

Summary
Neutrophils are a major component of the innate host response, and the outcome of the interaction between the oral microbiota and neutrophils is a key determinant of oral health status. The composition of the oral microbiome is very complex and different in health and disease. Neutrophils are constantly recruited to the oral cavity, and their protective role is highlighted in cases where their number or functional responses are impeded, resulting in different forms of periodontal disease. Periodontitis, one of the more severe and irreversible forms of periodontal disease, is a microbial-induced chronic inflammatory disease that affects the gingival tissues supporting the tooth. This chronic inflammatory disease is the result of a shift of the oral bacterial symbiotic community to a dysbiotic more complex community. Chronic inflammatory infectious diseases such as periodontitis can occur because the pathogens are able to evade or disable the innate immune system. In this review, we discuss how human neutrophils interact with both the symbiotic and the dysbiotic oral community; an understanding of which is essential to increase our knowledge of the periodontal disease process.
Keywords: neutrophils, periodontitis, symbiotic, dysbiotic oral community
1 | Introduction
As humans, we are constantly exposed to bacterial challenges through the air we inhale, our skin and what we eat. However, we can maintain a healthy life due to our innate immune system which has evolved to protect us from the constant threat of infection provoked by the continuous exposure to foreign microbes at each of the accessible entry zones in our body. Not all the encounters that we have with these foreign ‘animalcules’ (initial designation attributed to bacteria by van Leeuwenhoek) result in a threat to our health. Moreover, the symbiotic interaction between the host and commensal microbiota has gained more and more attention lately and proven to be beneficial to our health.1,2 A delicate balance exists between the host and its associated, highly complex microbiota to maintain homeostasis and health.3 Even though the oral cavity is an example of an area of our body which is heavily colonized by a diverse microbial community, its interaction with the host immune system remains poorly understood when compared to studies done with the gut microbiome.4,5
Neutrophils are the main innate immune cell that is recruited in very high numbers to the oral cavity and are responsible for ensuring a healthy periodontal tissue.6–8 The junctional epithelium, a specialized tissue that surrounds the tooth, generates a chemotactic interleukin (IL)-8 gradient due to its close contact with the oral biofilm community, which results in constant recruitment of neutrophils to the gingival sulcus.5,9 Neutrophil presence is associated with protection against periodontal disease since increased periodontitis severity has been observed in congenital diseases like leukocyte adhesion deficiency, Chediak-Higashi syndrome, Papillon-Lefèvre syndrome and chronic/cyclic neutropenia which are all conditions in which neutrophil recruitment is compromised.10 However, in chronic inflammatory diseases such as periodontitis, pathogens have evolved mechanisms to evade neutrophil clearance, which results in neutrophil accumulation in the periodontal tissue, and potential release of the neutrophil arsenal into the extracellular space causing tissue damage, and in severe cases, bone loss.11 For example, while the oxidative burst response mounted by neutrophils is an efficient strategy for clearance of infection with anaerobic bacteria, several periodontal pathogens such as Porphyromonas gingivalis are resistant to oxidative killing.11,12 Additionally, hyperactive/primed neutrophils can also predispose individuals to develop periodontitis. These neutrophils' enhanced response is characterized by the release of reactive oxygen intermediates, several cationic peptides, and enzymes such as matrix metalloproteinases (MMPs) that results in increased tissue damage and positions the neutrophil as a perpetrator of periodontitis.13,14 Thus, a delicate balance between neutrophil function and bacterial challenge has to be maintained to ensure periodontal health. This review discusses the role of human neutrophils in maintaining a healthy oral mucosa and evidence associated to how the cell interaction with periopathogens can shift the homeostatic balance into a dysbiotic environment promoting a chronic inflammatory scenario—leading to periodontitis (Fig. 1).
Figure 1.
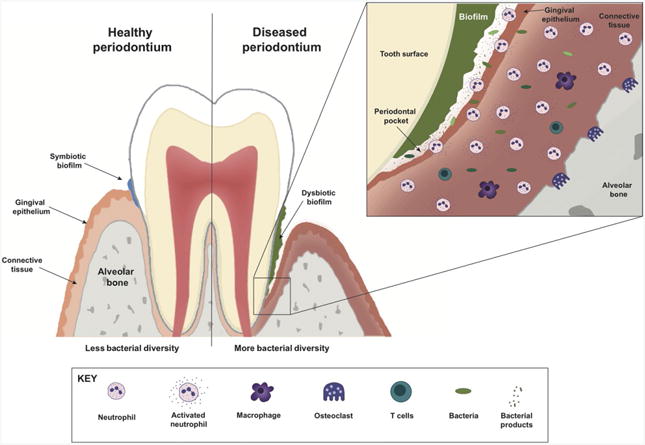
A schematic of the host response in a healthy and diseased periodontium. The local oral microbiota of a healthy periodontal site (left) is characterized by low bacterial diversity and quantity. In this homeostatic scenario, the symbiotic biofilm enhances the recruitment of neutrophils which mount a controlled acute inflammatory response. However, in the case of periodontitis (right) genetic predisposing conditions of the host, and/or environmental risk factors favor the colonization of exogenous pathogens, which results in an increase in bacterial diversity, formation and overgrowth of a dysbiotic bacterial biofilm, with massive recruitment and neutrophil infiltration invading the gingival epithelium, and the crevicular fluid. The result of this chronic inflammation involves both the innate and adaptive immune cells which results in periodontal lesions (right, and inset) with connective tissue and alveolar bone destruction
2 | Neutrophils and Oral Health: The Result of a Harmonious Coexistence
In the oral cavity, the sentinel traffic of neutrophils through the junctional epithelium into the gingival space is enhanced by the peaceful partnership established with the indigenous symbiotic oral community.15 Animal studies using germ-free mice (GF) demonstrate neutrophils patrolling the junctional epithelium; however, the number of cells is significantly enhanced by colonization with commensal bacteria.15,16 Neutrophil migration to this area is facilitated by the high porosity of the junctional epithelium and the IL-8 chemotactic gradient generated locally that guides the high number of neutrophils from the blood vessels toward the crevicular fluid. Here, neutrophils will form a protective wall between the oral community colonizing the tooth and the junctional epithelium6,7,9 (Fig. 2A).
Figure 2.
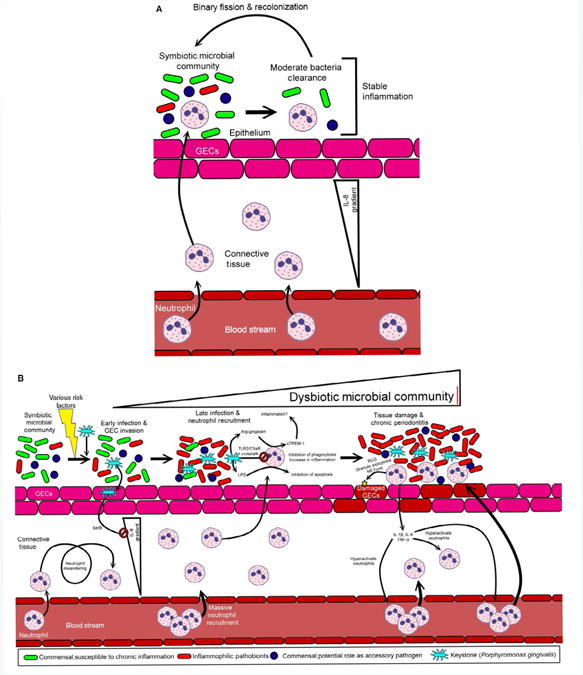
Neutrophil response in periodontal health and disease. (A) A symbiotic microbial community adheres to the gingival epithelial cells (GECs). As the bacterial burden increases, neutrophils regularly exit the blood stream entering the connective tissue layer beneath the epithelium. Some of these neutrophils will transverse the epithelium to kill some of the associated microbes—reducing the bacteria concentration. This process occurs without inflammation or tissue damage. (B) Following environmental stresses (smoking, poor diet, injury etc.) the keystone pathogen, Porphyromonas gingivalis, can colonize the symbiotic microbial community. Following colonization, the GECs are invaded by P. gingivalis which shuts down the IL-8 production via SerB resulting in local chemokine paralysis. Neutrophils enter the connective tissue, but are not initially directed to the infection. This causes many neutrophils to accumulate in the connective tissue. As some neutrophils traverse the epithelium in late infection, their killing mechanisms are hindered by multiple mechanisms including TLR2/C5aR crosstalk. Lipopolysaccharide (LPS) from P. gingivalis inhibits apoptosis of neutrophils leading to excessive inflammation, and P. gingivalis' gingipains may increase inflammation by accumulation of sTREM-1. Neutrophil degranulation, reactive oxygen species (ROS) production and NETosis damage GECs resulting in cytokine (TNF-α, IL-6, and IL-1β) secretion by the GECs and the activated neutrophils. This further activates circulating neutrophils and results in large neutrophil chemotaxis to the periodontal plaque. A cycle of tissue damage followed by neutrophil activation/recruitment followed by more tissue damage ensues. Inflammophilic pathobionts take advantage of the inflammation while susceptible commensals are eradicated
Nonetheless, microbiota can also influence other aspects of neutrophil functions. It is now recognized that the presence of the resident microbiota also contributes to the steady state of neutrophil production since studies in GF mice reveal a significant decrease in neutrophil numbers.17 The encounters that neutrophils have with the metabolites or byproducts that are generated by the local microbial community, such as short-chain fatty acids, are important in maintaining both systemic and tissue immunity.2,18,19 Moreover, the microbiota contributes to the active phenotype of aged neutrophils in circulation and participate in their removal which provides some novel insight into why these cells have such a short life span in circulation.20
In periodontal health, the interaction between neutrophils and the indigenous, or commensal, symbiotic microbial community is tightly controlled to prevent tissue damage. Studies comparing GF mice and specific-pathogen free (SPF) mice reveal that the absence of the oral commensal microbiota has no impact on the structure of the gingival tissue; this is in stark contrast to the significant role that the gut commensal microbiota has on the structural formation of the intestinal tissue.16 As shown in knockout mice, the chemotactic receptor CXCR2 facilitates neutrophil recruitment to the periodontal tissue. This was found since neutrophils are absent in the junctional epithelium of CXCR2 knockout mice while the cells are present in the blood vessels. Interestingly, both CXCL1 and CXCL2, ligands of the CXCR2 receptor, are expressed in the junctional epithelium of GF mice; however, only a significant increase on CXCL2 expression levels, not of CXCL1, is observed in the epithelium of SPF mice when compared to the GF. The presence of the indigenous oral bacterial community enhanced neutrophil recruitment to the periodontal tissue by selectively stimulating the expression of the potent neutrophil chemokine CXCL2.16 In a recent study, Greer et al.21 compare the expression levels of CXCL2 and neutrophil location in different areas of the junctional epithelium across the tooth, among GF, SPF and GF mice which were gavaged with single commensal oral bacteria like Streptococcus sp. or Lactobacillus sp.21 Neutrophils show a similar location pattern in the periodontium of the SPF mice and bacteria-gavaged GF mice, but this is distinct from the pattern seen in GF mice. Moreover, a positive correlation exists between the neutrophil location and the expression levels of CXCL2 in the junctional epithelium of bacteria-colonized animals. In the presence of the indigenous oral bacterial community, both the numbers of neutrophils as well as the levels of the CXCL2 chemokine are enhanced in the interdental region of the periodontal tissue. This provides novel and valuable information about how the commensal organisms modulate the innate immune response to maintain homeostasis in the oral cavity.21
From both mice and human studies, the protective role that neutrophils play in preservation of the oral health is well established, since low neutrophil counts as well as deficiency in neutrophil functional responses have been associated with the clinical manifestation of periodontal disease.13,22 Neutrophils are very efficient phagocytic cells and have a vast and diverse arsenal of antimicrobial resources to cope with pathogens which will fight back with their own very sophisticated artillery as well.23,24 An important and efficient antimicrobial mechanism against bacteria challenge is the respiratory burst response, with high consumption of oxygen, that results in the production of reactive oxygen species (ROS) through the activation of the NADPH oxidase complex.23 A rare genetic disorder known as chronic granulomatous disease (CGD) characterized by mutations in the NADPH oxidase which results in neutrophils defective in mounting a respiratory burst response, prones these patents early in life to recurrent infections.25,26 CGD patents can present with higher bacteria colonization and gingivitis compared to normal healthy controls; however, these patents do not manifest with severe periodontitis.6,13,27
First, it has been suggested that the inability of CGD neutrophils to mount a robust respiratory burst response might result in less tissue damage which could explain why these patents are less susceptible to periodontitis.13 In addition, emerging evidence has emphasized the importance of salivary effector functions in the preservation of a homeostatic environment, especially in the context of CGD. Not only does saliva diligently control the number of bacteria that accumulates on the tooth surface via several components with antimicrobial activity, but in response to saliva, neutrophils from healthy and CGD patents can form neutrophil extracellular traps (NETs).28 Besides the respiratory burst response and granule release, NET formation is another host-defense mechanism that neutrophils can deploy to detain and/or kill pathogens. During NET formation, neutrophils release their nuclear content to form a net of decondensed chromatin that is decorated with antimicrobial granule proteins.29,30
It is important to note that not all NETs are created equal; since their initial discovery in 2004 which describes that neutrophil stimulation with phorbol myristate acetate (PMA) will induce NET formation after 2–3 hours, ultimately resulting in a new mechanism of cell death known as NETosis,29 recent publications have unveiled the finesse and versatility that can be achieved by this neutrophil response which appears to be more tailored than initially believed. Depending on the stimuli that neutrophils encounter, NET formation can be dependent or independent of the activation of the NADPH oxidase and ROS generation.31 In addition, neutrophils can customize the timing and outcome of NETosis. In response to viable bacteria and fungi, neutrophils undergo vital NETosis where NETs form very quickly (in less than 30 minutes) and the cell remains viable; contrary to PMA-induced NETosis.31 In the recent study by Mohanty et al.28, yet another mechanism for NET formation is described. Saliva-induced NETs, triggered by the interaction between sialyl Lewisx and L-selectin, had strong antimicrobial activity against oral bacteria. Notably, saliva-induced NETs form independently of the NADPH oxidase, are resistant to DNAses. Although they did not contain elastase,28 they did incorporate other neutrophil antimicrobial components like calgranulin and hCAP-18 that have known antimicrobial activities against oral bacteria.6 In conclusion, CGD neutrophils' saliva-induced NETs with antimicrobial activity toward the oral bacteria could be an important mechanism of how neutrophils maintain homeostasis in the oral mucosa of these patents despite lacking a successful respiratory burst.
Ultimately, neutrophils will release their chromatin DNA to form NETs in an attempt to control the normal symbiotic oral microbiota and maintain homeostasis.28 However, this beneficial response can be impaired by systemic and local diseases. The saliva from patents with Behçet disease or recurrent aphthous stomatitis, who present with ulcers on their oral mucosa but possess neutrophils with normal functional responses and do not suffer from neutropenia, do not have the ability to induce NETs. Furthermore, the saliva from recurrent aphthous stomatitis individuals collected during periods of no mouth ulcers are able to induce NETs; however, that antimicrobial ability is significantly reduced or absent in saliva collected during peak periods of aphthous ulcers.28 One possible explanation for this phenomenon is that the oral microbial community present in recurrent aphthous stomatitis patents is different from the one present in age and gender-matched controls.32 Nevertheless, at this point, we cannot answer what is the root cause of the change in the oral microbial community in these individuals. Does the mucosal lesion trigger the microbial change? Or, is it the modified microbial community what triggers the ulcers? In these patents, the saliva collected 24 hours before the ulceration event fail to induce NETs due to loss of sialyl Lewis28; a plausible explanation could be that the modified oral bacterial community present in the ulcer phase is releasing higher amounts of sialidases which manifests as a loss of the protective mucins in the saliva during those ulcerous periods. Yet, it is unknown whether those oral bacteria, the producers of sialidases, are absent or in lower abundance during the periods of healthy oral mucosa. More research is needed to answer some of the above questions, but the answers will be critical to provide fundamental information needed to comprehend the interplay between human neutrophils and the symbiotic oral microbiota.
In addition to NET formation and the respiratory burst response, neutrophils have another potent antimicrobial strategy to combat pathogens.33 These versatile innate immune cells are members of the granulocyte family of leukocytes, and as such have different granules which are involved in bacterial killing both intracellular and extracellularly, as well as during each step of neutrophils journey from the blood vessels to the infected and/or inflamed tissue.23,34 Neutrophils have four different types of granules, which can be differentiated based on their protein content and on their functional response. They can either be recruited to the bacteria-containing phagosome or stimulated to undergo exocytosis and release their matrix content to the extracellular environment.35 Neutrophil granules are mobilized in a hierarchical way in which each granule requires increasingly potent stimulation to induce its exocytosis. Secretory vesicles are the easiest granule to undergo exocytosis, followed by gelatinase granules, then specific granules, and finally azurophil granules which will require a very strong stimuli to induce its mobilization.24 The diverse repertoire of proteins and receptors present at the membrane of each granule subtype as well as within the granule lumen highlights the important role each granule plays in the different neutrophil responses.36–38 The azurophil granule, also called peroxidase positive due to the presence of myeloperoxidase (MPO) on its lumen, possesses most of the ‘heavy artllery’ to combat microorganisms. The α-defensins, the different serine proteases like neutrophil elastase (NE), proteinase 3 (PR3), cathepsin G, and neutrophil serine protease 4 (NSP4) are stored in this granule subtype.35,39
Papillon-Lefèvre syndrome (PLS), is a rare genetic disorder caused by a defect on the gene that encodes for the cysteine protease cathepsin C (CTSC). Clinically, it manifest as hyperkeratosis and severe periodontitis early on in the lives of affected individuals.10,22 This is partially because the affected protein, CTSC, is an important enzyme that cleaves the N-terminal on the different NSPs, a key step that is required for the activation and proper biological function of the NSPs.40,41 Thus, neutrophils from PLS patents lack all the serine proteases normally stored in azurophil granules; which will compromise some of the neutrophil antimicrobial activity, but ultimately does not result in more susceptibility to bacterial infection besides the severe periodontitis.40 This raises the question, why is bacterial susceptibility limited to the periodontium of those affected by PLS? It has been suggested that one of the causes is the minimal activity of PR3, which is one of the four serine proteases present in neutrophil azurophil granules. PR3 is an indispensable serine protease that cleaves hCAP18 to generate an important host-defense peptide LL-37, which has a strong antimicrobial activity toward oral bacterium such as Aggregatibacter actinomycetemcomitans,42 and also toward the keystone periodontal pathogen P. gingivalis.43 High levels of hCAP18, the precursor of LL-37, are detected in the crevicular fluids of PLS patents but not the mature antimicrobial peptide.44 Although, the absence of the NSPs and of LL-37 in PLS patents is correlated with the severity of periodontal disease, the disease progresses because of several factors.43–45 When neutrophils from PLS patents are challenged ex vivo with PMA, or opsonized S. aureus, or the heat-killed oral pathogen A. actnomycetemcomitans significantly higher levels of pro-inflammatory cytokines, as well as of matrix metalloprotease-9, are detected in comparison to healthy controls.46 Also, PLS neutrophils show a higher respiratory burst response upon PMA stimulation compared to control healthy donors, with deficient chemotactic activity toward both MIP-1α and the formylated bacterial peptide formyl- methionyl-leucyl-phenylalanine (fMLF).46 This led Roberts et al.46 to propose that the hyperactive neutrophil phenotype present in PLS patents, together with the deficient chemotactic ability, will halt neutrophils in the connective tissue and contribute to gingival tissue damage and promote inflammation—both hallmarks of periodontal disease.
CGD and PLS are often compared as examples of rare genetic diseases affecting different neutrophil antimicrobial defense mechanisms but with opposite manifestations in relation to periodontal disease.13,47 Despite the fact that both conditions share suboptimal neutrophil responses, CGD patents have no predisposition to periodontal disease, whereas PLS patents manifest severe periodontitis. Neutrophils from both CGD and PLS patents will induce NET formation when cells are exposed to saliva.28 This provides an important, basal antimicrobial defense response against symbiotic microbiota that ultimately helps maintain the homeostatic environment in the oral mucosa. When the conditions in the oral cavity change from symbiotic to a dysbiotic microbiota, the hyperactive neutrophil phenotype described in PLS patents in combination with the absence of serine proteases, lack of an active LL-37, and an inefficient chemotactic capacity prolongs their stay in the connective tissue contributing to tissue destruction and progressing to severe periodontitis.46 In contrast, neutrophils from CGD patents have impaired ROS generation, but intact oxygen-independent antimicrobial responses and normal chemotactic response, allowing them to mount a more effective fight toward the dysbiotic oral microbiota and cause less tissue destruction.
Overall, several host-derived mediators contribute to a healthy periodontium. The specialized junctional epithelium participates in the release of antimicrobial peptides, like β-defensins and LL-37, which helps to maintain less diverse bacterial community that has been associated with a healthy periodontium. Bacterial presence constantly stimulates the release of IL-8 from the gingival epithelium, which generates a chemotactic gradient that continually guides neutrophils to the gingival sulcus (Fig. 2A). In addition, the local oral symbiotic microbial community induces the production of IL-1β,48 which can prime the immune system and generates a state of mild inflammation which is linked to promoting immune fitness.2,49
3 | Neutrophils and Periodontal Disease: The Result of a Discordant Coexistence
3.1 | Periodontitis: a chronic, dysbiotic, polymicrobial inflammatory disease
It is estimated that approximately 46% of the adult population in the United States who are at age 30 or older suffer from periodontitis with Hispanics showing the highest prevalence when compared to non-Hispanics individuals.50 Periodontitis is a complex chronic inflammatory disease of the periodontium, the tissue that surrounds and supports the teeth. If left unattended, periodontitis results in an increase rate of bone resorption around the tooth root area, with consequential loss of the tooth.9,51 Treatment for this chronic inflammatory disease is targeted to remove the dysbiotic biofilm and includes scaling, root planning and curettage (to remove the biofilm formed in the periodontal pocket), a regimen of broad spectrum antibiotics and surgery in severe cases. Unfortunately, these treatments are only a temporary solution to control and prevent the disease progression into a massive destruction of the periodontium leading to bone loss (Fig. 1, inset). Moreover, periodontal disease and periodontal pathogens have also been linked to increased risk for serious systemic conditions such as atherosclerosis, diabetes, and rheumatoid arthritis.9
In the oral cavity, the tooth surface offers a suitable ecological niche for bacteria colonization and biofilm formation resulting in a varied polymicrobial community.51 Specifically, the diverse environment present in the oral cavity allows the microbial communities to find the best ecological niche that fits their growth requirements, resulting in the formation of unique microbial biofilm communities associated to the oral location.52 Yet until recently, research into periodontal disease has focused primarily on a relatively small group of putative pathogens including P. gingivalis, Tannerella forsythia, and Treponema denticola.11,53 However, a reappraisal of microbial etiology has marked a turning point in the field due to the development of culture-independent techniques that allow oral biologists to identify bacterial species directly from nucleic acids. Due to this, several newly recognized/appreciated organisms have been found to be elevated in periodontal disease as compared to health.54–56 Aas et al.57 show that a diverse and complex bacterial community is part of the healthy oral cavity after characterizing the bacterial profile of five clinically healthy individuals at nine different oral sites including the buccal epithelium, the maxillary anterior vestibule, the dorsum and lateral tongue surface, the hard and soft palate, the tonsils, the tooth surfaces, and the subgingival plaque. Another report using 16S pyrosequencing and a higher number of patents (29 healthy controls and 29 individuals with chronic periodontitis) shows a differential bacterial community composition between healthy controls and disease patents with 100-300 bacterial species detected in a single individual.58 Some of the newly identified uncultivated bacterial species are clearly associated with disease such as Filifactor alocis,59 and some of the health-associated bacterial species are still present in periodontitis patents, however in lower numbers.58 The use of next-generation sequencing techniques provides sophisticated tools to achieve a more precise characterization of the complex composition of the inhabitants of the oral microbiome. Similarly, sequencing enables the identification of low abundance microorganisms and helps to understand their role in the progression from oral health to disease.58,60
Our general idea is that infectious diseases are caused or triggered by the action of a single foreign pathogen; however, it is clear that many human diseases with an infectious etiology, like periodontitis, are originated by the complex association and interaction of a diverse polymicrobial community.4,11,61,62 The initial paradigm of periodontits, established more than 20 years ago already contemplated the idea that the disease was not originated by a single pathogen and explained the progression and severity of the disease by a shift from a predominant oral gram-positive aerobic microbial community toward a gram-negative anaerobic community.11,63 In addition, the data obtained from oral biofilm studies using checkerboard DNA-DNA techniques link the different stages of the disease to a specific bacterial group or complex with the presence of the triad of bacteria composed by P. gingivalis, T. forsythia and T. denticola strongly associated with increase severity of periodontitis.64,65 The advance of new technology has revealed that the etiology of periodontitis is more complex than the initial paradigm. For example, high numbers of the gram-positive bacterium F. alocis are present in periodontal disease sites, whereas increased levels of the gram-negative uncultivated bacterium Veillonella sp. oral clone X042 are associated with healthy periodontal sites and classified as a beneficial bacterial species.66 Based on the current evidence, the development of periodontitis is associated with a shift in the symbiotic oral microbiota into a dysbiotic polymicrobial community.11,53,61,67 In a healthy gingival tissue, the local symbiotic bacterial community is less diverse and rich, with neutrophil recruitiment to clear the infection and resolving the inflammation with no collateral damage to the host4,61,68 (Fig. 2A). The progression from health to periodontitis is now explained as the transition from a symbiotic microbial community that due to several risks factors, such as smoking, changes in the diet, an immunocompromised host, tissue injury, or the colonization of the oral cavity by pathogenic bacteria such as the oral keystone pathogen P. gingivalis, can modify the oral ecosystem resulting in a dysbiotic polymicrobial community61 (Fig. 2B). As a result, the host response toward the highly rich and diverse dysbiotic microbial challenge is more robust and dysregulated, transitioning from a controlled/stable immune response into a non-resolving chronic inflammatory response4,61,68 (Fig. 2B).
The stability of the local oral microbial community reveals important information to understand the transition from health to disease. Over a 2-year period, 75% of the subgingival oral bacterial community was conserved for individuals grouped under a healthy stable periodontal status; however, a significant change to that local or indigenous bacterial community with loss or gain of bacterial species was seen in patents whose periodontal status worsen.66 However, none of those subjects who showed an increase in periodontitis severity within the 2 years period had a complete replacement of their bacterial community.66 It is clear that modifications in the bacteria-bacteria relationship, resulting in differential expression of bacterial genes or in the composition of the oral community associated with periodontal health, will lead to disease.69 The shift in the community can be explained by the combined effort of bacteria acting as ‘accessory pathogens’ helping ‘keystone pathogens’ to manipulate both the bacterial and host interactions providing an optimal niche for ‘pathobionts’ to flourish and outgrow in the new ecological system61,70 (Fig. 2B). The new paradigm to describe the etiology of periodontitis introduces a new concept of ‘nososymbiocity’ in which some members of the local or indigenous microbial community can participate and/or facilitate the establishment of exogenous keystone bacterial members leading to a synergistic dysbiotic community (reviewed in 61).
3.2 How human neutrophils cope with the ‘nososymbiocity’ of periodontitis
In the periodontal tissue, neutrophils are mainly found in the junctional epithelium and in the crevicular fluids where it has been described that neutrophils can form a protective ‘wall’ facing the oral plaque and protecting the epithelium.7 A study done almost 30 years ago demonstrated that different ultrastructural neutrophil morphology was seen in cells isolated from the crevicular fluid of patents with severe periodontitis.71 At that time, the transmission electron micrographs reveal neutrophils with no evidence of undergoing phagocytosis, cells with high number of phagocytic vacuoles, and cells with ‘ghost’ phagocytic vacuoles. Both cells which contain phagocytic vacuoles show less granule content overall.71 Moreover, high content of immunoglobulins, predominantly IgG, and complement component 3 is found in the crevicular fluid, forming immune complexes detectable inside neutrophils.71 This study indicates that not only the phagocytic clearance of the oral bacteria can induce neutrophil activation, but also the presence of immune complexes in the crevicular fluid is a potent cell activator. Furthermore, analysis of neutrophils collected from the periodontal pockets from 11 adult patents with periodontitis, without periodontal treatment for a year and with evidence of alveolar bone loss shows that necrosis, and not apoptosis, is the predominant form of cell death, which contributes to the tissue damage and prolongs the inflammation seen in periodontitis.72 A more recent study, which analyzed gingival biopsies and purulent crevicular exudates from patents with chronic periodontitis, reveals that bacteria in the subgingival plaque is constantly dispersed as a way to colonize new surfaces, and that the abundance of NETs in the crevicular exudates is more effective than phagocytosis in preventing bacteria adhesion and invasion of the gingival epithelium.73
To protect against the collateral damage that neutrophil antimicrobial serine proteases such as NE and PR3 may cause the host tissue, endogenous protease inhibitors are present in the gingival tissue such as secretory leukocyte protease inhibitor (SLPI) and elafin. Analysis of crevicular fluids from chronic periodontitis patents reveals significantly lower levels of SLPI with concomitant higher activity of both NE and PR3 activity compared to periodontal healthy controls.74 Moreover, the keystone periodontal pathogen, P. gingivalis, has several virulence factors that tip the balance in favor of dysbiosis and host tissue damage. An exemplary type of virulence factor is the secreted cysteine proteolytic enzymes called gingipains.75 One of the many properties that makes P. gingivalis gingipains so successful is the ability to inactivate the endogenous protease inhibitors.76 Crevicular fluids from periodontitis patents have a positive correlation of high NE and PR3 activity with the presence of P. gingivalis and a negative correlation with the levels of SLPI.74 Furthermore, in crevicular fluids collected from periodontitis patents, the levels of elafin, which is normally anchored to the extracellular matrix to protect it from degradation and to assure the enzyme activity to guard the host tissue integrity, show a positive correlation with P. gingivalis arginine-specific gingipain, Rgps.74 These findings suggest that the sheddase activity of P. gingivalis gingipains on elafin will compromise the protective function of the inhibitor and leave the host tissue more susceptible to degradation by neutrophil active proteases present in the gingival tissue.
Since neutrophils are equipped with a lethal arsenal of antimicrobial compounds, their response to control infection is finely tuned to diminish collateral damage to the host tissue. It would be very detrimental to the host to have circulating professional phagocytes, like neutrophils, which will respond and become fully activated independent of what type of enemy or threat they have to contain and control. Hence, neutrophils can fine tune the degree of response they mount upon a given dangerous situation. The level of cell activation is the result of a range of increasing degree of activation states instead of an on/of switch mechanism, with priming being a reversible step that modulates neutrophils graded response to an insult. Priming of neutrophils is understood as the enhanced functional response of the cell to a stimulus by previous exposure of the cell to a priming agent. Priming agents can be host-derived, like different cytokines or chemokines, such as TNFα, IL-1β, IFNα, IL-8, growth factors like GM-CSF, or bacterial-derived compounds like lipopolysaccharide (LPS).77 The regulation of priming is key in the host-defense response since it prepares neutrophils to mount an appropriate level of response when encountering a particular stimulus. Exposure of neutrophils to priming agents like TNFα will enhance not only the cells respiratory burst response, but also the release of neutrophil granules upon a subsequent stimulation. Neutrophil granule exocytosis contributes to TNFα priming of the respiratory burst response by increasing the expression of the NADPH oxidase membrane components at the neutrophil plasma membrane.78
Several studies have shown that circulating neutrophils isolated from patents diagnosed with community acquired pneumonia, or who suffered from trauma, or from a chronic inflammatory disease like rheumatoid arthritis have a primed phenotype and can mount an enhanced respiratory burst response upon exposure to a second stimulus.79–81 In neutrophils, the respiratory burst response induced by different stimuli can trigger the assembly of the NADPH oxidase complex at the phagosomal membrane or at the cell plasma membrane, and, upon enzyme activation, superoxide radicals are released either to the phagosomal lumen or outside the cell into the extracellular space. Neutrophils isolated from peripheral blood of chronic periodontts patents, have higher basal respiratory burst activity when compared to healthy matched individuals. Patent neutrophils present a primed phenotype determined by a significantly higher extracellular ROS production when challenged ex vivo with opsonized S. aureus, or non-opsonized oral pathogens such as Fusobacterium nucleatum or P. gingivalis.82 F. nucleatum is a gram-negative oral bacterium that acts as an important ‘accessory pathogen’ involved in the progression from a symbiotic to a dysbiotic oral community.83–85 A transcriptome analysis of human neutrophils challenged with F. nucleatum for 3 hours resulted in the differential regulation of 208 genes including some genes involved in the respiratory burst response as well as upregulation of ‘stress-response’ genes HSPA1A, HSPH1, and HSPA8, along with oxidative stress response genes like superoxide dismutase 2 and metallothioneins, which could be a cell protective mechanism to counterbalance the high ROS generation by the bacterial challenge.85 Another study shows that peripheral blood neutrophils isolated from patients with severe chronic periodontitis when challenged with IgG-opsonized S. aureus, or with complement opsonized zymosan or with a soluble stimulus like PMA have a 32% higher extracellular ROS production, measured by isoluminol, compared to healthy age and gender-matched individuals.86
The enhanced respiratory burst response by peripheral blood neutrophils isolated from periodontal disease patents when exposed to a second stimulation like S. aureus or PMA is an indication that those circulating cells are primed. High plasma levels of IL-8, GM-CSF, and IFNα are detected in periodontitis patents compared to age-matched healthy individuals. Both IL-8 and GM-CSF are responsible for the enhanced baseline release of ROS generation upon challenge of neutrophils from healthy individuals with the plasma collected from those periodontitis patents.87 Furthermore, when healthy neutrophils are incubated with plasma from periodontitis patents, the elevated levels of IFNα contribute to both the enhanced basal respiratory burst response and the priming of fMFL-stimulated superoxide release.87,88
Cytokine release by peripheral blood neutrophils isolated from healthy individuals, or from patents with chronic periodontitis before and after a successful non-surgical periodontal treatment, were determined after 18 hours of no challenge or cells challenged with Escherichia coli LPS, or opsonized S. aureus, or F. nucleatum, or with P. gingivalis. The release of IL-8, IL-6, TNFα, and IL-1β from unstimulated cells after 18 hours is similar between all the groups; however, a significantly higher release of all cytokines is seen by patent neutrophils upon the different bacterial challenge when compared to the response of neutrophils from healthy controls.89 There are no differences in the bacteria-stimulated cytokine response by neutrophils isolated from postperiodontal therapy patents compared to the levels detected previous to the periodontal therapy, which might indicate that the priming state of the circulating cells is still present after periodontal therapy or that neutrophils hyper-reactivity is imprinted or constitutive.89
Some periodontitis patents who do not respond to periodontal treatments, both performed at the dentist office and after a good home care, are classified as refractory periodontitis patents.90 The respiratory burst response was analyzed from oral neutrophils isolated from 13 patents with refractory periodontitis upon PMA stimulation and two groups are distinguished within the cohort, one group as high and another as low responders based on the percent of ROS production. The refractory periodontitis patents within the cohort classified as high responders also present more severe bone loss than the low responders; however, the bacterial plaque index and the age are not significantly different between these two groups in this patent cohort. The magnitude of ROS generation by the oral neutrophils is correlated with the disease severity and larger longitudinal studies might confirm if a hyperactive oral neutrophil phenotype could predict if patents would be refractory to treatment and more susceptible to disease progression.90
Significantly higher number of neutrophils are recruited into the oral cavity of chronic periodontitis patents when compared to healthy individuals, and a transcriptome analysis reveals significant changes in gene expression between the two groups.91 Oral neutrophils from chronic periodontitis patents show increased expression of Bcl-2, a pro-survival marker, and a decreased expression of Bax, a pro-apoptotc marker, both by transcriptome analysis and validation by immunoblot and flow cytometry compared to oral cells isolated from healthy controls.91 Furthermore, oral neutrophils from chronic periodontts patents have a differential enhanced surface expression of CD11b, CD63, CD66b, which is associated with an active neutrophil phenotype.92 All three surface markers are present in different neutrophil granule subtypes, and indicate that oral neutrophils are undergoing active granule exocytosis. The exocytosis of azurophil granule content into the extracellular space is tightly regulated to prevent tissue damage. The significant increase of CD63 surface expression, an azurophil granule marker, observed in oral neutrophils, not on peripheral blood neutrophils, from chronic periodontitis patents highlights the magnitude of cell activation and the contribution of it to inflict tissue damage in sites of chronic unresolved inflammation such as what occurs in the disease periodontium. Moreover, peripheral blood neutrophils from chronic periodontitis patents display reduced chemotactic ability toward IL-8 and fMLF when compared to cells isolated from age and gender-matched control individuals. Blood neutrophils isolated from those chronic periodontitis patents 3 months after non-surgical therapy show partial recovery with similar velocity and accuracy migration toward IL-8 but not to fMFL93 The reduced chemotactic ability paired with the hyperactive phenotype described for both circulating as well as oral neutrophils from chronic periodontitis patents further supports the role that these dysfunctional innate immune cells might have in collateral tissue damage and periodontal disease progression. The presence of primed neutrophils both in circulation and in the oral cavity described in periodontitis patents might shed some light to understand the proposed association between periodontal disease as well as periodontal pathogens with systemic inflammatory conditions such as atherosclerosis, diabetes, and rheumatoid arthritis.
3.3 | Neutrophil interaction with the terrible threes of periodontitis: the unresolved contest
3.3.1 | The first kid of the periodontal triad
T. denticola is an anaerobic, gram-negative motile spirochete that can be detected, albeit in low numbers, in the gingival plaque of healthy individuals; however, it is present in very high numbers in the subgingival periodontal pocket and is considered one of the major microorganisms associated with the dysbiotic biofilm formation in periodontal lesions.84 Moreover, T. denticola may contribute to the bacterial colonization of periodontal tissues by limiting neutrophil chemotaxis.94,95 IL-8 secretion by periodontal epithelial cells is an important mechanism by which neutrophils are recruited to the periodontal pockets, and epithelial cells fail to secrete IL-8 as a response to T. denticola exposure.94 Additionally, this pathogen is able to degrade IL-8 that is already present at the infection site, which disables the neutrophil chemotactic gradient. Combined with the impaired neutrophil chemotactic phenotype described for periodontitis patents, this discovery might help to understand the described neutrophil meandering and halt in the gingival tissue associated with the collateral tissue damage.15
Other studies suggest that different virulence factors of T. dent-cola may modulate key innate immune responses, contributing to the development of periodontal diseases.95–97
Several studies agree that the major outer sheath protein (Msp) of T. denticola is one of its most important virulence factors in contributing to the disease progression. This membrane protein modulates neutrophil signaling pathways involved in cytoskeletal dynamics that are relevant in chemotaxis and phagocytosis.98–100 Magalhães et al.100 determined that pretreatment with Msp from T. denticola has an inhibitory effect on murine neutrophil movement and polarity even when they were exposed to the potent chemoatractant fMLF in vivo. Similarly, Msp pretreatment impairs human neutrophil chemotaxis toward fMLF and phagocytosis of immunoglobulin G-coated polystyrene microspheres in vitro, by perturbing actin remodeling and reorganization.97 Puthengady et al.97 observed that pretreatment with T. denticola's Msp reduces transient cytosolic calcium concentrations in neutrophils. The presence of calcium is fundamental during the actin filament modifications that lead to cell motility; thus, by reducing cytosolic calcium, the bacteria can disrupt neutrophil movement.97
Another way Msp controls neutrophil cytoskeletal functions like migration, adhesion, and cell shape is by preventing the activation of Rac1, a small G protein from the Rho-GTPases family. Rac1 is critical for the formation of lamellipodia and the modulation of the phosphat-dylinositol-3 kinase in neutrophils, required for adequate cell polarization and chemotaxis.100,101 T. denticola Msp Rac1 inhibition is selective as indicated by an experiment in which under the same conditions the activation of other small G proteins, such as Rac2, Cdc42, and RhoA, is not affected by the Msp challenge.100 These data further support the concept that the disruption of cytoskeletal dynamics may serve as this pathogen's evasion mechanism.100 Lastly, T. denticola Msp also causes extracellular matrix degradation by stimulating the release of activated MMPs from neutrophils, an effect that might lead to periodontal tissue damage.96,102
Dentilisin, a surface lipoprotein-protease complex of T. denticola, is another important virulence factor of this periopathogen.102 Yamazaki et al.103 found that dentilisin activates the alternative complement pathways by hydrolyzing host proteins, such as complement component 3 (C3), and generate iC3b, which can bind to complement receptor 3 present on neutrophils' plasma membrane and result in cell activation. Challenge of human neutrophils with T. denticola, but not with the dentilisin-deficient mutant, results in ROS generation and release of MMP-9.103 Induction of neutrophils respiratory burst response with the high generation of ROS is a potent and effective antimicrobial weapon; however, despite the fact that T. denticola is recognized as a strict anaerobe, this bacterium is able to survive in the presence of ROS generated by the host.96 Treatment of T. denticola with the protease inhibitor chymostatin prevented complement activation, the bacteria-induced ROS generation and MMP-9 release by human neutrophils. T. denticola dentilisin-induced neutrophil activation contributes to periodontal tissue damage and the chronic inflammation of the periodontium.103
3.3.2 | The second kid of the periodontal triad
Tannerella forsythia is a gram-negative anaerobic rod that, together with P. gingivalis and T. denticola, form the triad of putative periodontal pathogens strongly associated with the severity of the disease. The presence of T. forsythia in the oral biofilm is considered a marker of periodontal disease progression, since it is strongly associated with the advanced and refractory forms of periodontitis and it is rarely cultured from healthy sites.104,105 Yet, despite being one of the main constituents in the pathogenesis of periodontal diseases, T. forsythia and its interaction with the immune response is arguably under-studied, probably due to its unique growth requirements.106–108
Gosling et al.106 showed that after subcutaneous immunization of mice with T. forsythia, lesions consist mainly of infiltrated neutrophils, granulomatous tissue, and peroxidase positive granules associated with neutrophil exocytosis. Neutrophil recruitiment peaked at day 1 after infection and decreased by day 7. These results suggest that unlike the other two putative periodontal pathogens, T. forsythia does not suppress neutrophil recruitiment to the infected site.106
The complement system is another key innate immune response for host immune protection from microorganisms.109 Activation of any of the three complement pathways will result in deposition of an active complement component C3b on bacterial surfaces leading to opsonization of the microorganism which facilitates its recognition by professional phagocytes like neutrophils. Besides contributing to a more efficient bacterial recognition, complement activation at terminal stages results in the formation of membrane attack complexes and the lysis of microbes.109 T. forsythia utilizes the metallopreoteinase virulence factors karilysin and mirolysin to modulate and evade the complement arm of the innate immune response. Incubation of T. forsythia with human serum proteins leads to the activation of only one of the three complement pathways, resulting in some opsonization of the bacteria.104,110
Jusko et al.104 showed that T. forsythia inhibits the classical and lectin pathways by degrading the initial complement components, such as the mannose-biding lectin, ficolin, and C4, whereas the classical pathway is inactivated at a later stage by degrading C5 molecules. Therefore, T. forsythia avoids complement-mediated killing by neutralizing the activated classical pathway, which ultimately allows the bacteria to remain viable after human complement exposure. Interestingly, the interaction of the bacterial virulence metalloproteinases, karilysin and mirolysin, with the host complement pathways still promotes the release of active C5a, an anaphylotoxin that forms chemotactic gradients to recruit neutrophils to the site of infection. C5a stimulates neutrophil chemotaxis into sites of infection, but exposure to high concentrations of C5a causes neutrophil activation and inefficient migration.104,110 P. gingivalis and T. forsythia are both found in the periodontal disease sites and there is a synergistic effect between both of the bacterial proteases, gingi-pains and karilysin, to prevent complement deposition.7 These two putative periodontal pathogens join forces to evade complement attack and promote high concentration of the complement anapyla-toxin C5a, which will induce neutrophil granule exocytosis and ROS which results in tissue disruption and leads to the progression of periodontal diseases.110
LL-37 is an important antimicrobial peptide secreted by neutrophils, usually present in high numbers in the gingival crevicular fluid (discussed above). It plays an important role in preventing the colonization of gingival tissue by pathogenic species.111 Incubation with purified karilysin results in LL-37 cleavage and the subsequent reduction of this peptide bactericidal activity—suggesting that this mechanism may contribute to protect not only T. forsythia but also other periodontal pathogens, such as P. gingivalis, against bacterial degradation by LL-37.111 Besides being bactericidal, LL-37 has potent ant-inflammatory activity; it binds to LPS, preventing both the stimulation of macrophages and the consequent release of pro-inflammatory mediators. Koziel et al.111 found that treatment with karilysin inactivates LL-37 capacity to neutralize LPS. Moreover, incubation with karilysin results in cleavage of the LL-37-LPS complex, causing the release of active LPS and therefore the secretion of pro-inflammatory cyto-kines such as TNF-α.111 This finding suggests that the inactivation of LL-37 by karilysin interferes with the ant-inflammatory function of this peptide, preserving local inflammation and promoting periodontal tissue damage.111
T. forsythia possesses a glycosylated S-layer on its cell surface that has been linked to bacterial resistance to innate immune clearance.106–108 In an oral gavage model, mice were infected either with a glycosylation deficient mutant or with a T. forsythia wildtype strain. Settem et al.108 found a significant increase in neutrophil recruitiment to the infected gingival space, as well as a limited Th17 response in mice inoculated with the glycosylation deficient mutant. Normally, the Th17 response is required for an efficient recruitiment of neutrophils; so, these findings suggest that the antigenic T. forsythia cell exterior favors bacterial survival in the periodontal pocket by blocking Th17-mediated neutrophil infiltration.108
The third most studied virulence factor of T. forsythia is serpins, which are serine protease inhibitors that are usually expressed in eukaryotic cells, but occasionally can also be found in prokaryotes.112 In the context of T. forsythia infection, serpins play an important role in maintaining homeostasis by regulating mechanisms like inflammation, complement activation and the inhibition of the neutrophil-derived proteases released for bacterial degradation. Specifically, the newly discovered serpin, miropin, inhibits the enzymatic activity of several neutrophil proteases present in the gingival crevicular fluid such as elastase and cathepsin G. This interference may mitigate bacterial clearance, promoting T. forsythia survival in the periodontal pocket.112
Illustrating yet another virulence factor of this periodontal pathogen to elude the innate host immune response is the finding that T. forsythia cultures contained significant levels of bacterial DNase as noted by Palmer et al.113 after finding zones of DNA hydrolysis surrounding T. forsythia colonies on plate cultures supplemented with mammalian DNA. It is known that some pathogens secrete DNase, an enzyme able to degrade nucleic acids, as a survival mechanism to escape NETs. It is tempting to speculate that T. forsythia could evade NETs antimicrobial and/or entrapping effect by use of its DNase to degrade the chromatin and of its karilysin to inactivate the antimicrobial peptide, LL-37, normally present in NETs.113,114 It is also possible that the presence of bacterial DNase can benefit T. forsythia in biofilm remodeling and contribute to bacteria dispersal and colonization of new surfaces.
3.3.3 | The third and most mischievous kid of the periodontal triad
P. gingivalis is a gram-negative anaerobic rod that has been called the ‘keystone pathogen’ in the onset of periodontitis.115 In this model, a low abundance of P. gingivalis will upset the balance between the oral microbiome and the immune system.115 Immune subversion must be acquired either through environmental stresses, diet or through the activity of P. gingivalis itself as previously discussed. The interactions between this species and the host immune system have been extensively studied.
To survive in the periodontal cavity, P. gingivalis must impair the neutrophil antimicrobial responses to prevent its killing while directing the neutrophils' many potent weapons at the host. P. gingivalis requires nutrients that can be taken from the host via the degradation of tissue.51,116 P. gingivalis is a very resourceful periodontal pathogen that makes use of enzymes, LPS and even other host cells to modulate neutrophil behavior.
As stated, P. gingivalis must be able to withstand the chronic inflammation that is characteristic of periodontal disease.116 Early studies on neutrophil-P. gingivalis interactions show neutrophils unable to effectively kill the bacteria intracellularly.117 Neutrophils isolated from periodontal disease patents demonstrate an even further reduced killing efficacy.118 Within the biofilm, P. gingivalis enjoys some camouflage from the immune system, but P. gingivalis also exists in the crevicular fluid in a planktionic or free-floating state.116 In this space, neutrophils can actively invade and phagocytose the bacteria.119 In periodontitis, recent studies have shown that the neutrophils are hyper-activated and undergo frequent NETosis in the crevicular fluid.119 Other than the crevicular fluid, the actual dental plaques are the main home for P. gingivalis.116 Neutrophils can mount a defense even against these biofilms by forming the previously mentioned ‘wall’ of neutrophils against the plaque.120 The neutrophils are highly impaired in their ability to phagocytose bacteria within the biofilm, and they turn to extracellular killing mechanisms instead.51 They undergo exocytosis of their different granule subtypes resulting in the release of antimicrobial peptides while generating ROS. The best way for P. gingivalis to avoid clearance by the neutrophils is to invade host cells such as gingival epithelial cells.121 Within these cells, P. gingivalis inhibits apoptosis and occupies its own replicative niche within autophagosomes.121,122
One of the major ways that P. gingivalis manipulates the neutrophils is through the process of local chemokine paralysis.123 Neutrophils are recruited to sites of infection by following a gradient of chemical signals generated from the resident tissue cells via a process called chemotaxis.9 In the case of periodontitis, gingival epithelial cells (GECs) secrete the chemoattractant IL-8 to recruit neutrophils.124 P. gingivalis inhibits the secretion of IL-8 from GECs with the ultimate effect of lowering the number of neutrophils that are called to the area.123 The mechanism for inhibition of IL-8 secretion is dependent on the P. gingivalis' serine phosphatase SerB following invasion of GECs by the pathogen.125 The synthesis and eventual secretion of IL-8 is mediated by the NF-κβ transcription factor.126 SerB dephosphorylates the p65 subunit of the NF-κB RelA/p65 homodimers that allow for IL-8 tran-scription–suppressing IL-8 synthesis125 (Fig. 2B).
P. gingivalis also utilizes very direct mechanisms against the neutrophil to hamper its chemotactic function.127 Bacterial products recovered from culture of P. gingivalis have an immobilizing effect on human neutrophils which may be due to the binding of non-chemotactic formylmethionyl peptides to the fMLF receptor.127,128 Interestingly, the inhibitory effect on neutrophil chemotaxis is not solely for the fMLF receptor, but C5a-mediated chemotaxis is also blocked.127,128 This could suggest two independent blocking agents or one with a more global effect on the neutrophil's chemotactic integrity.127 Succinic acid is a metabolic by-product of the Bacteriodes species under which P. gingivalis used to be classified, and it could be responsible for the previously described culture product effects.129 Succinic acid has been shown to inhibit chemotaxis and even reduce neutrophil respiratory burst by decreasing intracellular pH.129,130 Although succinic acid is produced by P. gingivalis, its major short-chain fatty acid metabolic by-product is butyric acid which has similar inhibitory effects in animal models, but has not yet been tested in human neutrophils.131
P. gingivalis' LPS also has a possible role in the control of cytokines and chemokines released.132 Inflammatory cytokine (IL-1β, TNFα, and IL-8) release from human neutrophils is significantly lower when stimulated with LPS from P. gingivalis compared to other gram-negative bacteria.132 The release of the IL-1 inhibitory molecule, IL-1ra, is significantly enhanced by neutrophils stimulated with P. gingivalis' LPS.132 Both TLR-2 and TLR-4 have been identified as possible receptors for LPS from P. gingivalis.133 Current evidence shows that LPS from P. gingivalis has an antagonistic role with TLR-4, and this may explain how the stimulation with LPS lowers the amount of inflammatory cytokines.133 There has been some evidence to the contrary showing that LPS from P. gingivalis actually increases IL-8 synthesis.134 It is possible that the IL-8 transcription is increased, but secretion is still inhibited. However, the study showing lower cytokine secretion does not show inhibition, but just less secretion than the others tested. The most likely explanation is that P. gingivalis' LPS has less affinity for the pattern recognition receptors than LPS from A. actnomycetemcomitans or E. coli. The LPS from P. gingivalis has also been implicated in bone resorption which is one of the primary symptoms of periodontal disease.135
Aside from the LPS effect on cytokine and chemokine secretion, it also delays apoptosis of differentiated HL60 cells.136 However, LPS has not yet been tested for this inhibition with isolated human neutrophils. Inhibition of apoptosis is a mechanism employed by other bacteria as well to induce further inflammation and tissue damage.137 The relationship between impairment and activation is complex because pathogens like P. gingivalis require inflammation to thrive, but they must also keep the inflammation under some control.115 If they do not impair neutrophil killing mechanisms or invoke their own defense mechanisms, then the exorbitant inflammation would result in its own clearance.
Other P. gingivalis produced products have also been shown to manipulate neutrophil function.138 These bacterial products recovered from culture lowered neutrophils ability to agglutinate which may correspond to control of granule exocytosis.138 Interestingly, there has been some evidence that P. gingivalis' products recovered from its growth culture, even alter neutrophil morphology.139 One bacterial product, trypsinlike protease (TLPase), plays a role in inhibition of phagocytosis.140 This inhibitory effect is more profound in neutrophils from periodontitis patents than healthy controls which may point to added effects from other pathogens and/or systemic alteration to the host's neutrophils.118,140 Neutrophils from periodontal disease patents are impaired in their ability to phagocytose bacteria, and TLPase from P. gingivalis may be one of the causes of this.140,141 Additionally, TLPase has been shown to increase the generation of superoxide anions and cleave the neutrophil formyl peptide receptor.142 Increasing the superoxide concentration in the extracellular environment will contribute to damage for both the host tissue and any sensitive bacteria.
Endogenous lipoxin A4 (LXA4) appears to inhibit the oxidative burst response and overall activation of human neutrophils when challenged by P. gingivalis.143 LXA4 is a eicosanoid that is heavily involved in the resolution of inflammation.144 Its presence in the crevicular fluid of periodontal disease patents and relative absence in asymptomatc patents has made it of particular interest.145 The generation of LXA4 occurs in response to the chronic inflammation found in the periodontal pocket.146,147 Further research in this area could explain how P. gingivalis takes advantage of host chemical mediators.
The Triggering Receptor Expressed on Myeloid cells 1 (TREM-1) is another endogenous factor that is believed to be manipulated by P. gingivalis.148 TREM-1 has been much more actively studied with human neutrophils. It is a membrane bound protein whose binding results in activation of the neutrophil.148 P. gingivalis affects TREM-1 in both a gingipain-dependent and independent manner. Gingipains (cysteine proteases) are discussed in more depth below, but they function in this case to cleave membrane bound TREM-1 to sTREM-1 (soluble). The whole bacteria increase the expression of TREM-1 as indicated by increased TREM-1 mRNA production. After longer incubation, the membrane expression of TREM-1 decreases while sTREM-1 increases due to Arg-gingipain cleavage of TREM-1 from the membrane.148 The increase in sTREM-1 is thought to increase the inflammatory signaling by increasing IL-8.148 P. gingivalis can also lower the sTREM-1 by Lys-gingipain activity.148 It has been hypothesized that the decision to upregulate Arg- vs Lys-gingipains may be due to nutrient availability. P. gingivalis would increase Arg-ginigpain to promote inflammation associated tissue damage, but upregulate Lys-gingipain if the inflammation becomes deleterious to the bacteria.127,148 However, it could also be a time-dependent regulation; sTREM-1 accumulates at late time points. The local chemokine paralysis and other neutrophil impairing functions of P. gingivalis tend to occur shortly after infection while the inflammation promoting functions occur later in the infection. P. gingivalis could be lowering inflammation initially to disturb the biofilm undetected and then promote inflammation once it has set itself up in the protected niches of the GECs and biofilms (Fig. 2B).
The actual mechanism for surviving the antimicrobial effect of ROS by P. gingivalis has not yet been fully determined.116 In the deep periodontal pocket, the environment is fairly anoxic which may in itself preclude neutrophils to mount an appropriate respiratory burst response.149 P. gingivalis lacks catalase, an enzyme which can consume the hydrogen peroxide (H2O2) produced in the phagosome as a result of the respiratory burst response and detoxify reactive oxygen radicals and protect the bacterium from oxygen-dependent damage, but it does express superoxide dismutase which catalyzes the dismutation of superoxide into H2O2 and provides some protection from oxygen-dependent damage.150 Another enzyme of P. gingivalis, alkyl hydroper-oxide reductase (Ahp), can metabolize H2O2 and provide some protection against the respiratory burst.151,152 However, it is still uncertain how much protection is really afforded to each of these enzymes against the copious amounts of ROS generated by neutrophils during an infection.
One of the major virulence factors produced by P. gingivalis is gingipains.153 Gingipains have been called the principal virulence factor created by P. gingivalis and are involved in modulation of leukocyte responses.153 For example, gingipain R cleaves proteinase-activated receptor on neutrophils resulting in neutrophil activation.154 These endopeptdases are also associated with tissue damage and heme acquisition.153 The heme acquired is used to form a black deposit on the bacteria which may be an oxidative sink to protect the bacteria from leukocyte-generated ROS.153,155 Both types of gingipains, the lysine-specific (Kgp) and the arginine-specific (RgpA), are primarily implicated in heme acquisition and deposition.153 Studies on gingipain activity and the presence of this hemin layer have shown that they are both very important to the bacteria's survival against ROS.156 Mutants that lack this layer show higher degrees of 8oxo-G DNA damage and subsequent attempts by the cell to repair the damage than the wild-type bacteria.156
Beyond the oxygen-dependent mechanisms employed by the neutrophil, they also utilize antimicrobial enzymes such as lysozyme and elastase to curb infection.157 Investigations of the exocytosis of neutrophil granules have shown no major azurophil granule exocytosis due to low levels of extracellular elastase.157 They also show a minimum induction of gelatinase granule exocytosis by measuring extracellular matrix metalloprotease-9.157 Additionally, gingipains are also involved in the breakdown of the neutrophils granule contents.158 Especially in the anoxic environment of the periodontal crevice, the degradation of oxygen-independent antimicrobial peptides contributes greatly to P. gingivalis' survival.
Another function of gingipains (in this case gingipain-1) produced by P. gingivalis is the cleavage of complement protein.159 Complement protein is important to both the opsonization process and the chemotaxis of neutrophils. The generation of complement component 5a (C5a) by gingipain cleavage of C5 results in C5a-mediated neutrophil recruitiment.159 Manipulation of complement and TLR-2 has been shown to occur in P. gingivalis mouse infection and modeled in human neutrophils to inhibit bacterial killing while simultaneously increasing the inflammation.160 Blocking the TLR2 and/or the C5aR in human (HL60) neutrophils results in an inhibition of bacterial killing.160 P. gingivalis acts to specifically uncouple the TLR2-MyD88 pathway which facilitates phagocytosis and subsequent bactericide from the TLR2-Mal-PI3K pathway which increases inflammation.160 This work has not yet been replicated using primary human neutrophils.
3.4 | Neutrophils and their new challenge: coping with the oral pathobionts
The inflamed periodontal tissue is an ideal environment for the growth of asaccharolytc periodontal pathogens because it is a constant source of essential nutrients coming from damaged tissue. Moreover, the inflamed environment with broken homeostasis favors the overgrowth of some bacterial species called ‘infammophilic’ or pathobionts which have a fitness advantage and will try to preserve inflammation at all cost.70 One such emerging pathogen is F. alocis, a gram-positive anaerobic rod. Although cultivable, this organism is slow-growing and difficult to detect by conventional culture-based methodologies. Nonetheless, it is becoming increasingly apparent that the presence of F. alocis is indicative of oral disease, as the organism has been detected significantly more frequently and in higher numbers at periodontal disease sites compared with healthy sites.59,66,161–165 Moreover, it has even been proposed that F. alocis should be included as a diagnostc indicator of disease.166 To date, studies on the pathogenic potential of F. alocis have established that the organism can form biofilms in vivo, preferentially colonizing the apical parts of the gingival pocket in close proximity to the soft tissues.167 Characterization of the different oral bacteria composition of different oral sites like saliva, supragingival and subgingival area, found several bacterial groups with strong co-occurrence pattern increasing together in disease sites and decreasing together in healthy sites implying synergism within the community.168 One of the co-occurrence group formed by eight members is center on F. alocis and some of the group members include two of the putative periodontal pathogens, P. gingivalis and T. forsythia, as well as some of the newly recognized periopathogen Peptoanaerobacter stomatis.168
In vitro, F. alocis can produce trypsin-like proteases169 and is resistant to oxidative stress.170 Study of the interaction with gingival epithelial cells has demonstrated that F. alocis is invasive, both in monoculture and synergistically with P. gingivalis, and it is pro-inflammatory by promoting the release of TNFα, IL-1β, and IL-6.171 Furthermore, F. alocis induces apoptosis in gingival epithelial cells by suppression of MEK1/2 and activation of caspase 3.171 In addition, we have recently shown that F. alocis is able to interact with other oral biofilm bacteria and accumulates in sites rich in F. nucleatum.172 F. alocis can establish colonization in a mouse subcutaneous chamber model, resulting in a quick acute inflammatory response with a peak of neutrophil infiltration 2 hours after infection with concomitant increase of proinflammatory cytokines such as TNFα, IL-6 and IL-1β, and control of the local infection with decreasing bacterial numbers by 2 hours which were completely reduced by 72 hours after infection.173 In addition, heat-killed F. alocis induces CXCL1 expression through NOD1 in mesothelial cells.174 We are beginning to understand the in vivo pathogenic properties of F. alocis and its ability to cause disease may not be solely restricted to the periodontal pocket. Mouse models have also shown the bacteria can spread to other tissues such as the spleen, lung, and kidney tissues with acute kidney damage observed resulting from inflammation.173
F. alocis survives in human neutrophils after 6 hours after challenge, preventing granule recruitiment to the bacteria-containing phagosome and failing to induce the respiratory burst response (J.E. Edmisson, C.L. Armstrong, S. Tian, A. Vashistha, J. Le, R.J. Lamont and S.M. Uriarte, unpublished observations). The studies published thus far begin to establish the pathogenic potential of this newly appreciated oral pathogen.
4 Concluding Remarks
The current evidence indicates that upon changes in the environment, such as smoking, diabetes, stress, antbiotic therapy, or colonization by keystone periodontal pathogens, the majority of the microbial composition identified in health will encounter a hostle niche and be outcompeted by the pathobionts, which have a fitness advantage and will flourish to promote inflammation that will sustain the dysbiotic community (Fig. 2B). Neutrophils are the most abundant leukocyte patrolling the oral cavity and the principal cell involved in preserving the host homeostasis by using its sophistcated ant-microbial artllery toward the constant microbial burden present in the mouth. A century has passed since the death of Elie Metchnikof, whose seminal discoveries of the importance of phagocytosis, inflammation, and the balance between homeostasis and disease, are some of the impressive legacies he lef as the father of innate immunity. Moreover, his remarkable discoveries of the role of the professional phagocytes, macrophages and ‘microphages’, as he initially referred to neutrophils, in phagocytosis and immunity are the steppingstione to the current interest in the gut microbiome.175
Exciting times are nigh since much more research is needed to understand the fascinating interplay between human neutrophils and the oral microbial community. By characterizing this interaction in health and disease, it will be possible to develop new therapeutc strategies to control what triggers the ‘nososymbiocity’ of periodontal disease.
Acknowledgments
This work was supported by a grant from The National Insttute of Dental and Craniofacial Research (DE024509 to S. M. U.). The authors want to thank Richard J. Lamont (University of Louisville) for his valuable critcs and suggestions for this paper. Also, the authors want to thank Irina Miralda (University of Louisville) for her comments and edits to this paper, as well as for her creative thinking for drawing figure 1 in this paper.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Belkaid Y. Tailored immunity at mucosae. Immunol Rev. 2014;260:5–7. doi: 10.1111/imr.12197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belkaid Y, Naik S. Compartimentalized and systemic control of tissue immunity by commensals. Nat Immunol. 2013;14:646–653. doi: 10.1038/ni.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brestoff JR, Arts D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol. 2013;14:676–684. doi: 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyle J, Chapple I. Molecular aspects of the pathogenesis of periodontitis. Periodontol. 2015;200069:7–17. doi: 10.1111/prd.12104. [DOI] [PubMed] [Google Scholar]
- 5.Curtis Michael A, Zenobia C, Darveau Richard P. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe. 2011;10:302–306. doi: 10.1016/j.chom.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zadeh HH, Nichols FC, Miyasaki KT. The role of the cell-mediated immune response to Actnobacillus actinomycetemcomitans and Porphyromonas gingivalis in periodontitis. Periodontol. 1999;200020:239–288. doi: 10.1111/j.1600-0757.1999.tb00163.x. [DOI] [PubMed] [Google Scholar]
- 7.Miyasaki KT. The neutrophil: mechanisms of controlling periodontal bacteria. J Periodontol. 1991;62:761–774. doi: 10.1902/jop.1991.62.12.761. [DOI] [PubMed] [Google Scholar]
- 8.Scott DA, Krauss JL. Neutrophils in periodontal inflammation. Front Oral Biol. 2012;15:56–83. doi: 10.1159/000329672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Micro. 2010;8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 10.Hart TC, Atkinson JC. Mendelian forms of periodontitis. Periodontol. 2007;200045:95–112. doi: 10.1111/j.1600-0757.2007.00233.x. [DOI] [PubMed] [Google Scholar]
- 11.Berezow AB, Darveau RP. Microbial shift and periodontitis. Periodontol. 2011;200055:36–47. doi: 10.1111/j.1600-0757.2010.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mydel P, Takahashi Y, Yumoto H, et al. Roles of the host oxidative immune response and bacterial antioxidant rubrerythrin during Porphyromonas gingivalis infection. PLoS Pathog. 2006;2:e76. doi: 10.1371/journal.ppat.0020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nussbaum G, Shapira L. How has neutrophil research improved our understanding of periodontal pathogenesis? J Clin Periodontol. 2011;38:49–59. doi: 10.1111/j.1600-051X.2010.01678.x. [DOI] [PubMed] [Google Scholar]
- 14.Ryder MI. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontol. 2010;200053:124–137. doi: 10.1111/j.1600-0757.2009.00327.x. [DOI] [PubMed] [Google Scholar]
- 15.Roberts FA, Darveau RP. Microbial protection and virulence in periodontal tissue as a function of polymicrobial communities: symbiosis and dysbiosis. Periodontol. 2015;200069:18–27. doi: 10.1111/prd.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zenobia C, Luo XL, Hashim A, et al. Commensal bacteria-dependent select expression of CXCL2 contributes to periodontal tissue homeostasis. Cell Microbiol. 2013;15:1419–1426. doi: 10.1111/cmi.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bugl S, Wirths S, Radsak MP, et al. Steady-state neutrophil homeostasis is dependent on TLR4/TRIF signaling. Blood. 2013;121:723–733. doi: 10.1182/blood-2012-05-429589. [DOI] [PubMed] [Google Scholar]
- 18.Devine DA, Marsh PD, Meade J. Modulation of host responses by oral commensal bacteria. J Oral Microbiol. 2015;7:1–4. doi: 10.3402/jom.v7.26941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clarke TB. Microbial programming of systemic innate immunity and resistance to infection. PLoS Pathog. 2014;10:e1004506. doi: 10.1371/journal.ppat.1004506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang D, Chen G, Manwani D, et al. Neutrophil ageing is regulated by the microbiome. Nature. 2015;525:528–532. doi: 10.1038/nature15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greer A, Irie K, Hashim A, et al. Site-specific neutrophil migration and CXCL2 expression in periodontal tissue. J Dent Res. doi: 10.1177/0022034516641036. pii: 0022034516641036. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hajishengallis E, Hajishengallis G. Neutrophil homeostasis and periodontal health in children and adults. J Dent Res. 2014;93:231–237. doi: 10.1177/0022034513507956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 24.Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. 2014;15:602–611. doi: 10.1038/ni.2921. [DOI] [PubMed] [Google Scholar]
- 25.Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetc, biochemical, and clinical features of chronic granulomatous disease. Medicine. 2000;79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Matute JD, Arias AA, Wright NAM, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114:3309–3315. doi: 10.1182/blood-2009-07-231498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soler-Palacin P, Margareto C, Llobet P, et al. Chronic granulomatous disease in pediatric patents: 25 years of experience. Allergol Immu-nopathol. 2007;35:83–89. doi: 10.1157/13106774. [DOI] [PubMed] [Google Scholar]
- 28.Mohanty Tirthankar T. A novel mechanism for NETosis provides ant-microbial defense at the oral mucosa. Blood. 2015;126:2128–2137. doi: 10.1182/blood-2015-04-641142. [DOI] [PubMed] [Google Scholar]
- 29.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 30.Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. 2012;198:773–783. doi: 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30:513–521. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 32.Bankvall M, Sjoberg F, Gale G, Wold A, Jontell M, Ostman S. The oral microbiota of patents with recurrent aphthous stomatitis. J Oral Microbiol. 2014;6:25739. doi: 10.3402/jom.v6.25739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolaczkowska E, Kubes P. Neutrophil recruitiment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 34.Soehnlein O, Weber C, Lindbom L. Neutrophil granule proteins tune monocytc cell function. Trends Immunol. 2009;30:538–546. doi: 10.1016/j.it.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Borregaard N, Sørensen OE, Theilgaard-Mönch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol. 2007;28:340–345. doi: 10.1016/j.it.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 36.Uriarte SM, Powell DW, Luerman GC, et al. Comparison of proteins expressed on secretory vesicle membranes and plasma membranes of human neutrophils. J Immunol. 2008;180:5575–5581. doi: 10.4049/jimmunol.180.8.5575. [DOI] [PubMed] [Google Scholar]
- 37.Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, McLeish KR. Proteomic analysis of human neutrophil granules. Mol Cell Proteomics. 2005;4:1503–1521. doi: 10.1074/mcp.M500143-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Rørvig S, Østergaard O, Heegaard NHH, Borregaard N. Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: correlation with transcriptome profiling of neutrophil precursors. J Leukoc Biol. 2013;94:711–721. doi: 10.1189/jlb.1212619. [DOI] [PubMed] [Google Scholar]
- 39.Perera NC, Wiesmüller KH, Larsen MT, et al. NSP4 is stored in azurophil granules and released by activated neutrophils as active endoprotease with restricted specificity. J Immunol. 2013;191:2700–2707. doi: 10.4049/jimmunol.1301293. [DOI] [PubMed] [Google Scholar]
- 40.Sorensen OE, Clemmensen SN, Dahl SL, et al. Papillon-Lefevre syndrome patent reveals species-dependent requirements for neutrophil defenses. J Clin Invest. 2014;124:4539–4548. doi: 10.1172/JCI76009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka D, Miyasaki KT, Lehrer RI. Sensitvity of Actinobacillus actinomycetemcomitans and Capnocytophaga spp. to the bactericidal action of LL-37: a cathelicidin found in human leukocytes and epithelium. Oral Microbiol Immunol. 2000;15:226–231. doi: 10.1034/j.1399-302x.2000.150403.x. [DOI] [PubMed] [Google Scholar]
- 43.Greer A, Zenobia C, Darveau RP. Defensins and LL-37: a review of function in the gingival epithelium. Periodontol. 2013;200063:67–79. doi: 10.1111/prd.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eick S, Puklo M, Adamowicz K, et al. Lack of cathelicidin processing in Papillon-Lefèvre syndrome patents reveals essential role of LL-37 in periodontal homeostasis. Orphanet J Rare Dis. 2014;9:1–11. doi: 10.1186/s13023-014-0148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gorr SU, Abdolhosseini M. Antimicrobial peptides and periodontal disease. J Clin Periodontol. 2011;38:126–141. doi: 10.1111/j.1600-051X.2010.01664.x. [DOI] [PubMed] [Google Scholar]
- 46.Roberts H, White P, Dias I, et al. Characterization of neutrophil function in Papillon-Lefèvre syndrome. J Leukoc Biol. 2016;100:1–12. doi: 10.1189/jlb.5A1015-489R. [DOI] [PubMed] [Google Scholar]
- 47.Hajishengallis G, Moutsopoulos NM, Hajishengallis E, Chavakis T. Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin Immunol. 2016;28:146–158. doi: 10.1016/j.smim.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dixon DR, Reife RA, Cebra JJ, Darveau RP. Commensal bacteria influence innate status within gingival tissues: a pilot study. J Periodontol. 2004;75:1486–1492. doi: 10.1902/jop.2004.75.11.1486. [DOI] [PubMed] [Google Scholar]
- 49.Darveau RP. The oral microbial consortum's interaction with the periodontal innate defense system. DNA Cell Biol. 2009;28:389–395. doi: 10.1089/dna.2009.0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eke PI, Dye BA, Wei L, et al. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol. 2015;86:611–622. doi: 10.1902/jop.2015.140520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. 2015;15:30–44. doi: 10.1038/nri3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paster BJ, Olsen I, Aas JA, Dewhirst FE. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol. 2006;200042:80–87. doi: 10.1111/j.1600-0757.2006.00174.x. [DOI] [PubMed] [Google Scholar]
- 53.Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol. 2012;27:409–419. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dewhirst FE, Chen T, Izard J, et al. The human oral microbiome. J Bacteriol. 2010;192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wade WG. Has the use of molecular methods for the characterization of the human oral microbiome changed our understanding of the role of bacteria in the pathogenesis of periodontal disease? J Clin Periodontol. 2011;38(Suppl 11):7–16. doi: 10.1111/j.1600-051X.2010.01679.x. [DOI] [PubMed] [Google Scholar]
- 56.Griffen AL, Beall CJ, Firestone ND, et al. CORE: a phylogenetically-curated 16S rDNA database of the core oral microbiome. PLoS ONE. 2011;6:e19051. doi: 10.1371/journal.pone.0019051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721–5732. doi: 10.1128/JCM.43.11.5721-5732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griffen AL, Beall CJ, Campbell JH, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012;6:1176–1185. doi: 10.1038/ismej.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar PS, Griffen AL, Barton JA, Paster BJ, Moeschberger ML, Leys EJ. New bacterial species associated with chronic periodontitis. J Dent Res. 2003;82:338–344. doi: 10.1177/154405910308200503. [DOI] [PubMed] [Google Scholar]
- 60.Duran-Pinedo AE, Frias-Lopez J. Beyond microbial community composition: functional activities of the oral microbiome in health and disease. Microbes Infect. 2015;17:505–516. doi: 10.1016/j.micinf.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hajishengallis G, Lamont RJ. Dancing with the stars: how choreographed bacterial interactions dictate nososymbiocity and give rise to keystone pathogens, accessory pathogens, and pathobionts. Trends Microbiol. 2016;24:477–489. doi: 10.1016/j.tim.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Short FL, Murdoch SL, Ryan RP. Polybacterial human disease: the ills of social networking. Trends Microbiol. 2014;22:508–516. doi: 10.1016/j.tim.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res. 1994;8:263–271. doi: 10.1177/08959374940080022001. [DOI] [PubMed] [Google Scholar]
- 64.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 65.Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontol. 2005;200038:135–187. doi: 10.1111/j.1600-0757.2005.00107.x. [DOI] [PubMed] [Google Scholar]
- 66.Kumar PS, Leys EJ, Bryk JM, Martinez FJ, Moeschberger ML, Griffen AL. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantiative 16S cloning and sequencing. J Clin Microbiol. 2006;44:3665–3673. doi: 10.1128/JCM.00317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med. 2015;21:172–183. doi: 10.1016/j.molmed.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Costalonga M, Herzberg MC. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol Lett. 2014;162:22–38. doi: 10.1016/j.imlet.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duran-Pinedo AE, Chen T, Teles R, et al. Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis. ISME J. 2014;8:1659–1672. doi: 10.1038/ismej.2014.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol. 2014;35:3–11. doi: 10.1016/j.it.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saito I, Komiyama K, Moro I, et al. Ultrastructural and immunocytochemical characterization of polymorphonuclear leukocytes from gingival crevice in man. J Periodontol. 1987;58:493–497. doi: 10.1902/jop.1987.58.7.493. [DOI] [PubMed] [Google Scholar]
- 72.Crawford JM, Wiltion JMA, Richardson P. Neutrophils die in the gingival crevice, periodontal pocket, and oral cavity by necrosis and not apoptosis. J Periodontol. 2000;71:1121–1129. doi: 10.1902/jop.2000.71.7.1121. [DOI] [PubMed] [Google Scholar]
- 73.Vitkov L, Klappacher M, Hannig M, Krautgartner WD. Extracellular neutrophil traps in periodontitis. J Periodontal Res. 2009;44:664–672. doi: 10.1111/j.1600-0765.2008.01175.x. [DOI] [PubMed] [Google Scholar]
- 74.Laugisch O, Schacht M, Guentsch A, et al. Periodontal pathogens affect the level of protease inhibitors in gingival crevicular fluid. Mol Oral Microbiol. 2012;27:45–56. doi: 10.1111/j.2041-1014.2011.00631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jan P, Aneta S, Takahisa I, James T. Gingipains, the major cysteine proteinases and virulence factors of Porphyromonas gingivalis: structure, function and assembly of multdomain protein complexes. Curr Protein Pept Sci. 2003;4:397–407. doi: 10.2174/1389203033487036. [DOI] [PubMed] [Google Scholar]
- 76.Potempa J, Pike RN. Corruption of innate immunity by bacterial proteases. J Innate Immun. 2009;1:70–87. doi: 10.1159/000181144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Swain SD, Rohn TT, Quinn MT. Neutrophil priming in host defense: role of oxidants as priming agents. Antoxid Redox Signal. 2002;4:69–83. doi: 10.1089/152308602753625870. [DOI] [PubMed] [Google Scholar]
- 78.Uriarte SM, Rane MJ, Luerman GC, et al. Granule exocytosis contributes to priming and activation of the human neutrophil respiratory burst. J Immunol. 2011;187:391–400. doi: 10.4049/jimmunol.1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fernandez-Botran R, Uriarte SM, Arnold FW, et al. Contrasting inflammatory responses in severe and non-severe community-acquired pneumonia. Inflammation. 2014;37:1158–1166. doi: 10.1007/s10753-014-9840-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brown GE, Silver GM, Reiff J, Allen RC, Fink MP. Polymorphonuclear neutrophil chemiluminescence in whole blood from blunt trauma patents with multiple injuries. J Trauma. 1999;46:297–305. doi: 10.1097/00005373-199902000-00017. [DOI] [PubMed] [Google Scholar]
- 81.Eggleton P, Wang L, Penhallow J, Crawford N, Brown KA. Differences in oxidative response of subpopulations of neutrophils from healthy subjects and patents with rheumatoid arthritis. Ann Rheum Dis. 1995;54:916–923. doi: 10.1136/ard.54.11.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mathews JB, Wright HJ, Roberts A, Cooper PR, Chapple IL. Hyper-activity and reactivity of peripheral blood neutrophils in chronic periodontitis. Clin Exp Immunol. 2007;147:255–264. doi: 10.1111/j.1365-2249.2006.03276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hasegawa Y, Mans JJ, Mao S, et al. Gingival epithelial cell transcriptional responses to commensal and opportunistic oral microbial species. Infect Immun. 2007;75:2540–2547. doi: 10.1128/IAI.01957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ximénez-Fyvie LA, Haffajee AD, Socransky SS. Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J Clin Periodontol. 2000;27:648–657. doi: 10.1034/j.1600-051x.2000.027009648.x. [DOI] [PubMed] [Google Scholar]
- 85.Wright HJ, Chapple ILC, Mathews JB, Cooper PR. Fusobacterium nucleatum regulation of neutrophil transcription. J Periodontal Res. 2011;46:1–12. doi: 10.1111/j.1600-0765.2010.01299.x. [DOI] [PubMed] [Google Scholar]
- 86.Fredriksson MI, Gustafsson AK, Bergström KG, Åsman BE. Constitutionally hyperreactive neutrophils in periodontitis. J Periodontol. 2003;74:219–224. doi: 10.1902/jop.2003.74.2.219. [DOI] [PubMed] [Google Scholar]
- 87.Dias IH, Mathews JB, Chapple IL, Wright HJ, Dunstion CR, Griifiths HR. Activation of the neutrophil respiratory burst by plasma from periodontitis patents is mediated by pro-inflammatory cytokines. J Clin Periodontol. 2011;38:1–7. doi: 10.1111/j.1600-051X.2010.01628.x. [DOI] [PubMed] [Google Scholar]
- 88.Wright HJ, Mathews JB, Chapple ILC, Ling-Mountord N, Cooper PR. Periodontitis associates with a type 1 IFN signature in peripheral blood neutrophils. J Immunol. 2008;181:5775–5784. doi: 10.4049/jimmunol.181.8.5775. [DOI] [PubMed] [Google Scholar]
- 89.Ling MR, Chapple IL, Mathews JB. Peripheral blood neutrophil cytokine hyper-reactivity in chronic periodontitis. Innate Immunity. 2015;21:714–725. doi: 10.1177/1753425915589387. [DOI] [PubMed] [Google Scholar]
- 90.Aboodi GM, Goldberg MB, Glogauer M. Refractory periodontitis population characterized by a hyperactive oral neutrophil phenotype. J Periodontol. 2010;82:726–733. doi: 10.1902/jop.2010.100508. [DOI] [PubMed] [Google Scholar]
- 91.Lakschevitz FS, Aboodi GM, Glogauer M. Oral neutrophil transcriptome changes result in a pro-survival phenotype in periodontal diseases. PLoS ONE. 2013;8:e68983. doi: 10.1371/journal.pone.0068983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lakschevitz FS, Hassanpour S, Rubin A, Fine N, Sun C, Glogauer M. Identfication of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Exp Cell Res. 2016;342:200–209. doi: 10.1016/j.yexcr.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 93.Roberts HM, Ling MR, Insall R, et al. Impaired neutrophil directional chemotactic accuracy in chronic periodontitis patents. J Clin Periodontol. 2015;42:1–11. doi: 10.1111/jcpe.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brissette CA, Pham TTT, Coats SR, Darveau RP, Lukehart SA. Treponema denticola does not induce production of common innate immune mediators from primary gingival epithelial cells. Oral Microbiol Immunol. 2008;23:474–481. doi: 10.1111/j.1399-302X.2008.00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dashper SG, Seers CA, Tan KH, Reynolds EC. Virulence factors of the oral spirochete Treponema denticola. J Dent Res. 2011;90:691–703. doi: 10.1177/0022034510385242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sela MN. Role of Treponema denticola in periodontal diseases. Crit Rev Oral Biol Med. 2001;12:399–413. doi: 10.1177/10454411010120050301. [DOI] [PubMed] [Google Scholar]
- 97.Puthengady Thomas B, Sun CX, Bajenova E, Ellen RP, Glogauer M. Modulation of human neutrophil functions in vitro by Treponema denticola major outer sheath protein. Infect Immun. 2006;74:1954–1957. doi: 10.1128/IAI.74.3.1954-1957.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Amin M, Ho ACS, Lin JY, Batista da Silva AP, Glogauer M, Ellen RP. Induction of de novo subcortical actin filament assembly by Treponema denticola major outer sheath protein. Infect Immun. 2004;72:3650–3654. doi: 10.1128/IAI.72.6.3650-3654.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Batista da Silva AP, Lee W, Bajenova E, McCulloch CAG, Ellen RP. The major outer sheath protein of Treponema denticola inhibits the binding step of collagen phagocytosis in fibroblasts. Cell Microbiol. 2004;6:485–498. doi: 10.1111/j.1462-5822.2004.00377.x. [DOI] [PubMed] [Google Scholar]
- 100.Magalhães MAO, Sun CX, Glogauer M, Ellen RP. The major outer sheath protein of Treponema denticola selectively inhibits Rac1 activation in murine neutrophils. Cell Microbiol. 2008;10:344–354. doi: 10.1111/j.1462-5822.2007.01045.x. [DOI] [PubMed] [Google Scholar]
- 101.Baker MJ, Pan D, Welch HCE. Small GTPases and their guanine-nucleotide exchange factors and GTPase-activating proteins in neutrophil recruitiment. Curr Opin Hematol. 2016;23:44–54. doi: 10.1097/MOH.0000000000000199. [DOI] [PubMed] [Google Scholar]
- 102.Godovikova V, Goetting-Minesky MP, Fenno JC. Composition and localization of Treponema denticola outer membrane complexes. Infect Immun. 2011;79:4868–4875. doi: 10.1128/IAI.05701-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yamazaki T, Miyamoto M, Yamada S, Okuda K, Ishihara K. Surface protease of Treponema denticola hydrolyzes C3 and influences function of polymorphonuclear leukocytes. Microbes Infect. 2006;8:1758–1763. doi: 10.1016/j.micinf.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 104.Jusko M, Potempa J, Karim AY, et al. A metalloproteinase karilysin present in the majority of Tannerella forsythia isolates inhibits all pathways of the complement system. J Immunol. 2012;188:2338–2349. doi: 10.4049/jimmunol.1101240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yoo JY, Kim HC, Zhu W, et al. Identification of Tannerella forsythia antigens specifically expressed in patents with periodontal disease. FEMS Microbiol Lett. 2007;275:344–352. doi: 10.1111/j.1574-6968.2007.00906.x. [DOI] [PubMed] [Google Scholar]
- 106.Gosling PT, Gemmell E, Carter CL, Bird PS, Seymour GJ. Immunohistological analysis of Tannerella forsythia-induced lesions in a murine model. Oral Microbiol Immunol. 2005;20:25–30. doi: 10.1111/j.1399-302X.2004.00188.x. [DOI] [PubMed] [Google Scholar]
- 107.Posch G, Sekot G, Friedrich V, et al. Glycobiology aspects of the periodontal pathogen Tannerella forsythia. Biomolecules. 2012;2:467–482. doi: 10.3390/biom2040467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Settem RP, Honma K, Nakajima T, et al. A bacterial glycan core linked to surface (S)-layer proteins modulates host immunity through Th17 suppression. Mucosal Immunol. 2013;6:415–426. doi: 10.1038/mi.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jusko M, Potempa J, Mizgalska D, et al. A metalloproteinase mirolysin of Tannerella forsythia inhibits all pathways of the complement system. J Immunol. 2015;195:2231–2240. doi: 10.4049/jimmunol.1402892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Koziel J, Karim AY, Przybyszewska K, et al. Proteolytic inactivation of LL-37 by karilysin, a novel virulence mechanism of Tannerella forsythia. J Innate Immun. 2010;2:288–293. doi: 10.1159/000281881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ksiazek M, Mizgalska D, Enghild JJ, Scavenius C, Thogersen IB, Potempa J. Miropin, a novel bacterial serpin from the periodontopathogen Tannerella forsythia, inhibits a broad range of proteases by using different peptide bonds within the reactive center loop. J Biol Chem. 2015;290:658–670. doi: 10.1074/jbc.M114.601716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Palmer LJ, Chapple ILC, Wright HJ, Roberts A, Cooper PR. Extracellular deoxyribonuclease production by periodontal bacteria. J Periodontal Res. 2012;47:439–445. doi: 10.1111/j.1600-0765.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- 114.Amulic B, Hayes G. Neutrophil extracellular traps. Curr Biol. 2011;21:R297–R298. doi: 10.1016/j.cub.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 115.Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10:717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hajishengallis G, Lamont RJ. Breaking bad: manipulation of the host response by Porphyromonas gingivalis. Eur J Immunol. 2014;44:328–338. doi: 10.1002/eji.201344202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Odell EW, Wu PJ. Susceptibility of Porphyromonas gingivalis and P. asaccharolytica to the non-oxidative killing mechanisms of human neutrophils. Arch Oral Biol. 1992;37:597–601. doi: 10.1016/0003-9969(92)90121-n. [DOI] [PubMed] [Google Scholar]
- 118.Eick S, Pfister W, Sigusch B, Straube E. Phagocytosis of periodontopathogenic bacteria by crevicular granulocytes is depressed in progressive periodontitis. Infection. 2000;28:301–304. doi: 10.1007/s150100070023. [DOI] [PubMed] [Google Scholar]
- 119.Vitkov L, Klappacher M, Hannig M, Krautgartner WD. Neutrophil fate in gingival crevicular fluid. Ultrastruct Pathol. 2010;34:25–30. doi: 10.3109/01913120903419989. [DOI] [PubMed] [Google Scholar]
- 120.Guentsch A, Puklo M, Preshaw PM, et al. Neutrophils in chronic and aggressive periodontitis in interaction with Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans. J Periodontal Res. 2009;44:368–377. doi: 10.1111/j.1600-0765.2008.01113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mysak J, Podzimek S, Sommerova P, et al. Porphyromonas gingivalis: major periodontopathic pathogen overview. J Immunol Res. 2014;2014:476068. doi: 10.1155/2014/476068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Belanger M, Rodrigues PH, Dunn WA, Jr, Progulske-Fox A. Autophagy: a highway for Porphyromonas gingivalis in endothelial cells. Autophagy. 2006;2:165–170. doi: 10.4161/auto.2828. [DOI] [PubMed] [Google Scholar]
- 123.Darveau RP, Belton CM, Reife RA, Lamont RJ. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 1998;66:1660–1665. doi: 10.1128/iai.66.4.1660-1665.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tonetti MS, Imboden MA, Lang NP. Neutrophil migration into the gingival sulcus is associated with transepithelial gradients of inter-leukin-8 and ICAM-1. J Periodontol. 1998;69:1139–1147. doi: 10.1902/jop.1998.69.10.1139. [DOI] [PubMed] [Google Scholar]
- 125.Takeuchi H, Hirano T, Whitmore SE, Morisaki I, Amano A, Lamont RJ. The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-kappaB RelA/p65. PLoS Pathog. 2013;9:e1003326. doi: 10.1371/journal.ppat.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 127.Olsen I, Hajishengallis G. Major neutrophil functions subverted by Porphyromonas gingivalis. J Oral Microbiol. 2016;8:30936. doi: 10.3402/jom.v8.30936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Van Dyke TE, Bartholomew E, Genco RJ, Slots J, Levine MJ. Inhibition of neutrophil chemotaxis by soluble bacterial products. J Periodontol. 1982;53:502–508. doi: 10.1902/jop.1982.53.8.502. [DOI] [PubMed] [Google Scholar]
- 129.Rotstein OD, Pruett TL, Fiegel VD, Nelson RD, Simmons RL. Succinic acid, a metabolic by-product of Bacteroides species, inhibits polymorphonuclear leukocyte function. Infect Immun. 1985;48:402–408. doi: 10.1128/iai.48.2.402-408.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rotstein OD, Nasmith PE, Grinstein S. The bacteroides by-product succinic acid inhibits neutrophil respiratory burst by reducing intracellular pH. Infect Immun. 1987;55:864–870. doi: 10.1128/iai.55.4.864-870.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vinolo MA, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem. 2011;22:849–855. doi: 10.1016/j.jnutbio.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 132.Yoshimura A, Hara Y, Kaneko T, Kato I. Secretion of IL-1 beta, TNF-alpha, IL-8 and IL-1ra by human polymorphonuclear leukocytes in response to lipopolysaccharides from periodontopathic bacteria. J Periodontal Res. 1997;32:279–286. doi: 10.1111/j.1600-0765.1997.tb00535.x. [DOI] [PubMed] [Google Scholar]
- 133.Yoshimura A, Kaneko T, Kato Y, Golenbock DT, Hara Y. Lipopolysaccharides from periodontopathic bacteria Porphyromonas gingivalis and Capnocytophaga ochracea are antagonists for human toll-like receptor 4. Infect Immun. 2002;70:218–225. doi: 10.1128/IAI.70.1.218-225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sugita N, Kimura A, Matsuki Y, Yamamoto T, Yoshie H, Hara K. Activation of transcription factors and IL-8 expression in neutrophils stimulated with lipopolysaccharide from Porphyromonas gingivalis. Inflammation. 1998;22:253–267. doi: 10.1023/a:1022344031223. [DOI] [PubMed] [Google Scholar]
- 135.Iino Y, Hopps RM. The bone-resorbing activities in tissue culture of lipopolysaccharides from the bacteria Actinobacillus actinomycetemcomitans, Bacteroides gingivalis and Capnocytophaga ochracea isolated from human mouths. Arch Oral Biol. 1984;29:59–63. doi: 10.1016/0003-9969(84)90043-8. [DOI] [PubMed] [Google Scholar]
- 136.Murray DA, Wilton JMA. Lipopolysaccharide from the periodontal pathogen Porphyromonas gingivalis prevents apoptosis of HL60-derived neutrophils in vitro. Infect Immun. 2003;71:7232–7235. doi: 10.1128/IAI.71.12.7232-7235.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schwartz JT, Barker JH, Kaufman J, Fayram DC, McCracken JM, Allen LA. Francisella tularensis inhibits the intrinsic and extrinsic pathways to delay constitutive apoptosis and prolong human neutrophil lifespan. J Immunol. 2012;188:3351–3363. doi: 10.4049/jimmunol.1102863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wilton JM, Hurst TJ, Carman RJ, Macey MG. Effects of Porphyromonas gingivalis culture products on human polymorphonuclear leukocyte function. FEMS Microbiol Immunol. 1990;2:285–293. doi: 10.1111/j.1574-6968.1990.tb03531.x. [DOI] [PubMed] [Google Scholar]
- 139.Scragg MA, Turton LR, Newman HN, Williams DM. Effects of Porphyromonas gingivalis culture products on the morphology of peripheral blood polymorphonuclear leucocytes from periodontitis patents and healthy subjects. J Clin Periodontol. 1995;22:585–590. doi: 10.1111/j.1600-051x.1995.tb00809.x. [DOI] [PubMed] [Google Scholar]
- 140.Wilton J, Hurst T, Scot E. Inhibition of polymorphonuclear leucocyte phagocytosis by Porphyromonas gingivalis culture products in patents with adult periodontitis. Arch Oral Biol. 1993;38:285–289. doi: 10.1016/0003-9969(93)90134-8. [DOI] [PubMed] [Google Scholar]
- 141.Carvalho RPM, Mesquita JS, Bonomo A, Elsas PX, Colombo APV. Relationship of neutrophil phagocytosis and oxidative burst with the subgingival microbiota of generalized aggressive periodontitis. Oral Microbiol Immunol. 2009;24:124–132. doi: 10.1111/j.1399-302X.2008.00484.x. [DOI] [PubMed] [Google Scholar]
- 142.Lala A, Amano A, Sojar HT, Radel SJ, Denardin E. Porphyromonas gingivalis trypsin-like protease: a possible natural ligand for the neutrophil formyl peptide receptor. Biochem Biophys Res Commun. 1994;199:1489–1496. doi: 10.1006/bbrc.1994.1399. [DOI] [PubMed] [Google Scholar]
- 143.Börgeson E, Lönn J, Bergström I, et al. Lipoxin A4 inhibits Porphyromonas gingivalis-induced aggregation and reactive oxygen species production by modulating neutrophil-platelet interaction and CD11b expression. Infect Immun. 2011;79:1489–1497. doi: 10.1128/IAI.00777-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Maderna P, Godson C. Lipoxins: resolutionary road. Br J Pharmacol. 2009;158:947–959. doi: 10.1111/j.1476-5381.2009.00386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pouliot M, Clish CB, Petasis NA, Van Dyke TE, Serhan CN. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: a role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry. 2000;39:4761–4768. doi: 10.1021/bi992551b. [DOI] [PubMed] [Google Scholar]
- 146.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 147.Kantarci A, Van Dyke TE. Lipoxin signaling in neutrophils and their role in periodontal disease. Prostaglandins Leukot Essent Fatty Acids. 2005;73:289–299. doi: 10.1016/j.plefa.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 148.Bostanci N, Thurnheer T, Aduse-Opoku J, Curtis MA, Zinkernagel AS, Belibasakis GN. Porphyromonas gingivalis regulates TREM-1 in human polymorphonuclear neutrophils via its gingipains. PLoS ONE. 2013;8:e75784. doi: 10.1371/journal.pone.0075784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Thompson HL, Wilton JM. Effects of anaerobiosis and aerobiosis on interactions of human polymorphonuclear leukocytes with the dental plaque bacteria Streptococcus mutans, Capnocytophaga ochracea, and Bacteroides gingivalis. Infect Immun. 1991;59:932–940. doi: 10.1128/iai.59.3.932-940.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Choi JI, Takahashi N, Kato T, Kuramitsu HK. Isolation, expression, and nucleotide sequence of the sod gene from Porphyromonas gingivalis. Infect Immun. 1991;59:1564–1566. doi: 10.1128/iai.59.4.1564-1566.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Johnson NA, Liu Y, Fletcher HM. Alkyl hydroperoxide peroxidase subunit C (ahpC) protects against organic peroxides but does not affect the virulence of Porphyromonas gingivalis W83. Oral Microbiol Immunol. 2004;19:233–239. doi: 10.1111/j.1399-302X.2004.00145.x. [DOI] [PubMed] [Google Scholar]
- 152.Diaz PI, Zilm PS, Wasinger V, Corthals GL, Rogers AH. Studies on NADH oxidase and alkyl hydroperoxide reductase produced by Porphyromonas gingivalis. Oral Microbiol Immunol. 2004;19:137–143. doi: 10.1111/j.0902-0055.2004.00120.x. [DOI] [PubMed] [Google Scholar]
- 153.Sheets SM, Robles-Price AG, McKenzie RM, Casiano CA, Fletcher HM. Gingipain-dependent interactions with the host are important for survival of Porphyromonas gingivalis. Front Biosci. 2008;13:3215–3238. doi: 10.2741/2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lourbakos A, Chinni C, Thompson P, et al. Cleavage and activation of proteinase-activated receptor-2 on human neutrophils by gingipain-R from Porphyromonas gingivalis. FEBS Lett. 1998;435:45–48. doi: 10.1016/s0014-5793(98)01036-9. [DOI] [PubMed] [Google Scholar]
- 155.Smalley JW, Silver J, Marsh PJ, Birss AJ. The periodontopathogen Porphyromonas gingivalis binds iron protoporphyrin IX in the mu-oxo dimeric form: an oxidative buffer and possible pathogenic mechanism. Biochem J. 1998;331(Pt 3):681–685. doi: 10.1042/bj3310681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Johnson NA, McKenzie R, McLean L, Sowers LC, Fletcher HM. 8-oxo-7,8-dihydroguanine is removed by a nucleotide excision repair-like mechanism in Porphyromonas gingivalis W83. J Bacteriol. 2004;186:7697–7703. doi: 10.1128/JB.186.22.7697-7703.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ding Y, Haapasalo M, Kerosuo E, Lounatmaa K, Kotiranta A, Sorsa T. Release and activation of human neutrophil matrix metallo- and serine proteinases during phagocytosis of Fusobacterium nucleatum, Porphyromonas gingivalis and Treponema denticola. J Clin Periodontol. 1997;24:237–248. doi: 10.1111/j.1600-051x.1997.tb01837.x. [DOI] [PubMed] [Google Scholar]
- 158.How KY, Song KP, Chan KG. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front Microbiol. 2016;7:53. doi: 10.3389/fmicb.2016.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wingrove JA, DiScipio RG, Chen Z, Potempa J, Travis J, Hugli TE. Activation of complement components C3 and C5 by a cysteine proteinase (gingipain-1) from Porphyromonas (Bacteroides) gingivalis. J Biol Chem. 1992;267:18902–18907. [PubMed] [Google Scholar]
- 160.Maekawa T, Krauss JL, Abe T, et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe. 2014;15:768–778. doi: 10.1016/j.chom.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hutter G, Schlagenhauf U, Valenza G, et al. Molecular analysis of bacteria in periodontitis: evaluation of clone libraries, novel phylotypes and putative pathogens. Microbiology. 2003;149:67–75. doi: 10.1099/mic.0.25791-0. [DOI] [PubMed] [Google Scholar]
- 162.Kumar PS, Griffen AL, Moeschberger ML, Leys EJ. Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. J Clin Microbiol. 2005;43:3944–3955. doi: 10.1128/JCM.43.8.3944-3955.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Colombo APV, Boches SK, Cotton SL, et al. Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J Periodontol. 2009;80:1421–1432. doi: 10.1902/jop.2009.090185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Colombo APV, Bennet S, Cotton SL, et al. Impact of periodontal therapy on the subgingival microbiota of severe periodontitis: comparison between good responders and individuals with refractory periodontitis using the human oral microbe identification microarray. J Periodontol. 2012;83:1279–1287. doi: 10.1902/jop.2012.110566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shaddox LM, Huang H, Lin T, et al. Microbiological characterization in children with aggressive periodontitis. J Dent Res. 2012;91:927–933. doi: 10.1177/0022034512456039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dahlen G, Leonhardt A. A new checkerboard panel for testing bacterial markers in periodontal disease. Oral Microbiol Immunol. 2006;21:6–11. doi: 10.1111/j.1399-302X.2005.00243.x. [DOI] [PubMed] [Google Scholar]
- 167.Schlafter S, Riep B, Griffen AL, et al. Filifactor alocis – involvement in periodontal biofilms. BMC Microbiol. 2010;10:1–13. doi: 10.1186/1471-2180-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chen H, Liu Y, Zhang M, et al. A Filifactor alocis-centered cooccurrence group associates with periodontitis across different oral habitats. Sci Rep. 2015;5:9053. doi: 10.1038/srep09053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Aruni AW, Roy F, Sandberg L, Fletcher HM. Proteome variation among Filifactor alocis strains. Proteomics. 2012;12:3343–3364. doi: 10.1002/pmic.201200211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Aruni AW, Roy F, Fletcher HM. Filifactor alocis has virulence attributes that can enhance its persistence under oxidative stress conditions and mediate invasion of epithelial cells by Porphyromonas gingivalis. Infect Immun. 2011;79:3872–3886. doi: 10.1128/IAI.05631-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Moffatt CE, Whitmore SE, Griffen AL, Leys EJ, Lamont RJ. Filifactor alocis interactions with gingival epithelial cells. Mol Oral Microbiol. 2011;26:365–373. doi: 10.1111/j.2041-1014.2011.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang Q, Wright CJ, Dingming H, Uriarte SM, Lamont RJ. Oral community interactions of Filifactor alocis in vitro. PLoS ONE. 2013;8:e76271. doi: 10.1371/journal.pone.0076271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wang Q, Jotwani R, Le J, et al. Filifactor alocis infection and inflammatory responses in the mouse subcutaneous chamber model. Infect Immun. 2014;82:1205–1212. doi: 10.1128/IAI.01434-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Marchesan J, Jiao YZ, Schaff RA, et al. TLR4, NOD1 and NOD2 mediate immune recognition of putative newly identified periodontal pathogens. Mol Oral Microbiol. 2015;31:243–258. doi: 10.1111/omi.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gordon S. Phagocytosis: an immunobiologic process. Immunity. 2016;44:463–475. doi: 10.1016/j.immuni.2016.02.026. [DOI] [PubMed] [Google Scholar]