. RESULTS AND DISCUSSION
Our primary aim was to determine the ability of the LC 21 T FT-ICR MS/MS platform to identify and characterize intact proteins and proteoforms from complex biological samples. We pursued a prototypical sample of whole-cell lysate generated from a well-characterized human colorectal cancer cell line17,18 and fractionated into discrete molecular weight (MW) ranges by use of GELFrEE at 10% T.19 GELFrEE fractions 1–8, containing proteins between approximately 5 and 50 kDa (Figure S-1), were collected from three biological replicates (hereafter designated as DLD-1 A, B, and C) and divided into sample sets for two experiments: (1) analysis of DLD-1 GELFrEE fractions by single-injection, top-down LC–MS/MS at 21 T (Sample Set 1, DLD-1 A) and (2) MS2 fragmentation method comparison (Sample Set 2, DLD-1 B and C). All samples were subjected to reverse-phase LC–MS/MS on a 21 T FT-ICR mass spectrometer with the goal of maximum protein identification and proteoform characterization in the minimum possible time. Data were searched concurrently against forward and decoy databases on the TDPortal with an average time requirement of 16 h per .raw file; the number of proteins and proteoforms identified in each biological replicate is shown in Table S-1. The C-score, a recently introduced metric that quantifies confidence in proteoform identification and characterization, can be interpreted as follows: Those C-scores below 3 indicate a proteoform that has been neither confidently identified nor characterized, those C-scores between 3 and 40 indicate a proteoform that has been confidently identified but not fully characterized, and those C-scores above 40 indicate a proteoform that has been confidently identified and extensively characterized.20 Proteoforms identified with C-scores of 40 or better from DLD-1 A, B, and C are listed in Tables S-2, S-3, and S-4, respectively.
3.1. Single Injections of GELFrEE Fractions Analyzed at 21 T
For Sample Set 1, single injections of DLD-1 A GELFrEE fractions 1–8 were subjected to LC–MS/MS analysis utilizing an 85 min LC gradient and top 2 data-dependent CID fragmentation. The resulting eight .raw files were searched against the database of candidate human proteoforms, which resulted in the identification of 580 unique proteins (defined here by UniProt accession numbers) observed as 1820 unique proteoforms (defined here by Proteoform Record, PFR; Consortium for Top-Down Proteomics Proteoform Repository http://repository.topdownproteomics.org/) at 1% FDR. The number of proteoforms identified per injection ranged from 141 to 593 (Figure 1A). Figure 1B shows the distribution of instantaneous q-values21 for unique proteins (with a median q-value of 5 × 10−19), and Figure 1C shows the distribution of C-scores20 for unique proteoforms identified from Sample Set 1. A total of 792 proteoforms (44%) had C-scores of 40 or above and 445 (24%) had C-scores between 3 and 40, indicating that a total of 1237 proteoforms were identified and at least partially characterized from a single GELFrEE separation of 400 μg of whole-cell lysate protein analyzed by eight LC–MS/MS runs. Of the 1820 proteoforms reported, 1360 matched to a proteoform in the database within 2.2 Da. The remaining 460 proteoforms were identified with mass shifts corresponding to multiple undocumented PTMs, artificial adducts, or cleavage events.
Figure 1.

Protein and proteoform identification and scoring metric distributions. Data from Sample Set 1 (DLD-1 A). (A) Total number of proteins (red) and proteoforms (blue) identified at 1% FDR per single LC–MS/MS injection of each of the eight 10% GELFrEE fractions of whole cell lysate. (B) −log (q-value) distribution for the 580 unique proteins (unique UniProt accession numbers) identified at 1% FDR. (C) C-score distribution for the corresponding 1820 unique proteoforms. For (B) and (C), each bin comprises the sum of identifications with scores between that bin and the one to the left, save for the lowest, which comprises the sum of the identifications between that number and zero.
Representative data obtained from a single DLD-1 A GELFrEE fraction (fraction 4) are shown in Figure 2. The total ion chromatogram (TIC) is shown (Figure 2A) along with single-scan MS1 spectra, which show the charge-state distributions of proteins eluting over 10 selected peaks. The protein identities were manually validated, and global q-values and C-scores are given. The data acquisition period ranged from 1.67 to 3.17 s/spectrum for MS1 spectra, which was collected as the sum of three 381 ms transient acquisitions (3 μS) to increase S/N for accurate deconvolution and further analysis.22 CID MS2 elapsed scan period ranged from 1 to 3 s and was collected as a single 381 ms transient (1 μS) following four fragment ion fills of the MSD. Maximum injection period was set to 500 ms for both scan types; up to 2 s of the quoted scan period was used solely for precursor ion accumulation. A single duty cycle (1 MS1, 2 MS2) was typically between 3 and 6 s. A total of 2422 spectra were acquired across the chromatogram shown (10–100 min). A total of 248 unique UniProt entries (observed as 561 proteoforms) were detected at a 1% FDR in this single experiment. A histogram giving the MW distribution of observed proteoforms is displayed in Figure 2B, which is consistent with the expected MW range based on the stained gel shown in Figure S-1.
Figure 2.

LC–MS/MS of a single injection of GELFrEE fraction at 21 T. Data from DLD-1 A (Sample Set 1) GELFrEE fraction 4. (A) Total ion chromatogram (TIC). Ten example identifications are indicated (with UniProt and PFR numbers) along with single-scan broadband mass spectra, q-values, and C-scores. (B) Histogram depicting the molecular weight distribution of proteoforms identified in the fraction; 248 unique UniProt entries (observed as 561 proteoforms) were detected at a 1% FDR in this single LC–MS/MS analysis. (C) Single-scan CID MS2 spectrum taken at ~41 min (green label in TIC shown in panel A) identified as translationally controlled tumor protein, p23. (D) Top: fragment mass error (ppm) versus m/z and sequence coverage obtained from (C). Bottom: 54 fragments were matched to the putative sequence within a 10 ppm mass tolerance (RMS error 0.72 ppm, S/N ≈ 500–5:1), yielding ~17% sequence coverage.
A single-scan CID MS2 spectrum (1.23 s elapsed scan time) of a protein eluting at ~41 min (indicated in green in the chromatogram) is shown in Figure 2C. The precursor ion was [M+16H]16+ with S/N of ~41:1 (range for all charge states was 102:1 to 5:1). This protein was detected with ~2.1 ppm intact mass error following deconvolution and was identified as translationally-controlled tumor protein (p23, UniProt P13693, ~20 kDa) with no modifications. A total of 54 fragment ions were matched to the putative sequence with an RMS error of 0.72 ppm (10 ppm allow tolerance; Figure 2D). The most abundant isotopologues in each multiplet matched to the putative sequence had S/N ratios from ~486:1 to ~5:1 and are given in Table S-5 along with observed RP and mass error. Taken together, these data exemplify the wealth of intact protein and proteoform information that can be extracted from a single injection of a complex biological sample analyzed by LC-FT-ICR MS/MS at 21 T.
3.2. CID and ETD Fragmentation Analyzed at 21 T
In addition to collision-induced (ion trap CID/beam-CID) fragmentation, the 21 T FT-ICR mass spectrometer is equipped with a front-end reagent ion source that is used to ionize fluoranthene for ETD fragmentation in the high pressure cell of a Velos Pro dual cell ion trap assembly.10 For sample set 2 (DLD-1 B and C), we compared the number of proteins and proteoforms identified by CID versus ETD fragmentation (performed as technical duplicates). Note that ETD is typically carried out with a smaller precursor ion population than CID because precursor ions are confined to the smaller, rear section of the trap as reagent ion is introduced.9,23 The use of small precursor ion population combined with the charge-destructive nature of ETD and increased sequence coverage afforded by ETD24,25 dilutes the remaining ion signal of any single fragment ion. Such reduced signal necessitates the use of more fragment ion fills of the MSD to achieve S/N comparable to CID MS2 spectra. Therefore, 10–12 fragment ion fills were used for ETD MS2. At a 500 ms maximum injection time per fragment ion fill, acquisition of one ETD spectrum was observed to take up to 9 s, as opposed to just 3 s for CID. Up to approximately 3-fold lower duty cycle was observed, with fewer spectra collected overall.
Figure 3 summarizes the results from Sample Set 2; four injections each (two biological replicates, DLD-1 B and C, two technical replicates - CID/ETD) of GELFrEE fractions 1–8 (32 total LC–MS/MS runs). A combined total of 538 unique proteins and 2476 proteoforms were identified at an FDR of 1% by the TDPortal-supported searches. Venn diagrams comparing the proteins and proteoforms identified within each biological replicate (DLD-1 B or C) by CID or ETD fragmentation can be seen in Figure S-2. Instantaneous q-value distributions for the proteins as well as C-score and MW distributions for the proteoforms identified within DLD-1 B or C can be seen in Figures S-3 and S-4. For the DLD-1 B and C datasets, 75 and 72% of the unique proteins were detected by both CID and ETD, with CID identifying slightly more protein entries than ETD. At the proteoform level, only 36 and 35%, respectively, were identified by both fragmentation techniques, and CID identified up to 228 (13%) more unique proteoforms than ETD. We believe that these discrepancies are due to both the longer time required to obtain ETD MS2 spectra and to the complexity of the samples studied, i.e., that more proteoforms remain to be discovered within each sample.
Figure 3.
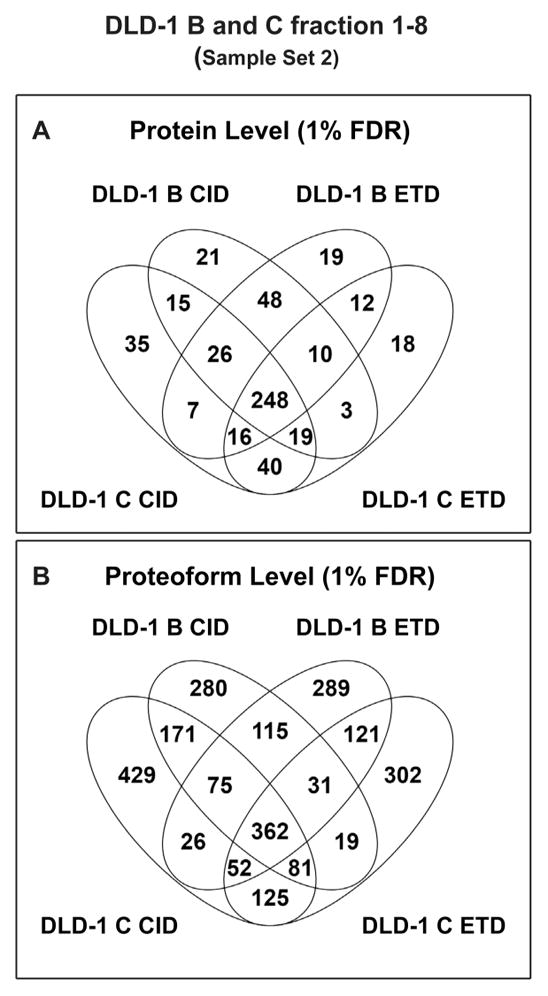
Differential protein and proteoform identification by CID and ETD. (A) Venn diagram showing the proteins (unique UniProt accession numbers) identified within Sample Set 2 (DLD-1 B and C, GELFrEE fractions 1–8). 248 identified proteins were observed in all four replicate analyses, but CID and ETD identified distinct subsets of unique proteins. (B) Venn diagram of proteoforms (unique PFRs) identified within Sample Set 2. 362 proteoforms were observed in all four replicate analyses, but CID and ETD resulted in the identification of discrete subsets of unique proteoforms.
Representative MS2 data from Sample Set 2 are shown in Figure 4. Single-scan CID and ETD MS2 spectra are shown for [M+26H]26+ (1.5 s scan acquisition) and [M+29H]29+ (3.3 s scan acquisition), respectively, of the 20 kDa protein peptidyl-prolyl cis–trans isomerase B (UniProt P23284, PFR 1364), taken from two replicate injections of fraction 5 (DLD-1 C). Zoom insets are shown and fragment ions are labeled. In both cases, the protein was detected with ≤2 ppm intact mass error and identified with high confidence (q-value ≤ 5 × 10−49). Note the greater degree of sequence information from the ETD MS2 spectrum (51% of possible bond cleavages) compared with the CID MS2 spectrum (21% of possible bond cleavages). As can be seen in Figures S-3 and S-4, C-scores for proteoforms identified by ETD fragmentation skewed higher than those identified by CID. ETD fragmentation also resulted in higher overall median q-values for proteins identified within DLD-1 B (3 × 10−17) and DLD-1 C (3 × 10−20) compared with those identified by CID fragmentation (1 × 10−13 and 2 × 10−14, respectively). This is not surprising because ETD tends to fragment large, highly charged precursors more efficiently than CID, resulting in higher sequence coverage and enabling more precise PTM site localization.25,26
Figure 4.

CID and ETD MS2 spectra of peptidyl-prolyl cis–trans isomerase B. (Top) Single-scan CID (Left) and ETD (Right) MS2 spectra of a precursor identified as peptidyl-prolyl cis–trans isomerase B (UniProt P23284 residues 34–216, PFR 1354). Expanded views (Center) of 50 m/z regions (denoted by gray brackets) reveal the wealth of identified fragments. Colored fragment labels indicate multiplets identified by manual inspection only. * = neutral loss of water from nearby fragment; ‡ = coisolated chemical noise/activation window. Sequence coverage maps and confidence metrics determined by TDPortal are given (Bottom).
3.3. Analysis of Larger Proteins
More than half of predicted human proteins are >30 kDa,14 so it is crucial that top-down methods improve at higher mass. We expect that the high-mass RP and dynamic range afforded by 21 T FT-ICR MS will push the boundaries of protein identification and characterization to well above 30 kDa. Toward that goal, we used DLD-1 cell lysate as a test case. For Sample Set 1 (DLD-1 A), the observed molecular weight distribution of unique proteoforms identified at 1% FDR is shown in Figure 5A. A total of 228 proteoforms with MW greater than 30 kDa were identified at 1% FDR; this represents ~13% of the total number of unique proteoforms within that sample set. All proteoforms larger than 30 kDa were observed in fractions 5–8 alone (four total injections for DLD-1 A). Confidence metrics for proteoforms identified at 1% FDR are shown in Figure 5B, in which C-scores (red) and log (q-values) (blue) are plotted against proteoform MW. As expected, confidence metrics for proteoforms larger than 30 kDa were lower than those for smaller proteoforms, which we believe can be at least partially attributed to the need for better chromatographic resolution of larger proteins. A large number of proteins were observed to coelute during analyses of fractions 7 and 8, resulting in very low (<10) S/N in the MS1 spectra and chimeric MS2 spectra, which confounded subsequent data analysis. We plan to investigate the use of chromatographic methods better optimized for the larger proteins that we expect to observe in fractions 9–12 (~50–100 kDa).
Figure 5.

Proteoform molecular weight distribution and scoring metrics as a function of molecular weight. Data from Sample Set 1 (DLD-1 A). (A) Molecular weight distribution of proteoforms identified at 1% FDR. 1820 proteoforms were identified at 1% FDR from just 8 LC–MS/MS runs (1 injection each of GELFrEE fractions 1–8). Proteoforms are grouped by monoisotopic mass (kDa). (B) Scatter plot depicting C-score (red) and log (q-value) (blue) as a function of proteoform molecular weight.
Typical results for proteins larger than 30 kDa are shown in Figure 6. MS1 data depicting charge-state distributions for three proteins are shown (Figure 6B) along with zoom insets that show selected isotopic distributions (right). Two of the three distributions shown were resolved in a single MS1 scan (3 μS, green and red). Resolution and charge-state determination for the 54 kDa protein required post-FT spectral averaging (average of 20 scans). We were able to determine these protein identities via de novo sequencing from the corresponding CID MS2 spectra (example shown in Figure 6C). All results were consistent with the identifications returned by subsequent database searching via TDPortal.
Figure 6.

Resolved isotopic distributions of proteins with MW > 30 kDa. (A) Total ion chromatogram obtained by LC–MS/MS of DLD-1 A GELFrEE fraction 8. (B) Single-scan mass spectra show protein charge-state distributions (650–1500 m/z, Left) for three chromatographic peaks and zoom insets of single charge states with resolved isotopic distributions (Right). Resolution and charge-state determination for the 54 kDa protein required post-FT spectral averaging of 20 scans. (C) CID MS2 spectrum of [M+55H]55+ ions of keratin type 1 cytoskeletal 18 protein. Zoom insets provide expanded views of the region indicated with gray bracket. Colored fragment labels indicate multiplets identified by manual inspection only. (D) Sequence coverage map and confidence metrics for the identification of keratin type 1 by TDPortal.
4. CONCLUSIONS
The 21 T FT-ICR instrument at the National High Magnetic Field Laboratory is particularly well equipped for high-throughput top-down proteomics due to the high RP obtained per unit time and the use of an external MSD to perform multiple precursor or fragment ion fills prior to high-resolution mass analysis. Multiple fills provide a necessary boost in S/N, which, when combined with the transient acquisition rate and RP achieved at 21 T, enable acquisition of high-quality MS2 spectra that facilitate intact protein identification on a chromatographic time scale. Here we demonstrate high-throughput proteoform identification by LC-FT-ICR MS/MS at 21 T. Initial experiments produced unparalleled results on the basis of number of identified proteins and proteoforms (580 UniProt entries expressed as 1820 unique proteoforms at 1% FDR for DLD-1 A) per total number of injections (eight individual LC–MS/MS runs) compared with previous top-down proteomics studies.14,27,28 For example, the largest previous top-down proteomics study focused on proteins <30 kDa and achieved 1063 unique proteins at 1% FDR from 423 LC–MS/MS runs.27,29 Here >50% of the coverage (on the protein entry level) was achieved in <2% of the number of comparable LC runs, demonstrating that LC-21 T FT-ICR MS/MS is the current state of the art method for high-throughput top-down proteomics.
We also compared CID and ETD in terms of optimal scan rate, number of proteoform identifications, and characterization confidence. Here we demonstrate that ETD outperformed CID with regard to confidence in protein identification and proteoform characterization (defined here with the scoring metrics, q-value, and C-score, respectively), which is consistent with prior studies.14,24,26,30,31 However, a smaller precursor ion population is used,9,23 and total ion current is diluted among more fragment ion channels, which requires more fragment ion fills of the MSD prior to high-resolution mass analysis. Ultimately, this results in a longer acquisition period required for ETD MS2 spectra. We observed that CID outperformed ETD with regard to total number of proteoforms identified. This, we presume, is due to a combination of faster spectral acquisition rates and higher S/N observed in CID MS/MS spectra, as total ion current tends to be diluted through fewer, more specific fragment ion channels.25,26,32,33 In the future, we intend to explore ways to increase the initial precursor ion population for ETD23 to lessen the need for more fragment ion fills and increase spectral acquisition rate. We have also implemented ultraviolet photodissociation34 within the ICR cell and plan to assess its utility for high-throughput proteoform identification and characterization.
Analysis of intact proteins becomes more challenging as protein mass increases. Ion current is distributed among more charge states, isotopologues, adducts, and fragment ion channels.22 Additionally, space-charge capacity in the linear trap scales inversely with m/z35–37 and data analysis is more difficult. These challenges are reflected by the focus of several recent studies on proteins <30 kDa or on the analysis of whole cell lysates derived from prokaryotes, which typically have proteomes comprising a lower molecular weight range.29,38–41 We were able to identify 372 proteoforms larger than 30 kDa at 1% FDR with only 20 LC injections (GELFrEE fractions 5–8, all bioreplicates, duplicates removed), setting new efficiency targets for isotopically resolved proteoform analysis. Furthermore, we believe that we can improve performance by optimizing the chromatographic separation of proteins larger than 30 kDa. We expect that improved sample resolution and optimal MS2 transient summing should combine with the high mass accuracy and ultrahigh RP achieved at 21 T to further improve the number of identifications for proteins up to or exceeding 60 kDa on a time scale that remains compatible with chromatography. Increasing the average MW range of the human proteome available to investigation by high-throughput top-down proteomics by even 20 kDa could facilitate the discovery of potentially thousands of new proteoforms, a significant portion of which might have direct clinical relevance in human disease or become dysregulated in human cancers.
Supplementary Material
Acknowledgments
We thank the Hunt Laboratory at the University of Virginia, and Thermo Fisher Scientific (San Jose, CA) for help with ETD hardware and software and Chad Weisbrod for helpful discussion. This work was supported by the National Science Foundation Cooperative Agreement No. DMR-1157490 and the State of Florida as well as by the National Institute of General Medical Science (P41GM108569) for the National Resource for Translational and Developmental Proteomics (NRTDP) based at Northwestern University. Further support was provided by the computational resources and staff for the Quest high performance computing facility at Northwestern University, which is jointly supported by the Office of the Provost, the Office for Research, and Northwestern University Information Technology.
ABBREVIATIONS
- MS
mass spectrometry
- PTM
post-translational modification
- S/N
signal-to-noise ratio
- RP
resolving power
- FT-ICR
Fourier-transform ion cyclotron resonance
- T
tesla
- MSD
multipole storage device
- MS/MS
tandem mass spectrometry
- ETD
electron-transfer dissociation
- CID
collision-induced dissociation
- LC
liquid chromatography
- FDR
false discovery rate
- %T
total acrylamide and bis-acrylamide monomer in g/100 mL
- GELFrEE
gel-eluted liquid fraction entrapment electrophoresis
- MS1
precursor mass spectrum
- MS2
fragment ion spectrum
- AGC
automatic gain control
- μS
microscans
- q
ion trap CID excitation q value
- MW
molecular weight
- PFR
proteoform record number
- TIC
total ion chromatogram
Footnotes
ORCID
Paul M. Thomas: 0000-0003-2887-4765
Neil L. Kelleher: 0000-0002-8815-3372
The authors declare the following competing financial interest(s): Some authors are involved with software commercialization activities.
All raw spectrum files (.raw), search engine files (.tdreport), and the flat file (.txt) used for the creation of the database against which data were searched have been uploaded to the UCSD MassIVE repository with the identifier/username MSV000079978 and can be accessed here: ftp://massive.ucsd.edu/MSV000079978/. The TDViewer application, which allows one to view the contents of the .tdreport files, is available for download at http://topdownviewer.northwestern.edu/
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.6b00696.
Figure S-1, Silver-stained gel image of DLD-1 A protein fractions. Figure S-2, Differential protein and proteoform identification by CID and ETD. Figure S-3, Score and MW Distributions of DLD-1 B Proteins and Proteoforms. Figure S-4, Score and MW Distributions of DLD-1 C Proteins and Proteoforms. Table S-1, Number of unique proteins and proteoforms identified at 1% FDR from three biological replicates. Table S-5, List of fragments matched to putative sequence of p23 (UniProt P13693) by TDPortal (shown in Figure 2C). (PDF) Tables S-2, S-3, and S-4, Proteoforms identified at 1% FDR with C-scores of 40 or better from each biological replicate (DLD-1 A, B, and C, respectively). (XLSX)
References
- 1.Nielsen ML, Savitski MM, Zubarev RA. Extent of modifications in human proteome samples and their effect on dynamic range of analysis in shotgun proteomics. Mol Cell Proteomics. 2006;5:2384–2391. doi: 10.1074/mcp.M600248-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Schluter H, Apweiler R, Holzhutter HG, Jungblut PR. Finding one’s way in proteomics: a protein species nomenclature. Chem Cent J. 2009;3 doi: 10.1186/1752-153X-3-11. 11-153X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dang X, Scotcher J, Wu S, Chu RK, Tolic N, Ntai I, Thomas PM, Fellers RT, Early BP, Zheng Y, Durbin KR, Leduc RD, Wolff JJ, Thompson CJ, Pan J, Han J, Shaw JB, Salisbury JP, Easterling M, Borchers CH, Brodbelt JS, Agar JN, Pasa-Tolic L, Kelleher NL, Young NL. The first pilot project of the consortium for top-down proteomics: a status report. Proteomics. 2014;14:1130–1140. doi: 10.1002/pmic.201300438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marshall AG, Hendrickson CL, Jackson GS. Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spectrom Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 5.Hendrickson CL, Quinn JP, Kaiser NK, Smith DF, Blakney GT, Chen T, Marshall AG, Weisbrod CR, Beu SC. 21 T Fourier Transform Ion Cyclotron Resonance Mass Spectrometer: A National Resource for Ultrahigh Resolution Mass Analysis. J Am Soc Mass Spectrom. 2015;26:1626–1632. doi: 10.1007/s13361-015-1182-2. [DOI] [PubMed] [Google Scholar]
- 6.Kaiser NK, Savory JJ, Hendrickson CL. Controlled ion ejection from an external trap for extended m/z range in FT-ICR mass spectrometry. J Am Soc Mass Spectrom. 2014;25:943–949. doi: 10.1007/s13361-014-0871-6. [DOI] [PubMed] [Google Scholar]
- 7.Schaub TM, Hendrickson CL, Horning S, Quinn JP, Senko MW, Marshall AG. High-performance mass spectrometry: Fourier transform ion cyclotron resonance at 14.5 T. Anal Chem. 2008;80:3985–3990. doi: 10.1021/ac800386h. [DOI] [PubMed] [Google Scholar]
- 8.Anderson LC, Karch KR, Ugrin SA, Coradin M, English AM, Sidoli S, Shabanowitz J, Garcia BA, Hunt DF. Analyses of Histone Proteoforms Using Front-end Electron Transfer Dissociation-enabled Orbitrap Instruments. Mol Cell Proteomics. 2016;15:975–988. doi: 10.1074/mcp.O115.053843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Earley L, Anderson LC, Bai DL, Mullen C, Syka JE, English AM, Dunyach JJ, Stafford GC, Jr, Shabanowitz J, Hunt DF, Compton PD. Front-end electron transfer dissociation: a new ionization source. Anal Chem. 2013;85:8385–8390. doi: 10.1021/ac401783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 12.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 13.Zamdborg L, LeDuc RD, Glowacz KJ, Kim YB, Viswanathan V, Spaulding IT, Early BP, Bluhm EJ, Babai S, Kelleher NL. ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucleic Acids Res. 2007;35:W701–6. doi: 10.1093/nar/gkm371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benjamini Y, Yekutieli D. The Control of the False Discovery Rate in Multiple Testing under Dependency. Ann Statist. 2001;29:1165–1188. [Google Scholar]
- 16.Higdon R, Haynes W, Kolker E. Meta-analysis for Protein Identification: A Case Study on Yeast Data. OMICS. 2010;14:309–314. doi: 10.1089/omi.2010.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demory Beckler M, Higginbotham JN, Franklin JL, Ham AJ, Halvey PJ, Imasuen IE, Whitwell C, Li M, Liebler DC, Coffey RJ. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol Cell Proteomics. 2013;12:343–355. doi: 10.1074/mcp.M112.022806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halvey PJ, Wang X, Wang J, Bhat AA, Dhawan P, Li M, Zhang B, Liebler DC, Slebos RJ. Proteogenomic analysis reveals unanticipated adaptations of colorectal tumor cells to deficiencies in DNA mismatch repair. Cancer Res. 2014;74:387–397. doi: 10.1158/0008-5472.CAN-13-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran JC, Doucette AA. Gel-eluted liquid fraction entrapment electrophoresis: an electrophoretic method for broad molecular weight range proteome separation. Anal Chem. 2008;80:1568–1573. doi: 10.1021/ac702197w. [DOI] [PubMed] [Google Scholar]
- 20.LeDuc RD, Fellers RT, Early BP, Greer JB, Thomas PM, Kelleher NL. The C-score: a Bayesian framework to sharply improve proteoform scoring in high-throughput top down proteomics. J Proteome Res. 2014;13:3231–3240. doi: 10.1021/pr401277r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Compton PD, Zamdborg L, Thomas PM, Kelleher NL. On the scalability and requirements of whole protein mass spectrometry. Anal Chem. 2011;83:6868–6874. doi: 10.1021/ac2010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riley NM, Mullen C, Weisbrod CR, Sharma S, Senko MW, Zabrouskov V, Westphall MS, Syka JE, Coon JJ. Enhanced Dissociation of Intact Proteins with High Capacity Electron Transfer Dissociation. J Am Soc Mass Spectrom. 2016;27:520–531. doi: 10.1007/s13361-015-1306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coon JJ, Ueberheide B, Syka JE, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc Natl Acad Sci U S A. 2005;102:9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JE, Shabanowitz J, Hunt DF. The utility of ETD mass spectrometry in proteomic analysis. Biochim Biophys Acta, Proteins Proteomics. 2006;1764:1811–1822. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coon JJ. Collisions or electrons? Protein sequence analysis in the 21st century. Anal Chem. 2009;81:3208–3215. doi: 10.1021/ac802330b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Catherman AD, Durbin KR, Ahlf DR, Early BP, Fellers RT, Tran JC, Thomas PM, Kelleher NL. Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Mol Cell Proteomics. 2013;12:3465–3473. doi: 10.1074/mcp.M113.030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahlf DR, Compton PD, Tran JC, Early BP, Thomas PM, Kelleher NL. Evaluation of the compact high-field orbitrap for top-down proteomics of human cells. J Proteome Res. 2012;11:4308–4314. doi: 10.1021/pr3004216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durbin KR, Fornelli L, Fellers RT, Doubleday PF, Narita M, Kelleher NL. Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa. J Proteome Res. 2016;15:976–982. doi: 10.1021/acs.jproteome.5b00997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Udeshi ND, Compton PD, Shabanowitz J, Hunt DF, Rose KL. Methods for analyzing peptides and proteins on a chromatographic timescale by electron-transfer dissociation mass spectrometry. Nat Protoc. 2008;3:1709–1717. doi: 10.1038/nprot.2008.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui W, Rohrs HW, Gross ML. Top-down mass spectrometry: recent developments, applications and perspectives. Analyst. 2011;136:3854–3864. doi: 10.1039/c1an15286f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wysocki VH, Tsaprailis G, Smith LL, Breci LA. Mobile and localized protons: a framework for understanding peptide dissociation. J Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 33.Reid GE, Wu J, Chrisman PA, Wells JM, McLuckey SA. Charge-state-dependent sequence analysis of protonated ubiquitin ions via ion trap tandem mass spectrometry. Anal Chem. 2001;73:3274–3281. doi: 10.1021/ac0101095. [DOI] [PubMed] [Google Scholar]
- 34.Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS. Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J Am Chem Soc. 2013;135:12646–12651. doi: 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Louris JN, Brodbelt-Lustig JS, Graham Cooks R, Glish GL, van Berkel GJ, McLuckey SA. Ion isolation and sequential stages of mass spectrometry in a quadrupole ion trap mass spectrometer. Int J Mass Spectrom Ion Processes. 1990;96:117–137. [Google Scholar]
- 36.Schwartz JC, Senko MW, Syka JE. A two-dimensional quadrupole ion trap mass spectrometer. J Am Soc Mass Spectrom. 2002;13:659–669. doi: 10.1016/S1044-0305(02)00384-7. [DOI] [PubMed] [Google Scholar]
- 37.Scherperel G, Reid GE. Emerging methods in proteomics: top-down protein characterization by multistage tandem mass spectrometry. Analyst. 2007;132:500–506. doi: 10.1039/b618499p. [DOI] [PubMed] [Google Scholar]
- 38.Ansong C, Wu S, Meng D, Liu X, Brewer HM, Deatherage Kaiser BL, Nakayasu ES, Cort JR, Pevzner P, Smith RD, Heffron F, Adkins JN, Pasa-Tolic L. Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proc Natl Acad Sci U S A. 2013;110:10153–10158. doi: 10.1073/pnas.1221210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiu L, Valeja SG, Alpert AJ, Jin S, Ge Y. Effective protein separation by coupling hydrophobic interaction and reverse phase chromatography for top-down proteomics. Anal Chem. 2014;86:7899–7906. doi: 10.1021/ac501836k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cannon JR, Cammarata MB, Robotham SA, Cotham VC, Shaw JB, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS. Ultraviolet photodissociation for characterization of whole proteins on a chromatographic time scale. Anal Chem. 2014;86:2185–2192. doi: 10.1021/ac403859a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Savaryn JP, Toby TK, Catherman AD, Fellers RT, LeDuc RD, Thomas PM, Friedewald JJ, Salomon DR, Abecassis MM, Kelleher NL. Comparative top down proteomics of peripheral blood mononuclear cells from kidney transplant recipients with normal kidney biopsies or acute rejection. Proteomics. 2016;16:2048–2058. doi: 10.1002/pmic.201600008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.